

Abstract
We reviewed mechanisms that determine reactive oxygen species (redox) homeostasis, redox information signaling and metabolic/regulatory function of autocrine insulin signaling in pancreatic β cells, and consequences of oxidative stress and dysregulation of redox/information signaling for their dysfunction. We emphasize the role of mitochondrion in β cell molecular physiology and pathology, including the antioxidant role of mitochondrial uncoupling protein UCP2. Since in pancreatic β cells pyruvate cannot be easily diverted towards lactate dehydrogenase for lactate formation, the respiration and oxidative phosphorylation intensity are governed by the availability of glucose, leading to a certain ATP/ADP ratio, whereas in other cell types, cell demand dictates respiration/metabolism rates. Moreover, we examine the possibility that type 2 diabetes mellitus might be considered as an inevitable result of progressive self-accelerating oxidative stress and concomitantly dysregulated information signaling in peripheral tissues as well as in pancreatic β cells. It is because the redox signaling is inherent to the insulin receptor signaling mechanism and its impairment leads to the oxidative and nitrosative stress. Also emerging concepts, admiting participation of redox signaling even in glucose sensing and insulin release in pancreatic β cells, fit in this view. For example, NADPH has been firmly established to be a modulator of glucose-stimulated insulin release.
1. Why to Deal with Redox Homeostasis in Pancreatic β Cells
Due to its complex health and economic sequels as well as steadily increasing prevalence, type 2 diabetes mellitus (T2DM) represents one of the serious burdens of the 21th century. Its pathogenesis is complex and different factors may prevail in individual cases. The typical feature of progressed T2DM is insulin resistance as well as β cell dysfunction [1, 2]. Excellent recent reviews cover gathered knowledge of all aspects of pancreatic β cell biology, development, molecular physiology, and medical aspects [1–19]. A great progress has been achieved in understanding molecular mechanism of physiological phenomena [1–5], etiology of T2DM and medical aspects or treatment [6–9], microscopic anatomy of human islets of Langerhans [10], and their in vivo imaging [11], as well as in understanding β cell biology, specifically longevity and development and differentiation of β cells [13–19]. This paper attempts to focus on aspects that determine β cell dysfunction and possess a common denominator in oxidative stress origin and/or dysregulated information signaling, while emphasizing dysregulated redox signaling. The impact is striking, since redox signaling is the inherent part of β cell physiology and contributes to, for example, insulin secretion. We left out of scope β cell development, cell cycle, longevity and differentiation, attempts to produce β cells from stem cells, and extensive description of pathophysiology. We had a chance only to touch a topic of information signaling and its dysregulation as it substantiates a subject for another extensive review. Due to the same reason, we leave also the themes of nitrosative stress and lipotoxicity which are, however, closely related to the oxidative stress. In turn, we focus more closely on some recent emerging aspects, such as possible signal modulating role of mitochondrial uncoupling protein UCP2 [20], reviewing and emphasizing the role of mitochondrion in β cell molecular physiology as well as pathology. Likewise, we focus on action of insulin itself on the β cell, that is, autocrine insulin secretion and its link to the redox homeostasis. We strictly distinguish mitochondrial and cytosolic sources of reactive oxygen species (ROS) and their antioxidant defense, and when possible, we distinguish also mitochondrial versus cytosolic redox regulations.
Why to deal with redox homeostasis in pancreatic β cells at all? Are not the above described emerging aspects of β cell biology superior to our focus? The answer lies in the necessity to establish, whether T2DM is the inevitable result of progressive self-accelerating oxidative stress [21, 22] and concomitant progressively dysregulated information signaling, including redox signaling that both lead to diabetic complications. This view becomes even more skeptical, when one realizes that redox signaling manifested by transient ROS burst at least locally is an inherent part of numerous molecular mechanisms, some of which will be reviewed here. Thus redox signaling is inherent to mechanism of insulin receptor signaling and emerging concepts admit its role even during glucose sensing and insulin release in pancreatic β cells [4]. For example, the role of NADPH has been already firmly established in modulation of insulin release. One may consider NADPH as antioxidant since it is an important metabolite usually shifting redox homeostasis towards the reduced state. However, when used by NADPH oxidases to produce ROS, it becames an evil of the pro-oxidant side. It is not surprising that H2O2 is another such key molecule and in further description we shall recognize many more metabolites and proteins with the Janus angel/devil double face.
2. Mitochondrial Generation and Scavenging of Reactive Oxygen Species (ROS) in Pancreatic β Cells
2.1. Mitochondrial ROS Sources
Likewise in other cell types, mitochondrial respiratory chain is the main source of superoxide (O2 ●−, and its conjugated acid-hydroperoxyl radical, HO2 ●, pKa 4.9) in mitochondrion of pancreatic β cells [21]. Specifically, Complex I, an H+-pumping NADH: quinone oxidoreductase, produces maximum superoxide only when both electron transport and H+ pumping are retarded [22, 23]. H+ pumping may be attenuated by highly established electrochemical gradient of protons at IMM (termed proton motive force, Δp, when expressed in mV units) or inhibited by oxidative stress-related mutations of ND5 subunit (or other mitochondrion-coded subunits) [22]. Intermediate O2 ●− formation results from fully reduced flavin as reported for isolated Complex I [24–26]. Binding of rotenone and similar inhibitors in proximity to the Q-site (a ubiquinone binding site) highly retards electron transport throughout the peripheral arm of Complex I. This was originally ascribed to the formation of longer-lived semiquinone species having a higher probability of reacting with oxygen which thus would form O2 ●− [27]. Detailed mechanism of O2 ●− formation within Complex I and its relation to H+-pumping have yet to be established. It is well recognized, however, that nearly all Complex I-produced O2 ●− is released to the matrix compartment [27]. Complex III, a ubiquinol-cytochrome c reductase, contributes to O2 ●− generation by autooxidation of the ubisemi-quinone anion radical (UQ●−) within so-called Q cycle [21, 27, 28], while it releases O2 ●− about equally to both sides of the inner mitochondrial membrane (IMM) [28, 29].
A fast electron flux via the whole respiratory chain at a high substrate pressure (NADH/NAD+ ratio) produces more O2 ●− than under conditions, when slower flux occurs at the same relative retardation (same oxidation/reduction states). Hence, in intact respiratory chain, mostly effectors that retard cytochrome c turnover between Complex III and IV (cytochrome c oxidase), slow down Q cycle or Q migration between Complex I and III, accelerate superoxide production [30].
2.2. Mild Uncoupling Attenuates Mitochondrial ROS Generation
The oxidative phosphorylation (OXPHOS) terminating at ATP synthase (Complex V) is driven by the proton motive force, Δp, formed by the respiratory chain H+ pumping at Complex I, III, and IV. The IMM part of ATP synthase, so-called FOATPase, thus consumes the adequate portion of Δp in non-dormant mitochondrion in a state, historically termed state-3. In vivo cell mitochondrial respiration is governed by the metabolic state and/or availability of substrates, and one can recognize various states-3 differing by distinct respiration rates, depending on the substrate load. A state-4, never existing in cells, is then given by zero ATP synthesis, hence by zero H+ backflux via the FOATPase. Respiration and H+ pumping at state-4 is given only by the other protonophores, either of protein character or by the native H+ permeability of IMM. As in mitochondrion Δp is predominantly in a form of IMM electrical potential, it is valid that ΔΨm is maximum at state-4 at the maximum substrate load. The H+ backflux excluding FOATPase is termed an H+ leak. Also other proteins may contribute to the H+ leak, such as the ADP/ATP carrier. A short-cut of proton circuit within IMM is also known as uncoupling and can be physiologically provided also by mitochondrial uncoupling proteins (UCPs) [20, 31–34]. When UCP-mediated protonophore activity plus IMM H+ leak does not overwhelm the FOATPase protonophoric activity, ATP synthesis, hence OXPHOS, still takes place. Such a mild uncoupling (mild in contrast to a complete uncoupling by agents termed uncouplers) is, however, beneficial in terms of lowering mitochondrial O2 ●− formation. The O2 ●− formation in both Complex I [22] and Complex III [27–29, 35] is diminished by mild uncoupling. Due to a relative predominance of mitochondrial ROS source within the cell, one can predict that even accumulated oxidative stress might be attenuated by mild uncoupling. Note, however, that oxidative stress originating from irreversible changes due to mutated subunits encoded by mitochondrial DNA (mtDNA) cannot be improved by mild uncoupling [22]. An example is given by certain mutations of ND5 subunit of Complex I (ensuring H+ pumping in intact wt form) that inhibit H+ pumping and lead to increased O2 ●− formation. Such a block is not withdrawn by uncoupling [22]. In conclusion, the retardation of H+ pumping which accelerates Complex I O2 ●− formation rater initiates further turn of a vicious spiral of self-accelerated oxidative stress.
2.3. Uncoupling Protein UCP2 Attenuates Mitochondrial ROS Generation
Among five UCP isoforms, UCP2 was identified in pancreatic β cells and it has been deduced that UCP2 exerts an important antioxidant role in β cells while preventing excessive superoxide formation within the respiratory chain [20]. There are still, however, controversies, on how UCP2-mediated uncoupling is initiated, since mutually incompatible models for the uncoupling mechanism of UCP2 (or other UCPs) have been developed [36–39]. In fact, the functional roles of UCP2 that were originally suggested—including the attenuation of ROS production [40–42], regulation of GSIS [42–44] (see Section 3.1), and regulation of Ca2+ levels in mitochondria [45–47]—are in dispute. We have previously documented the fatty acid (FA) cycling model [39] using reconstituted UCPs into liposomes [37, 38, 48–53] and black lipid membranes [54–57] and demonstrated that transport of polyunsaturated FAs (PUFAs), including hydroperoxy FAs [52] is faster [51, 56, 57]. According to the FA cycling model, FA anions are the true substrates transported by UCP2 and other UCPs [53, 57]. After protonation on the trans-side of the IMM, protonated FAs are internalized into the lipid bilayer core and subsequently flip to the cis-side of IMM and thus carry a proton across the membrane [38]. Opposing models have postulated a pathway that requires only protons, for which FAs are enhancers of basal H+ transport [36, 40]. Lipid peroxidation products, for example, 4-hydroxy-2-nonenal, may also act as enhancers of proton transport by chemical modification of UCPs [58]; however, recently, we have provided an evidence that they do so only when FAs are present (Pohl E, Jabůrek M, et al., unpublished).
In pancreatic β cells it has been observed that the UCP2-mediated mild uncoupling decreases the yield of ATP from glucose [43, 59]. Further studies suggested superoxide activation of UCP2-mediated uncoupling on the basis of observation of elevated ΔΨm in islets treated with a superoxide dismutase (SOD) mimetic manganese [III] tetrakis (4-benzoic acid) porphyrin (MnTBAP) or overexpressing MnSOD, absent in islets from UCP2 KO mice [60]. Upon presumed inhibition of UCP2-mediated uncoupling by Genipin, ΔΨm increased in wt islets but not in UCP2 KO islets [61]. UCP2 overexpression in INS-1 cells attenuated IL1β-induced ROS formation [62]. With UCP2 silencing, a mild uncoupling in mitochondria isolated from INS-1E cells was linked to UCP2, while accounting for up to 30% of H+ leak [63]. UCP2-mediated uncoupling was detectable also in intact INS-1E cells as compared to those silenced for UCP2 [64]. In turn, Galetti et al. could not demonstrate any effect of UCP2 overexpression on mitochondrial coupling in INS-1 cells, neither after oleate addition [65]. Chronic absence of UCP2 in UCP2 KO mice of three highly congenic strain backgrounds caused oxidative stress reflected by decreased GSH/GSSG ratio in blood or examined tissues while their islets had elevated levels of antioxidant enzymes and increased nitrotyrosine content [66]. Pancreatic β cells from UCP2 KO mice had chronically higher ROS when compared to wt mice, as estimated by dihydro-dichlorofluorescein diacetate fluorescent probe (CM-H2DCFDA, further abbreviated DCF) [67]. Mice with selective knock-out of UCP2 in pancreatic β cells (UCP2BKO mice) exhibited somewhat increased glucose-induced ΔΨm [20]. UCP2BKO mice had also elevated intracellular ROS levels as determined by DCF [20]. These results comply with the antioxidant function of UCP2-mediated mild uncoupling. UCP2 may also modulate redox signaling, if could be effectively switched on and off.
2.4. Mitochondrial Superoxide Dismutases and Glutathione Peroxidases
O2 ●− in the matrix is converted to H2O2 by matrix MnSOD [68], while O2 ●− released to the intramembrane space (IMS) is partly dismuted by IMS CuZnSOD [69, 70]. Any residual O2 ●− which diffuses into the cytosol is similarly converted by the cytosolic CuZnSOD. If any mitochondrial O2 ●− can reach the extracellular space, it is then detoxified by extracellular CuZnSOD (SOD3). Once H2O2 is produced, it can easily penetrate through membranes, thanks to its uncharged property and poor reactivity. H2O2 can be degraded by peroxisomal catalase (rare in mitochondria with exception of the heart) and by three isoforms of selenium-dependent glutathione peroxidases (GPX) which, with a cofactor glutathione, convert H2O2 to water and also convert free FAOOHs to their corresponding hydroxy acids (FAOH). The fourth isoform, GPX4, specifically acts on hydroperoxy groups of peroxidized phospholipid side chains and on cholesterol hydroperoxides [71, 72]. Resulted GSSG is reduced to GSH by glutathione reductase with NADPH as a cofactor. Mitochondria have their own GSH pools independent of the cytosolic GSH pool. Both key mitochondrial ROS detoxifying enzymes MnSOD and GPX were demonstrated to be essential not only for balancing redox homeostasis but also for insulin secretion [73, 74].
3. Mitochondrial Redox Homeostasis and Glucose-Stimulated Insulin Secretion (GSIS)
3.1. Tuned Oxidative Phosphorylation (OXPHOS) as Determinant of Glucose-Stimulated Insulin Secretion (GSIS) but Also Mitochondrial ROS Generation
The intimately specific feature of pancreatic β cells lies in glucose sensing through the oxidative phosphorylation [75–79]. Respiration and OXPHOS rates, leading to a certain ATP/ADP ratio, are governed by the availability of glucose, whereas in most other cell types, cell demand dictates respiration/metabolism rates and the ATP/ADP ratio. It is because of a specific enzyme/regulation pattern of β cells. At first, unlike in numerous other cell types, pyruvate cannot be diverted towards lactate dehydrogenase for lactate formation in β cells. Consequently, glucose cannot be metabolized by anaerobic glycolysis, which provides so-called Warburg phenotype in cancer cells and under physiological cell responses to hypoxia and other adaptations [80, 81]. Thus nearly 100% of glucose is metabolized by OXPHOS in β cells (likewise in hepatocytes and numerous differentiated OXPHOS cells). The pattern of pyruvate dehydrogenase kinase genes is surely responsible for this. Thus, β cell PDK1 and PDK3 are “constitutively blocked” [82], and PDK2 is “inefficient” so that it does not phosphorylate PDH E1α subunit of pyruvate dehydrogenase (PDH), hence does not inhibit its activity. At low basal glucose, PDH is 90% active, whereas at maximum glucose PDH is inhibited only by 22% [82]. Also hexokinase IV (glucokinase) in β cells is not inhibited by glucose-6-phosphate like in for example, skeletal muscle cells [83]. The lack of such a feedback inhibition of glycolysis directly connects glycolysis to pyruvate. Finally, the human glucose transporter GLUT1 or rodent GLUT2 are not dependent on insulin [84, 85], so glucose in β cell cytosol is proportional to bloodstream glucose [86]. This is perfect setting for a sensor.
Consequently, glucose metabolism in β cells is finely adjusted to the blood glucose levels [87]. At starvation with ~3 mM glucose levels, β cell respiration is relatively low, as well as the intensity of ATP synthesis, corresponding to the established state-3[Glc = 3 mM] [88–90]. The ΔΨm is still lower than would be at state-4 with 3 mM glucose. Increasing glucose intake into β cells may increase up to OXPHOS-saturating ~12 to 15 mM glucose, when maximum OXPHOS takes places with the established state-3max, maximum respiration and maximum ΔΨm [90]. The resulting increased ATP/ADP ratio in the cell cytosol initiates closure of plasma membrane ATP-sensitive K+ channels [1, 91, 92], leading to plasma membrane depolarization and opening of voltage-sensitive Ca2+ channels [1, 93]. Increased cytosolic Ca2+ initiates insulin granule exocytosis [3, 94–96]. The above description represents a simplified schema of glucose-stimulated insulin secretion (GSIS). It has been hypothesized that β cells maintain a relatively high [ATP]/[ADP] value even in low glucose and that glucose metabolism leads to dramatically decreased free ADP with only modestly increased ATP [97]. If a high [ATP]/[ADP] ratio exists even at low glucose levels, as a result, the total adenine nucleotide concentration is unchanged during a glucose-induced elevation.
GSIS was also reported to be modulated or accelerated by other metabolic pathways related to mitochondria, such as phosphocreatine shuttle, additional Ca2+ signaling due to glutamate metabolism [98, 99], citrate export [100], phosphoenolpyruvate [101], and pyruvate cycling [102, 103]. A common denominator in these modulations is NADPH, the role of which on insulin secretion has yet to be established. Overall, GSIS possesses also a component due to the autocrine function of insulin (see Sections 3.3 and 5.2).
3.2. ROS Generation Dependence on Glucose
For cells not completely depleted of glucose we hypothesize (Figure 1) that the release of superoxide to the mitochondrial matrix upon the GSIS onset is diminished with regard to release rates at lower glucose concentrations. GSIS should simultaneously result in a decrease of the mitochondrial oxidative stress. The incremental increase of electron flow through the respiratory chain is not high at ~3 mM glucose, and its raise due to a further glucose intake is relatively lower when compared with the effect of H+ backflow via the FO part of ATP synthase that elevates respiration (classic respiratory control for isolated mitochondria). Thus the effect of elevated OXPHOS intensity prevails and ROS production is attenuated. This should be valid also for decrease of mitochondrial ROS formation with decreasing ADP, hence increasing ATP [97] and has been experimentally observed [104]. In turn, at extensive glucose depletion, the effect of substrate load (a directly proportional increase in superoxide formation, e.g., on Complex I, with increasing NADH or respiration) should overcome the suppressing role of H+ returning via FOATPase at higher intensity of OXPHOS. Hence, experimentally, results of increasing mitochondrial ROS upon GSIS might be observed using DCF [105] or other means [106, 107] as well as increasing reducing equivalents [108].
Figure 1.
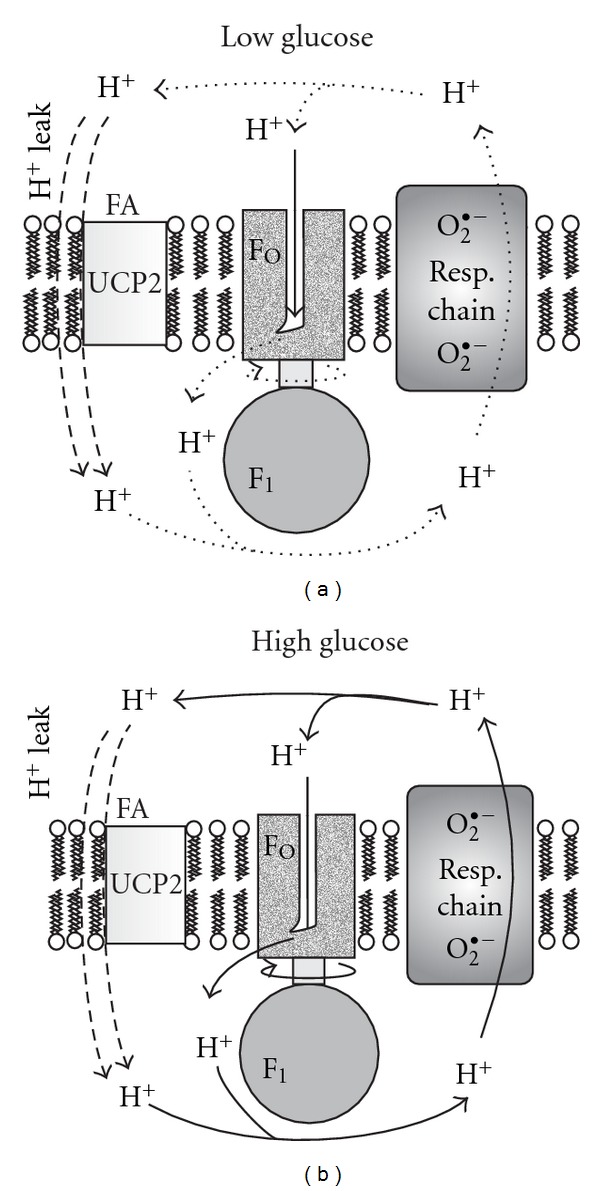
Mitochondrial superoxide formation should decrease upon a sudden glucose increase. Schemas depict the basic proton circuit within the inner mitochondrial membrane at low and high glucose, while higher rates, and therefore higher respiration rates, are depicted by thicker arrows. Thus the right schema depicts situation at a sudden glucose intake when state-3 respiration is maximum, as well as ATP synthesis, and hence also H+ backflux through the FO part of ATP synthase. Under these conditions superoxide formation within the respiratory chain complexes I and III is low (depicted by smaller fonts) and should be lower than at low glucose (left schema) at slower coupled respiration rate, where much higher superoxide should be formed (depicted by bigger fonts).
Since H2O2 of mitochondrial origin may readily access cytosol, one may report on mitochondrial ROS contribution, when measuring cytosolic ROS sensitive to mitochondrial inhibitors [105]. As explained above, a various extent of glucose depletion may provide distinct outcome in ROS assays, which are further dependent on the employed probe. Thus using dihydroethidium fluorescent monitoring in primary rat β cells, Martens et al. have found that unlike in non β cells, oxidative stress diminishes with increasing glucose upon GSIS [109]. ROS decrease monitored by DCF in isolated Langerhans islets upon GSIS has also been indicated [110]. Other laboratories have reported increases in ROS upon GSIS [105–107]. Note, that insulin secretion in INS1 cells was also induced by exogenous H2O2 and diethyl maleate [111], or by mono-oleoyl-glycerol [112] which both elevate intracellular H2O2.
3.3. Autocrine Insulin and Mitochondrial ROS Generation
Autocrine insulin has acute (4 hour) effects on GSIS in healthy humans [113]. Studies of Poderoso group have pointed out an emerging role of mitochondrial NO synthase (mtNOS) activated upon insulin signaling via the Akt-2/protein-kinase-B-mediated phosphorylation in skeletal muscle [114]. Released nitric oxide, a freely permeable radical, NO●, having a half-life of 1 to 10 s, causes a mild oxidative and nitrosative stress but also transiently diminishes respiration. In skeletal muscle and liver NO● can facilitate conversion of glucose to glycogen. Experimentally, it has been proven by a sustained insulin dosage that the insulin-Akt-2-mtNOS pathway mediates NO● burst in skeletal muscle [114]. Also, nitric oxide donors increase glucose uptake in primary human skeletal muscle cells [115]. Signaling via phosphatidylinositol-3-kinase (PI3K) (and hence downstream Akt-2 signaling) was responsible for insulin receptor activation by nonpeptidyl mimetic L-783,281 which inhibited GSIS as well as basal insulin secretion in human islets of Langerhans [116]. Also a direct observation in isolated mitochondria that insulin signaling regulates mitochondrial function in β cells has been reported [117].
Since pancreatic β cells contain a functional insulin receptor [117–120], an acute autocrine insulin signaling may lead to the similar acute effects as in skeletal muscle and liver, besides chronic positive effects on stimulation of β cell proliferation [118], hence being beneficial for regulation of adult β cell mass. Transgenic mice lacking insulin receptor in pancreatic β cells (βIRKO mice) exhibited increased apoptosis, decreased proliferation, and reduced β cell mass [119]. The insulin receptor has also been found essential for islet compensatory growth response to insulin resistance [120]. There are two arms of autocrine insulin signaling via insulin receptor, the Raf-1 kinase arm and the Akt kinase arm. Insulin stimulates primary β cell proliferation via Raf-1 kinase and suppresses apoptosis. The Akt arm increases β cell mass and improves glucose tolerance. A signalosome complex of glucokinase, pro-apoptotic protein, Bcl-2-associated death promoter, BADS, and protein kinase A has been reduced in βIRKO mice, thus linking a lack of autocrine insulin with development of type 2 diabetes [117].
If mtNOS is indeed activated upon insulin signaling in β cells, the predicted outcome may substantiate different roles than in skeletal muscle cells and hepatocytes, just due to impossibility to switch to a partial aerobic glycolysis and provide a spectrum of anaplerotic pathways. The released NO● may transiently inhibit Complex I and cytochrome c oxidase. NO● may also reacting with superoxide, thus forming peroxynitrite which can further act against otherwise diminishing mitochondrial superoxide production.
4. Key Players Contributing to Cytosolic ROS Homeostasis in Pancreatic β Cells
4.1. Cytosolic ROS Sources in Pancreatic β Cells
Among the cytosolic ROS sources in pancreatic β cells, a family of NADPH oxidases (NOX) is the most important as the major plasma membrane or cytosolic superoxide sources. They catalyze the one-electron reduction of oxygen to generate O2 ●−, while utilizing NADPH as electron donor. Isoforms NOX1, NOX2, and NOX4 may play a significant role in β cells [121, 122], hypothetically related to GSIS regulation and cell integrity [123]. Decreased NOX2 expression may contribute to regulatory mechanisms diminishing ROS upon high levels of metabolism [123]. NOX enzymes are composed by six hetero-subunits, which must associate, usually in a stimulus-dependent manner [124], with exception of constitutively assembled NOX4. Malic enzyme conversion of malate to pyruvate [125] or mitochondrial shuttles [100–103] may provide NADPH for NOX enzymes, since there is a relatively low pentose phosphate pathway activity in β cells. The catalytic core is formed by the two integral membrane protein subunits gp91phox and one p22phox plus by flavocytochrome b558. Additional subunits, p67phox, p47phox, p40phox, and the small GTPase Rac, are located in the cytosol during the resting state and upon activation assemble with the core [124]. Enzyme activation is initiated by p47phox phosphorylation through various protein kinases, such as protein kinase C (PKC) [124, 126, 127]. The upregulation of gp91phox and p22phox was demonstrated in β cells from rodent models of type 2 diabetes [128]. Another ROS source may be provided by cytochrome 450 enzymes such as CYP2E1 which determines mechanism of ketone-stimulated insulin release in pancreatic β cells [129]. Peroxisomes in β cells contribute also to endoplasmic reticulum (ER) stress [130].
4.2. Cytosolic Redox Buffers and Antioxidant Enzymes in Pancreatic β Cells
Redox buffers and antioxidant enzymes detoxify the produced ROS and frequently exert specific roles in β cell ROS homeostasis. Thus an increased antioxidant output from the pentose-phosphate pathway was suggested to decrease ROS upon GSIS [131]. Acute reduction in ROS by glucose was correlated with the increased pentose-phosphate pathway activity [132]. Catalase, glutathione peroxidase (GPX), and superoxide dismutase (SOD1 or CuZnSOD) represent the three of most important intracellular antioxidant enzymes, a primary defense system. However, the expression and activity of antioxidant enzymes is low in rodent β cells compared to other organs [132]. This property increases their susceptibility to an oxidative insult. When compared to liver content, pancreatic islets contain only 1% catalase, 2% GPX and 29% SOD1 activites [73, 133–135]. β cells posses also low repair machinery for oxidatively damaged DNA [136]. In turn, β cells are rich in peroxidase-based antioxidant defenses, such as glutaredoxin and thioredoxin [137]. Human β cells seem to be less prone to oxidative stress than are rodent β cells, possibly because they have greater catalase and SOD activity [138]. Yet, GPX activity is poorly detectable in human islets [139]. Besides vitamin E (α-tocopherol), ascorbate, and uric acid, among small antioxidant molecules, glutathione provides an important mechanism protecting the β cells against oxidative damage [140, 141]. Glutathione, present in mM concentrations, is kept in the reduced state (GSH) by glutathione reductase. GSH transfers its reducing equivalents to ascorbate, GPX, and glutaredoxins.
The main protein antioxidant defense is composed of disulfide reductases namely, thioredoxin (TRX), glutaredoxin (GRX), peroxiredoxins (PRX) and glutamate-cysteine ligase. Thioredoxin represents a disulfide reductase for protein sulfhydryl groups, maintaining proteins in the reduced state [142]. Thioredoxin reductase uses electrons from NADPH and regenerates oxidized TRX. Similarly, glutaredoxin reductase-2 [143] reduces H2O2 or hydroperoxy-FA lipid chains to water or hydroxyFA lipid chains, respectively, on expense of conversion of GSH to oxidized glutathione GSSG, which is regenerated by glutathione reductase. Peroxiredoxins are a family of thiol peroxide reductases which uses TRX or other thiol-containing proteins to clear H2O2 or lipid peroxides [144]. Peroxiredoxin reaction product is sulfenic acid. At the TRX shortage, peroxiredoxin is inactivated to PRX-SO2 [145], which can be reversed by sulfiredoxins, at expense of ATP, yielding PRX-SOH.
Interestingly, being localized at peri-plasma membrane cytosol, glutaredoxin GRX1 has been also implicated in modulation of Ca2+-dependent insulin exocytosis, which was suppressed by GRX1 silencing [143]. The stimulatory action of NADPH on the exocytotic machinery was found to correlate with ~30% inhibition in whole-cell Ca2+ currents. Upon GRX1 silencing, NADPH did not amplify insulin release, but still inhibited Ca2+ currents [143].
4.3. Redox Information Signaling
The deviation in redox state towards pro-oxidation or reduction is always given by balance between production of ROS and antioxidant defense. Since in pancreatic β cells mainly NADPH-dependent systems operate, such as the thioredoxin or glutaredoxin system, the elevated ROS may activate stress-sensitive second messengers such as p38MAPK, JNK [146], and PKC [147]. Also, the transcription factors MAF-A and PDX1 participating in β cell proliferation and insulin biosynthesis were shown to be sensitive to oxidative stress [148, 149]. Moreover, the evidence of redox signaling exists in GSIS of pancreatic β cells. First, the exocytosis of insulin, namely, soluble NSF attachment protein receptor (SNARE) complex, is significantly reduced upon H2O2 treatment [150]. NADPH as an important component of antioxidant defense system was also proposed as a parallel mediator GSIS or modulator of canonical GSIS mechanism, since an increase in the NADPH pool is usually accompanied by the increase in insulin granule exocytosis [137]. In the β cell cytosol, NADPH is formed by reduction of its oxidized counterpart NADP via pyruvate cycling pathways mediated by cytosolic malic enzyme (ME1) and cytosolic isocitrate dehydrogenase (IDH1) as well as via glucose-6-phosphate dehydrogenase, the rate limiting enzyme of the pentose phosphate pathway shuttle. In mitochondria, NADPH is regenerated via NADP dependent reduction mediated by ME3 and mitochondrial IDH2 as well as via nicotinamide nucleotide transhydrogenase [151]. All above reactions change only reduced/oxidized form of NADPH, without changes in total NADPH and NADP pool. This can be performed through NAD kinase whose single cytosolic isoform was found to regulate insulin secretion in β cells [152]. NAD kinase was found to be activated by glucose stimulated increase in Ca2+. Because NAD kinase is cytosolic, the produced NADP(H) can be used by other NADP/NADP(H) dependent enzymes. Also NADPH oxidases (implicated in GSIS) belong to NADPH consuming enzymes working in pro-oxidant mode. In conclusion, the redox couple NAD(P)/NAD(P)H plays an important role for GSIS. A pro-oxidative state can also induce ER stress, which can further impair β cell function by activation of PERK to decrease insulin synthesis [153].
5. Oxidative Stress in Pancreatic β Cells and Its Role in Type 2 Diabetes
5.1. Type 2 Diabetes Mellitus
The progressed T2DM is manifested by both insulin resistance in peripheral tissues as well as β cell dysfunction [1, 2, 6–9, 154–157]. Insulin resistance in skeletal muscle and fat tissue, increased liver gluconeogenesis, abnormal secretion of incretins and impaired central regulation of food intake and energy expenditure are indicated for T2DM [1, 2, 6–9, 153–159]. Overt hyperglycemia results mainly from an interaction between insulin resistance in the peripheral tissues and failing insulin secretory capacity. In both cases, the metabolic abnormalities typical for diabetes are linked to insufficient β cell mass, which is unconditional in type 1 DM (T1DM) or may be only relative in T2DM. The impaired glucose tolerance and later diabetes are manifested in the progressed stage, when already pancreatic β cells cannot defeat increased metabolic demands and their function fails. It is proposed that impaired GSIS might be a primary cause or, alternatively, it may result from the globally deregulated metabolism.
A clear disproportion between fuel intake and energy expenditure in T2DM etiology suggests participation of a metabolic disorder. This may be reflected also by impairment of redox homeostasis, impairment of insulin signaling, and redox signaling and dysfunctions at mitochondrial level in both, primarily (or earlier) in peripheral insulin-sensitive tissue, but also may play a significant role in failing pancreatic β cells [153–159]. Description of emerging role of redox signaling and ROS in β cell biogenesis and maintenance of β cell mass and in its loss in diabetes is out scope of this paper. However, we emphasize that progressive oxidative stress does not represent only chronic exposure to ROS per se, leading to oxidation of proteins, lipids, and DNA, notably mitochondrial DNA that results in further turn of self-accelerating metabolic deterioration. Progressive oxidative stress also impairs redox signaling [4, 131, 149, 160–163], insulin signaling [1, 2, 74, 164], autocrine insulin signaling [117–120], and housekeeping mechanisms of cells, namely autophagy and mitochondria-specific autophagy, mitophagy [5, 165–167], besides initiating an inappropriate apoptosis [168–170]. Another component of oxidative stress comes from intake of excessive fatty acids and lipid peroxidation products, generally termed as lipotoxicity [88, 171–173]. Yet another component results from elevated blood glucose as is known as glucotoxicity affecting β cells as well [174]. It can be mentioned that distinct early events of impairment may converge upon T2DM progress towards the same consequences (Figure 2).
Figure 2.
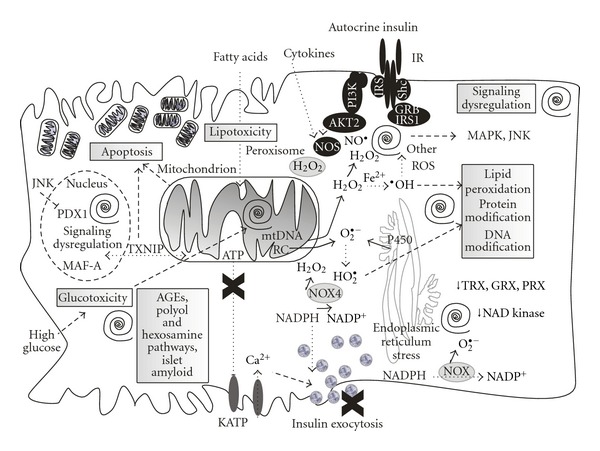
Vicious spirals of repeating self-accelerating oxidative stress and dysregulated redox and information signaling as possible causes of type 2 diabetes. Schema of cell events that occur at type 2 diabetes development, as related to oxidative stress and impaired redox homeostasis and signaling, dysfunctional insulin signaling in peripheral tissues, and autocrine insulin signaling in failing pancreatic β cells. Considered “vicious spirals” (depicted by black spirals) of progressive oxidative stress leading to oxidation of proteins, lipids, and DNA, notably mitochondrial DNA, all resulting in further turn of self-accelerating metabolic deterioration and specifically impairment of the glucose sensing. Progressive oxidative stress also impairs redox signaling and autocrine insulin signaling which further deteriorates fitness of β cells and their housekeeping mechanisms, specifically autophagy and mitochondria-specific autophagy, mitophagy, besides initiating an inappropriate apoptosis. Another component of oxidative stress comes from the intake of excessive fatty acids and lipid peroxidation products, generally termed as lipotoxicity. Yet another component results from elevated blood glucose as is known as glucotoxicity further accelerating cell oxidative stress, impairing cell maintenance, dysregulating information signaling and leading to advanced glycation end products (AGEs), (yet further accelerating oxidative stress and other cell stresses), activating polyol pathway and thus again contributing to pro-oxidation redox homeostasis, activating hexosamine pathway and dysregulating crucial survival pathways including insulin receptor (autocrine) signaling, and finally enhancing glycosylation and forming antiparallel crossed β-pleated sheet structure called amylin-derived islet amyloid, promoting β cell cytotoxicity.
5.2. Mitochondrial Oxidative Stress
The impaired mitochondrial function belongs to key dysfunctions leading to insulin resistance and diabetes progression [1, 2, 4, 5, 123, 131, 132, 154–159, 175–191]. Typically, the decrease of extent between minimum and maximum OXPHOS leads to impaired GSIS. Mitochondrial dysfunction and excessive oxidative stress of mitochondrial origin may lead to lipid accumulation and peroxidation, shifts of the cellular redox balance towards oxidative stress, to the endoplasmic reticulum stress, and to a secondary inflammatory response [159]. All these events take place not only in peripheral tissues but also in β cells [156–159, 175–177]. Whereas energy metabolism of peripheral tissues in relation to T2DM etiology has been intensively investigated, β cell bioenergetics, metabolism, and pathogenesis related to T2DM are not fully understood.
The first target of oxidative stress in mitochondrion is mtDNA [158, 159, 177–184] and its maintenance proteins and proteins of mtDNA transcription and replication machinery [185, 186]. It is exemplified for Goto Kakizaki rats, a T2DM model, characterized by an onset of Langerhans islet pathology, indicated by islet hypertrophy with decreasing the number of insulin-secreting β cells [187, 188]. Indeed, degradation of mtDNA in the remaining β cells has been found [189, 190] as well as fragmented mitochondrial reticulum morphology [191]. Especially, mtDNA variants in the coding and control regions can have combined effects influencing T2DM development [178]. MtDNA encodes seven subunits of respiratory chain, Complex I ND1 to ND6 and ND4L, cyt b (subunit of Complex III), three subunits of Complex IV, that is, cytochrome c oxidase, subunits 1, 2, 3, and ATP synthase subunits 6 and 8, plus 22 tRNAs and two ribosomal RNAs. Certain mtDNA mutations in these mt genes should lead to oxidative stress and initiate β cell dysfunction [184], such as in the heart [192]. Thus an ATP8 subunit mutation has been associated with increased mitochondrial superoxide generation, impaired GSIS, and increased β cell mass adaptation [179]. Excessive ROS due to mtDNA mutation can induce apoptosis [180]. Similar to Goto Kakizaki rats, age-related decline in mtDNA copy number has been indicated in human Langerhans islets [183]. Nevertheless, a general link between clinically found mtDNA mutations and β cell dysfunction is not yet established (e.g., [182]). The reason lies in robust mtDNA genetics, when heteroplasmy has to exceed enormous threshold until deleterious outcomes arise. However, when the important feature of mtDNA genetics is impaired, such as impaired mtDNA maintenance protein transcription factor B1, mitochondrial, TFB1M, there is a risk of T2DM development [185]. Hence mitochondrial disorder of rather metabolic origin prevails in T2DM development [158, 177].
5.3. Cytosolic Oxidative Stress
Likewise in mitochondrion, when ROS sources exceed the antioxidant defense, oxidative stress prevails. Depending on the shift from the physiologically tolerant ROS homeostasis even mild oxidative stress may activate ROS-sensitive information signaling. Persistent oxidative stress is deleterious due to accumulation of oxidized proteins, lipids, and DNA. In pancreatic β cells an antioxidant defense can be considered as low. However, it may be set so to provide redox regulations involved in GSIS and β cell housekeeping processes.
Under diabetic conditions, oxidative stress markers have been frequently detected in β cells, such as 8-hydroxy-2′-deoxyguanosine (8-OHdG) reporting on DNA oxidation [136, 193, 194], 4-hydroxy-2,3-nonenal, that is, one of lipid peroxidation end-products [193, 195] or nitrotyrosine [194]. Studies using diabetic models showed improved insulin sensitivity by antioxidants [161]; however, antioxidant benefits to diabetic patient treatments may not be extrapolated to normal subjects for preventive purposes, since overly diminishing intracellular ROS by excessively high antioxidant enzyme activities deregulate GSIS.
As described in Section 4.2, β cell function may be easily impaired under yet mild oxidative stress. Such stress imposes also activation of ROS-sensitive second messengers, such as p38 mitogen-activated protein kinase, p38MAPK [146], or c-Jun N-terminal kinase, JNK/SAPK [148]. Activation of JNK pathway during oxidative stress results in decreased insulin gene expression by affecting the DNA binding activity of the epigenetic regulation of transcriptional factor pancreatic duodenal homeobox (PDX1). Thus, in turn, the beneficial effect of antioxidants on diabetic patient could be explained besides the protection of oxidative destructions of macromolecules also by maintenance of PDX1. PDX1 plays pivotal role in proliferation, survival, and function of β cells and activation of insulin gene expression [148, 196]. The epigenetic regulation of PDX1 involves histone acetylation of H3 and H4, which helps to remodel the chromatin in the PDX1 promotor to form more accessible structure for transcription and to maintain high level of functional PDX1 [196]. This promotes β cell differentiation and insulin synthesis for compensating insulin resistance. Also direct H2O2 effect on PDX1 was found to be induced through specific phosphorylation on Ser61 and/or Ser66, resulting in an increasing degradation rate and decreasing half-life of the protein [197]. PDX1 protein is also regulated through FOXA2 activator. SOD1 promotor was shown to contain four binding sites for FOXA2 [198, 199].
The cytokine-induced β cell dysfunction and apoptosis is also based on ROS-induced intracellular signaling pathways [200, 201]. Cytokine-generated ROS induce expression of inducible nitric oxide synthase (iNOS) which results in NO● release and translocation of nuclear factor-κB (NFκB). In turn, NFκB induces NADPH oxidase as a major cytosolic ROS source. Recently, thiredoxin-interacting protein (TXNIP) was found to shuttle between nucleus and mitochondrion to which migrates upon oxidative stress and promotes apoptosis via matrix ASK1-induced release of cytochrome c [202]. This complies with results of TXNIP-KO mice studies [203, 204], in which streptozocin treatment is 50-fold less prone to apoptosis [203]. TXNIP also mediates ER stress induced β cell death and has numerous implications in diabetes development [205, 206].
5.4. Consequences of Chronic High Glucose
One would need to conclude that some important physiological aspects and numerous cell regulations in pancreatic β cells are dependent on glucose [174]. Surprisingly concentrations outside of physiological stimulatory range of 3 to ~10 mM glucose (“GSIS range”) are deleterious especially when exposed to β cells at prolonged time. Thus, even low glucose can stimulate oxidative stress via AMPK activation [207]. Glucose at high end of GSIS range is one of the most important stimuli for β cell mass maintenance by stimulating proliferation, neogenesis and hypertrophy [174], most specifically via autocrine insulin signaling [117–120]. We may refer to glucotoxicity at much higher glucose levels, leading to effects that overwhelm the beneficial glucose “maintenance effects.” Thus, for example, hyperglycemia deteriorates β cells after islets transplantation [208]. However, a hallmark of glucotoxicity is that hyperglycemia causes a profound oxidative stress that is possible to attenuate by overexpression of proteins of antioxidant defense [148, 193, 209–211]. Activation of JNK and impairment of PDX1 function (cf. above) belong to one of mechanisms involved. Also ER stress comes from glucotoxicity [206]. The classic pathway of glucotoxicity comes from the spontaneous reactions of glucose and other sugars with amine residues of proteins, lipids, and nucleic acids forming so-called advanced glycation end products (AGE) [212]. Polyol pathway is activated when excess glucose is converted to sorbitol in the presence of aldose reductase, consuming NADPH and thus contributing to pro-oxidation state [213]. By increased flux of glucose via so-called hexosamine pathway, resulting in induction of O-glycosylation of signaling molecules, a crucial survival pathway is dysregulated leading to oxidative stress, as insulin receptor and insulin receptor substrates/PI3 kinase pathway [214]. Also elevated diacylglycerol under hyperglycemia activates protein kinase C which subsequently activates NADPH oxidase and ROS production [127]. Finally, persistent hyperglycemia/ROS exposure enhances glycosylation, thus unfolding of some proteins, lipids and nucleic acids, notably alters a 37 amino acid islet amyloid polypeptide (IAPP), termed amylin. Resulting antiparallel crossed β-pleated sheet structure called amylin-derived islet amyloid (ADIA) is sensitive to free radical polymerization and thus promotes β cell cytotoxicity [215–217].
6. Future Perspectives
In the future research it will be probably established whether T2DM is an inevitable disease and whether one may develop strategy to highly retard or completely exclude the pathological outcomes of progressive self-accelerating oxidative stress and nitrosative stress and concomitant dysregulated information signaling. The emerging role of redox signaling in GSIS and processes of molecular physiology of pancreatic β cells need to be elucidated as well. Unfortunately, neither targeted antioxidants might be able to defeat T2DM, since they simultaneously disrupt the inherent physiological redox signaling. Perhaps more focused strategies on yet unknown mechanisms will help to defeat T2DM world epidemy.
Acknowledgment
This work has been supported by Grant Agency of the Czech Republic, Grant no. P302/10/0346 to P. Ježek and P304/10/P204 to A. Dlasková.
References
- 1.Ashcroft FM, Rorsman P. Diabetes mellitus and the β cell: the last ten years. Cell. 2012;148(6):1160–1171. doi: 10.1016/j.cell.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gupta D, Krueger CB, Lastra G. Over-nutrition, obesity and insulin resistance in the development of β-cell dysfunction. Current Diabetes Reviews. 2012;8(2):76–83. doi: 10.2174/157339912799424564. [DOI] [PubMed] [Google Scholar]
- 3.Jewell JL, Oh E, Thurmond DC. Exocytosis mechanisms underlying insulin release and glucose uptake: conserved roles for Munc18c and syntaxin 4. American Journal of Physiology. 2010;298(3):R517–R531. doi: 10.1152/ajpregu.00597.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leloup C, Casteilla L, Carrière A, et al. Balancing Mitochondrial redox signaling: a key point in metabolic regulation. Antioxidants and Redox Signaling. 2011;14(3):519–530. doi: 10.1089/ars.2010.3424. [DOI] [PubMed] [Google Scholar]
- 5.Jung HS, Lee MS. Role of autophagy in diabetes and mitochondria. Annals of the New York Academy of Sciences. 2010;1201:79–83. doi: 10.1111/j.1749-6632.2010.05614.x. [DOI] [PubMed] [Google Scholar]
- 6.Gunasekaran U, Gannon M. Type 2 diabetes and the aging pancreatic beta cell. Aging. 2011;3(6):565–575. doi: 10.18632/aging.100350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin Y, Sun Z. Current views on type 2 diabetes. Journal of Endocrinology. 2010;204(1):1–11. doi: 10.1677/JOE-09-0260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tripathy D, Chavez AO. Defects in insulin secretion and action in the pathogenesis of type 2 diabetes mellitus. Current Diabetes Reports. 2010;10(3):184–191. doi: 10.1007/s11892-010-0115-5. [DOI] [PubMed] [Google Scholar]
- 9.Eckel RH, Kahn SE, Ferrannini E, et al. Endocrine Society; American Diabetes Association; European Association for the Study of Diabetes. Obesity and type 2 diabetes: what can be unified and what needs to be individualized? Diabetes Care. 2011;34(6):1424–1430. doi: 10.2337/dc11-0447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Veld P, Marichal M. Microscopic anatomy of the human islet of Langerhans. Advances in Experimental Medicine and Biology. 2010;654:1–19. doi: 10.1007/978-90-481-3271-3_1. [DOI] [PubMed] [Google Scholar]
- 11.Ichise M, Harris PE. Imaging of β-cell mass and function. Journal of Nuclear Medicine. 2010;51(7):1001–1004. doi: 10.2967/jnumed.109.068999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cnop M, Igoillo-Esteve M, Hughes SJ, Walker JN, Cnop I, Clark A. Longevity of human islet α- and β-cells. Diabetes, Obesity and Metabolism. 2011;13(Supplement 1):39–46. doi: 10.1111/j.1463-1326.2011.01443.x. [DOI] [PubMed] [Google Scholar]
- 13.Tavana O, Zhu C. Too many breaks (brakes): pancreatic β-cell senescence leads to diabetes. Cell Cycle. 2011;10(15):2471–2484. doi: 10.4161/cc.10.15.16741. [DOI] [PubMed] [Google Scholar]
- 14.Fernandez-Valverde SL, Taft RJ, Mattick JS. MicroRNAs in β-cell biology, insulin resistance, diabetes and its complications. Diabetes. 2011;60(7):1825–1831. doi: 10.2337/db11-0171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DeFronzo RA, Abdul-Ghani MA. Preservation of β-cell function: the key to diabetes prevention. Journal of Clinical Endocrinology and Metabolism. 2011;96(8):2354–2366. doi: 10.1210/jc.2011-0246. [DOI] [PubMed] [Google Scholar]
- 16.Levetan C. Distinctions between islet neogenesis and β-cell replication: implications for reversal of Type 1 and 2 diabetes. Journal of Diabetes. 2010;2(2):76–84. doi: 10.1111/j.1753-0407.2010.00074.x. [DOI] [PubMed] [Google Scholar]
- 17.Demeterco C, Hao E, Lee SH, Itkin-Ansari P, Levine F. Adult human β-cell neogenesis? Diabetes, Obesity and Metabolism. 2009;11(Supplement 4):46–53. doi: 10.1111/j.1463-1326.2009.01105.x. [DOI] [PubMed] [Google Scholar]
- 18.Jun HS. In vivo regeneration of insulin-producing β-cells. Advances in Experimental Medicine and Biology. 2010;654:627–640. doi: 10.1007/978-90-481-3271-3_27. [DOI] [PubMed] [Google Scholar]
- 19.Noguchi H. Pancreatic stem/progenitor cells for the treatment of diabetes. The Review of Diabetic Studies. 2010;7(2):105–111. doi: 10.1900/RDS.2010.7.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robson-Doucette CA, Sultan S, Allister EM, et al. Beta-cell uncoupling protein 2 regulates reactive oxygen species production, which influences both insulin and glucagon secretion. Diabetes. 2011;60(11):27110–27119. doi: 10.2337/db11-0132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ježek P, Hlavatá L. Mitochondria in homeostasis of reactive oxygen species in cell, tissues, and organism. International Journal of Biochemistry and Cell Biology. 2005;37(12):2478–2503. doi: 10.1016/j.biocel.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 22.Dlasková A, Hlavatá L, Ježek P. Oxidative stress caused by blocking of mitochondrial Complex I H+ pumping as a link in aging/disease vicious cycle. International Journal of Biochemistry and Cell Biology. 2008;40(9):1792–1805. doi: 10.1016/j.biocel.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 23.Dlasková A, Hlavatá L, Ježek J, Ježek P. Mitochondrial Complex I superoxide production is attenuated by uncoupling. International Journal of Biochemistry and Cell Biology. 2008;40(10):2098–2109. doi: 10.1016/j.biocel.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 24.Kussmaul L, Hirst J. The mechanism of superoxide production by NADH: ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(20):7607–7612. doi: 10.1073/pnas.0510977103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.King MS, Sharpley MS, Hirst J. Reduction of hydrophilic ubiquinones by the flavin in mitochondrial NADH:ubiquinone oxidoreductase (complex I) and production of reactive oxygen species. Biochemistry. 2009;48(9):2053–2062. doi: 10.1021/bi802282h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pryde KR, Hirst J. Superoxide is produced by the reduced flavin in mitochondrial complex I: a single, unified mechanism that applies during both forward and reverse electron transfer. Journal of Biological Chemistry. 2011;286(20):18056–18065. doi: 10.1074/jbc.M110.186841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brand MD, Affourtit C, Esteves TC, et al. Mitochondrial superoxide: production, biological effects, and activation of uncoupling proteins. Free Radical Biology and Medicine. 2004;37(6):755–767. doi: 10.1016/j.freeradbiomed.2004.05.034. [DOI] [PubMed] [Google Scholar]
- 28.Muller FL, Roberts AG, Bowman MK, Kramer DM. Architecture of the Qo site of the cytochrome bc 1 complex probed by superoxide production. Biochemistry. 2003;42(21):6493–6499. doi: 10.1021/bi0342160. [DOI] [PubMed] [Google Scholar]
- 29.Muller FL, Liu Y, van Remmen H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. Journal of Biological Chemistry. 2004;279(47):49064–49073. doi: 10.1074/jbc.M407715200. [DOI] [PubMed] [Google Scholar]
- 30.Ježek P, Plecitá-Hlavatá L. Mitochondrial reticulum network dynamics in relation to oxidative stress, redox regulation, and hypoxia. International Journal of Biochemistry and Cell Biology. 2009;41(10):1790–1804. doi: 10.1016/j.biocel.2009.02.014. [DOI] [PubMed] [Google Scholar]
- 31.Diano S, Horvath TL. Mitochondrial uncoupling protein 2 (UCP2) in glucose and lipid metabolism. Trends in Molecular Medicine. 2012;18(1):52–58. doi: 10.1016/j.molmed.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 32.Mailloux RJ, Harper ME. Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radical Biology and Medicine. 2011;51(6):1106–1115. doi: 10.1016/j.freeradbiomed.2011.06.022. [DOI] [PubMed] [Google Scholar]
- 33.Cannon B, Shabalina IG, Kramarova TV, Petrovic N, Nedergaard J. Uncoupling proteins: a role in protection against reactive oxygen species-or not? Biochimica et Biophysica Acta. 2006;1757(5-6):449–458. doi: 10.1016/j.bbabio.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 34.Ježek P, Žáčková M, Růžička M, Škobisová E, Jabůrek M. Mitochondrial uncoupling proteins—facts and fantasies. Physiological Research. 2004;53(Supplement 1):S199–S211. [PubMed] [Google Scholar]
- 35.Korshunov SS, Skulachev VP, Starkov AA. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Letters. 1997;416(1):15–18. doi: 10.1016/s0014-5793(97)01159-9. [DOI] [PubMed] [Google Scholar]
- 36.Klingenberg M, Echtay KS. Uncoupling proteins: the issues from a biochemist point of view. Biochimica et Biophysica Acta. 2001;1504(Supplement 1):128–143. doi: 10.1016/s0005-2728(00)00242-5. [DOI] [PubMed] [Google Scholar]
- 37.Ježek P, Žáčková M, Růžička M, Škobisová E, Jabůrek M. Mitochondrial Uncoupling Proteins—Facts and Fantasies. Physiological Research. 2004;53(1):S199–S211. [PubMed] [Google Scholar]
- 38.Ježek P, Engstová H, Žáčková M, et al. Fatty acid cycling mechanism and mitochondrial uncoupling proteins. Biochimica et Biophysica Acta. 1998;1365(1-2):319–327. doi: 10.1016/s0005-2728(98)00084-x. [DOI] [PubMed] [Google Scholar]
- 39.Skulachev VP. Fatty acid circuit as a physiological mechanism of uncoupling of oxidative phosphorylation. FEBS Letters. 1991;294(3):158–162. doi: 10.1016/0014-5793(91)80658-p. [DOI] [PubMed] [Google Scholar]
- 40.Shabalina IG, Nedergaard J. Mitochondrial (“mild”) uncoupling and ROS production: physiologically relevant or not? Biochemical Society Transactions. 2011;39(5):1305–1309. doi: 10.1042/BST0391305. [DOI] [PubMed] [Google Scholar]
- 41.Mattiasson G, Sullivan PG. The emerging functions of UCP2 in health, disease, and therapeutics. Antioxidants and Redox Signaling. 2006;8(1-2):1–38. doi: 10.1089/ars.2006.8.1. [DOI] [PubMed] [Google Scholar]
- 42.Krauss S, Zhang CY, Lowell BB. The mitochondrial uncoupling-protein homologues. Nature Reviews Molecular Cell Biology. 2005;6(3):248–261. doi: 10.1038/nrm1592. [DOI] [PubMed] [Google Scholar]
- 43.Zhang CY, Baffy G, Perret P, et al. Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, β cell dysfunction, and type 2 diabetes. Cell. 2001;105(6):745–755. doi: 10.1016/s0092-8674(01)00378-6. [DOI] [PubMed] [Google Scholar]
- 44.Parker N, Vidal-Puig AJ, Azzu V, Brand MD. Dysregulation of glucose homeostasis in nicotinamide nucleotide transhydrogenase knockout mice is independent of uncoupling protein 2. Biochimica et Biophysica Acta. 2009;1787(12):1451–1457. doi: 10.1016/j.bbabio.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Trenker M, Malli R, Fertschai I, Levak-Frank S, Graier WF. Uncoupling proteins 2 and 3 are fundamental for mitochondrial Ca2+ uniport. Nature Cell Biology. 2007;9(4):445–452. doi: 10.1038/ncb1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu Z, Zhang J, Zhao B. Superoxide anion regulates the mitochondrial free Ca2+ through uncoupling proteins. Antioxidants and Redox Signaling. 2009;11(8):1805–1818. doi: 10.1089/ars.2009.2427. [DOI] [PubMed] [Google Scholar]
- 47.Brookes PS, Parker N, Buckingham JA, et al. UCPs—unlikely calcium porters. Nature Cell Biology. 2008;10(11):1237–1240. doi: 10.1038/ncb1108-1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jabůrek M, Vařecha M, Gimeno RE, et al. Transport function and regulation of mitochondrial uncoupling proteins 2 and 3. Journal of Biological Chemistry. 1999;274(37):26003–26007. doi: 10.1074/jbc.274.37.26003. [DOI] [PubMed] [Google Scholar]
- 49.Garlid KD, Orosz DE, Modrianský M, Vassanelli S, Ježek P. On the mechanism of fatty acid-induced proton transport by mitochondrial uncoupling protein. Journal of Biological Chemistry. 1996;271(5):2615–2620. doi: 10.1074/jbc.271.5.2615. [DOI] [PubMed] [Google Scholar]
- 50.Ježek P, Modrianský M, Garlid KD. A structure-activity study of fatty acid interaction with mitochondrial uncoupling protein. FEBS Letters. 1997;408(2):166–170. doi: 10.1016/s0014-5793(97)00335-9. [DOI] [PubMed] [Google Scholar]
- 51.Žáčková M, Škobisová E, Urbánková E, Ježek P. Activating ω-6 polyunsaturated fatty acids and inhibitory purine nucleotides are high affinity ligands for novel mitochondrial uncoupling proteins UCP2 and UCP3. Journal of Biological Chemistry. 2003;278(23):20761–20769. doi: 10.1074/jbc.M212850200. [DOI] [PubMed] [Google Scholar]
- 52.Jabůrek M, Miyamoto S, Di Mascio P, Garlid KD, Ježek P. Hydroperoxy fatty acid cycling mediated by mitochondrial uncoupling protein UCP2. Journal of Biological Chemistry. 2004;279(51):53097–53102. doi: 10.1074/jbc.M405339200. [DOI] [PubMed] [Google Scholar]
- 53.Ježek P, Jabůrek M, Garlid KD. Channel character of uncoupling protein-mediated transport. FEBS Letters. 2010;584(10):2135–2141. doi: 10.1016/j.febslet.2010.02.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Urbánková E, Voltchenko A, Pohl P, Ježek P, Pohl EE. Transport kinetics of uncoupling proteins: analysis of UCP1 reconstituted in planar lipid bilayers. Journal of Biological Chemistry. 2003;278(35):32497–32500. doi: 10.1074/jbc.M303721200. [DOI] [PubMed] [Google Scholar]
- 55.Beck V, Jabůrek M, Breen EP, Porter RK, Ježek P, Pohl EE. A new automated technique for the reconstitution of hydrophobic proteins into planar bilayer membranes. Studies of human recombinant uncoupling protein 1. Biochimica et Biophysica Acta. 2006;1757(5-6):474–479. doi: 10.1016/j.bbabio.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 56.Beck V, Jabůrek M, Demina T, et al. High efficiency of polyunsaturated fatty acids in the activation of human uncoupling protein 1 and 2 reconstituted in planar lipid bilayers. FASEB Journal. 2007;21(4):1137–1144. doi: 10.1096/fj.06-7489com. [DOI] [PubMed] [Google Scholar]
- 57.Rupprecht A, Sokolenko EA, Beck V, et al. Role of the transmembrane potential in the membrane proton leak. Biophysical Journal. 2010;98(8):1503–1511. doi: 10.1016/j.bpj.2009.12.4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Echtay KS, Esteves TC, Pakay JL, et al. A signalling role for 4-hydroxy-2-nonenal in regulation of mitochondrial uncoupling. EMBO Journal. 2003;22(16):4103–4110. doi: 10.1093/emboj/cdg412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chan CB, De Leo D, Joseph JW, et al. Increased uncoupling protein-2 levels in β-cells are associated with impaired glucose-stimulated insulin secretion: mechanism of action. Diabetes. 2001;50(6):1302–1310. doi: 10.2337/diabetes.50.6.1302. [DOI] [PubMed] [Google Scholar]
- 60.Krauss S, Zhang CY, Scorrano L, et al. Superoxide-mediated activation of uncoupling protein 2 causes pancreatic β cell dysfunction. Journal of Clinical Investigation. 2003;112(12):1831–1842. doi: 10.1172/JCI19774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang CY, Parton LE, Ye CP, et al. Genipin inhibits UCP2-mediated proton leak and acutely reverses obesity- and high glucose-induced β cell dysfunction in isolated pancreatic islets. Cell Metabolism. 2006;3(6):417–427. doi: 10.1016/j.cmet.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 62.Produit-Zengaffinen N, Davis-Lameloise N, Perreten H, et al. Increasing uncoupling protein-2 in pancreatic beta cells does not alter glucose-induced insulin secretion but decreases production of reactive oxygen species. Diabetologia. 2007;50(1):84–93. doi: 10.1007/s00125-006-0499-6. [DOI] [PubMed] [Google Scholar]
- 63.Affourtit C, Brand MD. Uncoupling protein-2 contributes significantly to high mitochondrial proton leak in INS-1E insulinoma cells and attenuates glucose-stimulated insulin secretion. Biochemical Journal. 2008;409(1):84–93. doi: 10.1042/BJ20070954. [DOI] [PubMed] [Google Scholar]
- 64.Affourtit C, Jastroch M, Brand MD. Uncoupling protein-2 attenuates glucose-stimulated insulin secretion in INS-1E insulinoma cells by lowering mitochondrial reactive oxygen species. Free Radical Biology and Medicine. 2011;50(5):609–616. doi: 10.1016/j.freeradbiomed.2010.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Galetti S, Sarre A, Perreten H, Produit-Zengaffinen N, Muzzin P, Assimacopoulos-Jeannet F. Fatty acids do not activate UCP2 in pancreatic beta cells: comparison with UCP1. Pflugers Archive. 2009;457(4):931–940. doi: 10.1007/s00424-008-0548-8. [DOI] [PubMed] [Google Scholar]
- 66.Pi J, Bai Y, Daniel KW, et al. Persistent oxidative stress due to absence of uncoupling protein 2 associated with impaired pancreatic β-cell function. Endocrinology. 2009;150(7):3040–3048. doi: 10.1210/en.2008-1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee SC, Robson-Doucette CA, Wheeler MB. Uncoupling protein 2 regulates reactive oxygen species formation in islets and influences susceptibility to diabetogenic action of streptozotocin. Journal of Endocrinology. 2009;203(1):33–43. doi: 10.1677/JOE-09-0117. [DOI] [PubMed] [Google Scholar]
- 68.Melov S. Mitochondrial oxidative stress. Physiologic consequences and potential for a role in aging. Annals of the New York Academy of Sciences. 2000;908:219–225. doi: 10.1111/j.1749-6632.2000.tb06649.x. [DOI] [PubMed] [Google Scholar]
- 69.Inoue M, Sato EF, Nishikawa M, et al. Mitochondrial generation of reactive oxygen species and its role in aerobic life. Current Medicinal Chemistry. 2003;10(23):2495–2505. doi: 10.2174/0929867033456477. [DOI] [PubMed] [Google Scholar]
- 70.Okado-Matsumoto A, Fridovich I. Subcellular distribution of superoxide dismutases (SOD) in rat liver. Cu,Zn-SOD in mitochondria. Journal of Biological Chemistry. 2001;276(42):38388–38393. doi: 10.1074/jbc.M105395200. [DOI] [PubMed] [Google Scholar]
- 71.Nomura K, Imai H, Koumura T, Kobayashi T, Nakagawa Y. Mitochondrial phospholipid hydroperoxide glutathione peroxidase inhibits the release of cytochrome c from mitochondria by suppressing the peroxidation of cardiolipin in hypoglycaemia-induced apoptosis. Biochemical Journal. 2000;351(1):183–193. doi: 10.1042/0264-6021:3510183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang HP, Schafer FQ, Goswami PC, Oberley LW, Buettner GR. Phospholipid hydroperoxide glutathione peroxidase induces a delay in G1 of the cell cycle. Free Radical Research. 2003;37(6):621–630. doi: 10.1080/1071576031000088283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Grankvist K, Marklund SL, Taljedal IB. CuZn-superoxide dismutase, Mn-superoxide dismutase, catalase and glutathione peroxidase in pancreatic islets and other tissues in the mouse. Biochemical Journal. 1981;199(2):393–398. doi: 10.1042/bj1990393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Loh K, Deng H, Fukushima A, et al. Reactive oxygen species enhance insulin sensitivity. Cell Metabolism. 2009;10(4):260–272. doi: 10.1016/j.cmet.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Leibiger IB, Brismar K, Berggren PO. Novel aspects on pancreatic beta-cell signal-transduction. Biochemical and Biophysical Research Communications. 2010;396(1):111–115. doi: 10.1016/j.bbrc.2010.02.174. [DOI] [PubMed] [Google Scholar]
- 76.Reaven GM. Insulin secretory function in type 2 diabetes: does it matter how you measure it? Journal of Diabetes. 2009;1(3):142–150. doi: 10.1111/j.1753-0407.2009.00016.x. [DOI] [PubMed] [Google Scholar]
- 77.Wiederkehr A, Wollheim CB. Mitochondrial signals drive insulin secretion in the pancreatic β-cell. Molecular and Cellular Endocrinology. 2012;353(1-2):128–137. doi: 10.1016/j.mce.2011.07.016. [DOI] [PubMed] [Google Scholar]
- 78.Schiff M, Loublier S, Coulibaly A, Bénit P, Ogier de Baulny H, Rustin P. Mitochondria and diabetes mellitus: untangling a conflictive relationship? Journal of Inherited Metabolic Disease. 2009;32(6):684–698. doi: 10.1007/s10545-009-1263-0. [DOI] [PubMed] [Google Scholar]
- 79.Portha B, Lacraz G, Chavey A, et al. Islet structure and function in the GK rat. Advances in Experimental Medicine and Biology. 2010;654:479–500. doi: 10.1007/978-90-481-3271-3_21. [DOI] [PubMed] [Google Scholar]
- 80.Ježek P, Plecitá-Hlavatá L, Smolková K, Rossignol R. Distinctions and similarities of cell bioenergetics and the role of mitochondria in hypoxia, cancer, and embryonic development. International Journal of Biochemistry and Cell Biology. 2010;42(5):604–622. doi: 10.1016/j.biocel.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 81.Smolková K, Plecitá-Hlavatá L, Bellance N, Benard G, Rossignol R, Ježek P. Waves of gene regulation suppress and then restore oxidative phosphorylation in cancer cells. International Journal of Biochemistry and Cell Biology. 2011;43(7):950–968. doi: 10.1016/j.biocel.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 82.Akhmedov D, De Marchi U, Wollheim CB, Wiederkehr A. Pyruvate dehydrogenase E1α phosphorylation is induced by glucose but does not control metabolism-secretion coupling in INS-1E clonal β-cells. Biochimica et Biophysica Acta. 2012;1823(10):1815–1824. doi: 10.1016/j.bbamcr.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 83.Park JH, Kim SJ, Park SH, et al. Glucagon-like peptide-1 enhances glucokinase activity in pancreatic β-cells through the association of Epac2 with Rim2 and Rab3A. Endocrinology. 2012;153(2):574–582. doi: 10.1210/en.2011-0259. [DOI] [PubMed] [Google Scholar]
- 84.McCulloch LJ, van de Bunt M, Braun M, Frayn KN, Clark A, Gloyn AL. GLUT2 (SLC2A2) is not the principal glucose transporter in human pancreatic beta cells: implications for understanding genetic association signals at this locus. Molecular Genetics and Metabolism. 2011;104(4):648–653. doi: 10.1016/j.ymgme.2011.08.026. [DOI] [PubMed] [Google Scholar]
- 85.Coppieters KT, Wiberg A, Amirian N, Kay TW, von Herrath MG. Persistent glucose transporter expression on pancreatic beta cells from longstanding type 1 diabetic individuals. Diabetes Metabolism Research and Reviews. 2011;27(8):746–754. doi: 10.1002/dmrr.1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kaminski MT, Lenzen S, Baltrusch S. Real-time analysis of intracellular glucose and calcium in pancreatic beta cells by fluorescence microscopy. Biochimica et Biophysica Acta. 2012;1823(10):1697–1707. doi: 10.1016/j.bbamcr.2012.06.022. [DOI] [PubMed] [Google Scholar]
- 87.Merglen A, Theander S, Rubi B, Chaffard G, Wollheim CB, Maechler P. Glucose sensitivity and metabolism-secretion coupling studied during two-year continuous culture in INS-1E insulinoma cells. Endocrinology. 2004;145(2):667–678. doi: 10.1210/en.2003-1099. [DOI] [PubMed] [Google Scholar]
- 88.Liang Y, Buettger C, Berner DK, Matschinsky FM. Chronic effect of fatty acids on insulin release is not through the alternation of glucose metabolism in a pancreatic β-cell line (βHC9) Diabetologia. 1997;40(9):1018–1027. doi: 10.1007/s001250050783. [DOI] [PubMed] [Google Scholar]
- 89.Porterfield DM, Corkey RF, Sanger RH, Tornheim K, Smith PJS, Corkey BE. Oxygen consumption oscillates in single clonal pancreatic β-cells (HIT) Diabetes. 2000;49(9):1511–1516. doi: 10.2337/diabetes.49.9.1511. [DOI] [PubMed] [Google Scholar]
- 90.Špaček T, Šantorová J, Zacharovová K, et al. Glucose-stimulated insulin secretion of insulinoma INS-1E cells is associated with elevation of both respiration and mitochondrial membrane potential. International Journal of Biochemistry and Cell Biology. 2008;40(8):1522–1535. doi: 10.1016/j.biocel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 91.McTaggart JS, Clark RH, Ashcroft FM. The role of the KATP channel in glucose homeostasis in health and disease: more than meets the islet. Journal of Physiology. 2010;588(17):3201–3209. doi: 10.1113/jphysiol.2010.191767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bennett K, James C, Hussain K. Pancreatic β-cell KATP channels: hypoglycaemia and hyperglycaemia. Reviews in Endocrine and Metabolic Disorders. 2010;11(3):157–163. doi: 10.1007/s11154-010-9144-2. [DOI] [PubMed] [Google Scholar]
- 93.Rorsman P, Braun M, Zhang Q. Regulation of calcium in pancreatic α- and β-cells in health and disease. Cell Calcium. 2012;51(3-4):300–308. doi: 10.1016/j.ceca.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cai EP, Casimir M, Schroer SA, et al. In vivo role of focal adhesion kinase in regulating pancreatic β-cell mass and function through insulin signaling, actin dynamics, and granule trafficking. Diabetes. 2012;61(7):1708–1718. doi: 10.2337/db11-1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhu D, Zhang Y, Lam PP, et al. Dual role of VAMP8 in regulating insulin exocytosis and islet β Cell growth. Cell Metabolism. 2012;16(2):238–249. doi: 10.1016/j.cmet.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 96.Rosengren AH, Braun M, Mahdi T, et al. Reduced insulin exocytosis in human pancreatic β-cells with gene variants linked to type 2 diabetes. Diabetes. 2012;61(7):1726–1733. doi: 10.2337/db11-1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fridlyand LE, Philipson LH. Does the glucose-dependent insulin secretion mechanism itself cause oxidative stress in pancreatic β-cells. Diabetes. 2004;53(8):1942–1948. doi: 10.2337/diabetes.53.8.1942. [DOI] [PubMed] [Google Scholar]
- 98.Maechler P, Carobbio S, Rubi B. In beta-cells, mitochondria integrate and generate metabolic signals controlling insulin secretion. International Journal of Biochemistry and Cell Biology. 2006;38(5-6):696–709. doi: 10.1016/j.biocel.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 99.Casimir M, Lasorsa FM, Rubi B, et al. Mitochondrial glutamate carrier GC1 as a newly identified player in the control of glucose-stimulated insulin secretion. Journal of Biological Chemistry. 2009;284(37):25004–25014. doi: 10.1074/jbc.M109.015495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Joseph JW, Jensen MV, Ilkayeva O, et al. The mitochondrial citrate/isocitrate carrier plays a regulatory role in glucose-stimulated insulin secretion. Journal of Biological Chemistry. 2006;281(47):35624–35632. doi: 10.1074/jbc.M602606200. [DOI] [PubMed] [Google Scholar]
- 101.Stark R, Pasquel F, Turcu A, et al. Phosphoenolpyruvate cycling via mitochondrial phosphoenolpyruvate carboxykinase links anaplerosis and mitochondrial GTP with insulin secretion. Journal of Biological Chemistry. 2009;284(39):26578–26590. doi: 10.1074/jbc.M109.011775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Heart E, Cline GW, Collis LP, Pongratz RL, Gray JP, Smith PJS. Role for malic enzyme, pyruvate carboxylation, and mitochondrial malate import in glucose-stimulated insulin secretion. American Journal of Physiology. 2009;296(6):E1354–E1362. doi: 10.1152/ajpendo.90836.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jitrapakdee S, Wutthisathapornchai A, Wallace JC, MacDonald MJ. Regulation of insulin secretion: role of mitochondrial signalling. Diabetologia. 2010;53(6):1019–1032. doi: 10.1007/s00125-010-1685-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Koshkin V, Wang X, Scherer PE, Chan CB, Wheeler MB. Mitochondrial functional state in clonal pancreatic β-cells exposed to free fatty acids. Journal of Biological Chemistry. 2003;278(22):19709–19715. doi: 10.1074/jbc.M209709200. [DOI] [PubMed] [Google Scholar]
- 105.Leloup C, Tourrel-Cuzin C, Magnan C, et al. Mitochondrial reactive oxygen species are obligatory signals for glucose-induced insulin secretion. Diabetes. 2009;58(3):673–681. doi: 10.2337/db07-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bindokas VP, Kuznetsov A, Sreenan S, Polonsky KS, Roe MW, Philipson LH. Visualizing superoxide production in normal and diabetic rat islets of Langerhans. Journal of Biological Chemistry. 2003;278(11):9796–9801. doi: 10.1074/jbc.M206913200. [DOI] [PubMed] [Google Scholar]
- 107.Sakai K, Matsumoto K, Nishikawa T, et al. Mitochondrial reactive oxygen species reduce insulin secretion by pancreatic β-cells. Biochemical and Biophysical Research Communications. 2003;300(1):216–222. doi: 10.1016/s0006-291x(02)02832-2. [DOI] [PubMed] [Google Scholar]
- 108.Patterson GH, Knobel SM, Arkhammar P, Thastrup O, Piston DW. Separation of the glucose-stimulated cytoplasmic and mitochondrial NAD(P)H responses in pancreatic islet β cells. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(10):5203–5207. doi: 10.1073/pnas.090098797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Martens GA, Cai Y, Hinke S, Stangé G, Van de Casteele M, Pipeleers D. Glucose suppresses superoxide generation in metabolically responsive pancreatic β cells. Journal of Biological Chemistry. 2005;280(21):20389–20396. doi: 10.1074/jbc.M411869200. [DOI] [PubMed] [Google Scholar]
- 110.Lacraz G, Figeac F, Movassat J, et al. Diabetic β-cells can achieve self-protection against oxidative stress through an adaptive up-regulation of their antioxidant defenses. PLoS ONE. 2009;4(8, article e6500) doi: 10.1371/journal.pone.0006500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pi J, Bai Y, Zhang Q, et al. Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes. 2007;56(7):1783–1791. doi: 10.2337/db06-1601. [DOI] [PubMed] [Google Scholar]
- 112.Saadeh M, Ferrante TC, Kane A, Shirihai O, Corkey BE, Deeney JT. Reactive oxygen species stimulate insulin secretion in rat pancreatic islets: studies using mono-oleoyl-glycerol. PLoS One. 2012;7(1, article e30200) doi: 10.1371/journal.pone.0030200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bouche C, Lopez X, Fleischman A, et al. Insulin enhances glucose-stimulated insulin secretion in healthy humans. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(10):4770–4775. doi: 10.1073/pnas.1000002107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Finocchietto P, Barreyro F, Holod S, et al. Control of muscle mitochondria by insulin entails activation of Akt2-mtNOS pathway: imlpications for the metabolic syndrome. PLoS ONE. 2008;3(3, article e1749) doi: 10.1371/journal.pone.0001749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Henstridge DC, Drew BG, Formosa MF, et al. The effect of the nitric oxide donor sodium nitroprusside on glucose uptake in human primary skeletal muscle cells. Nitric Oxide. 2009;21(2):126–131. doi: 10.1016/j.niox.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 116.Persaud SJ, Asare-Anane H, Jones PM. Insulin receptor activation inhibits insulin secretion from human islets of Langerhans. FEBS Letters. 2002;510(3):225–228. doi: 10.1016/s0014-5793(01)03268-9. [DOI] [PubMed] [Google Scholar]
- 117.Liu S, Okada T, Assmann A, et al. Insulin signaling regulates mitochondrial function in pancreatic β-cells. PLoS ONE. 2009;4(11, article e7983) doi: 10.1371/journal.pone.0007983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Brennand K, Huangfu D, Melton D. All beta cells contribute equally to islet growth and maintenance. PLoS Biology. 2007;5(7, article e163) doi: 10.1371/journal.pbio.0050163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kulkarni RN, Brüning JC, Winnay JN, Postic C, Magnuson MA, Kahn CR. Tissue-specific knockout of the insulin receptor in pancreatic β cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell. 1999;96(3):329–339. doi: 10.1016/s0092-8674(00)80546-2. [DOI] [PubMed] [Google Scholar]
- 120.Okada T, Chong WL, Hu J, et al. Insulin receptors in β-cells are critical for islet compensatory growth response to insulin resistance. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(21):8977–8982. doi: 10.1073/pnas.0608703104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Oliveira HR, Verlengia R, Carvalho CRO, Britto LRG, Curi R, Carpinelli AR. Pancreatic β-cells express phagocyte-like NAD(P)H oxidase. Diabetes. 2003;52(6):1457–1463. doi: 10.2337/diabetes.52.6.1457. [DOI] [PubMed] [Google Scholar]
- 122.Uchizono Y, Takeya R, Iwase M, et al. Expression of isoforms of NADPH oxidase components in rat pancreatic islets. Life Sciences. 2006;80(2):133–139. doi: 10.1016/j.lfs.2006.08.031. [DOI] [PubMed] [Google Scholar]
- 123.Newsholme P, Gaudel C, Krause M. Mitochondria and diabetes. An intriguing pathogenetic role. Advances in Experimental Medicine and Biology. 2012;942:235–247. doi: 10.1007/978-94-007-2869-1_10. [DOI] [PubMed] [Google Scholar]
- 124.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiological Reviews. 2007;87(1):245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 125.Morgan D, Oliveira-Emilio HR, Keane D, et al. Glucose, palmitate and pro-inflammatory cytokines modulate production and activity of a phagocyte-like NADPH oxidase in rat pancreatic islets and a clonal beta cell line. Diabetologia. 2007;50(2):359–369. doi: 10.1007/s00125-006-0462-6. [DOI] [PubMed] [Google Scholar]
- 126.Newsholme P, Morgan D, Rebelato E, et al. Insights into the critical role of NADPH oxidase(s) in the normal and dysregulated pancreatic beta cell. Diabetologia. 2009;52(12):2489–2498. doi: 10.1007/s00125-009-1536-z. [DOI] [PubMed] [Google Scholar]
- 127.Inoguchi T, Li P, Umeda F, et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C-dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes. 2000;49(11):1939–1945. doi: 10.2337/diabetes.49.11.1939. [DOI] [PubMed] [Google Scholar]
- 128.Nakayama M, Inoguchi T, Sonta T, et al. Increased expression of NAD(P)H oxidase in islets of animal models of type 2 diabetes and its improvement by an AT1 receptor antagonist. Biochemical and Biophysical Research Communications. 2005;332(4):927–933. doi: 10.1016/j.bbrc.2005.05.065. [DOI] [PubMed] [Google Scholar]
- 129.Murdock DJL, Clarke J, Flatt PR, Barnett YA, Barnett CR. Role of CYP2E1 in ketone-stimulated insulin release in pancreatic B-cells. Biochemical Pharmacology. 2004;67(5):875–884. doi: 10.1016/j.bcp.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 130.Elsner M, Gehrmann W, Lenzen S. Peroxisome-generated hydrogen peroxide as important mediator of lipotoxicity in insulin-producing cells. Diabetes. 2011;60(1):200–208. doi: 10.2337/db09-1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Rebelato E, Abdulkader F, Curi R, Carpinelli AR. Control of the intracellular redox state by glucose participates in the insulin secretion mechanism. PLoS One. 2011;6(8, article e24507) doi: 10.1371/journal.pone.0024507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lenzen S. Oxidative stress: the vulnerable β-cell. Biochemical Society Transactions. 2008;36, part 3:343–347. doi: 10.1042/BST0360343. [DOI] [PubMed] [Google Scholar]
- 133.Lenzen S, Drinkgern J, Tiedge M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radical Biology and Medicine. 1996;20(3):463–466. doi: 10.1016/0891-5849(96)02051-5. [DOI] [PubMed] [Google Scholar]
- 134.Tiedge M, Lortz S, Drinkgern J, Lenzen S. Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes. 1997;46(11):1733–1742. doi: 10.2337/diab.46.11.1733. [DOI] [PubMed] [Google Scholar]
- 135.Modak MA, Datar SP, Bhonde RR, Ghaskadbi SS. Differential susceptibility of chick and mouse islets to streptozotocin and its co-relation with islet antioxidant status. Journal of Comparative Physiology B. 2007;177(2):247–257. doi: 10.1007/s00360-006-0126-3. [DOI] [PubMed] [Google Scholar]
- 136.Modak MA, Parab PB, Ghaskadbi SS. Pancreatic islets are very poor in rectifying oxidative DNA damage. Pancreas. 2009;38(1):23–29. doi: 10.1097/MPA.0b013e318181da4e. [DOI] [PubMed] [Google Scholar]
- 137.Ivarsson R, Quintens R, Dejonghe S, et al. Redox control of exocytosis: regulatory role of NADPH, thioredoxin, and glutaredoxin. Diabetes. 2005;54(7):2132–2142. doi: 10.2337/diabetes.54.7.2132. [DOI] [PubMed] [Google Scholar]
- 138.Welsh N, Margulis B, Borg LA, et al. Differences in the expression of heat-shock proteins and antioxidant enzymes between human and rodent pancreatic islets: implications for the pathogenesis of insulin-dependent diabetes mellitus. Molecular Medicine. 1995;1(7):806–820. [PMC free article] [PubMed] [Google Scholar]
- 139.Tonooka N, Oseid E, Zhou H, Harmon JS, Robertson RP. Glutathione peroxidase protein expression and activity in human islets isolated for transplantation. Clinical Transplantation. 2007;21(6):767–772. doi: 10.1111/j.1399-0012.2007.00736.x. [DOI] [PubMed] [Google Scholar]
- 140.Newsholme P, De Bittencourt PIH, O’Hagan C, De Vito G, Murphy C, Krause MS. Exercise and possible molecular mechanisms of protection from vascular disease and diabetes: the central role of ROS and nitric oxide. Clinical Science. 2009;118(5):341–349. doi: 10.1042/CS20090433. [DOI] [PubMed] [Google Scholar]
- 141.Krause MS, McClenaghan NH, Flatt PR, de Bittencourt PI, Murphy C, Newsholme P. L-Arginine is essential forpancreatic beta-cell functional integrity, metabolism and defense from inflammatory challenge. Journal of Endocrinology. 2011;211(1):87–97. doi: 10.1530/JOE-11-0236. [DOI] [PubMed] [Google Scholar]
- 142.Bachnoff N, Trus M, Atlas D. Alleviation of oxidative stress by potent and selective thioredoxin-mimetic peptides. Free Radical Biology and Medicine. 2011;50(10):1355–1367. doi: 10.1016/j.freeradbiomed.2011.02.026. [DOI] [PubMed] [Google Scholar]
- 143.Reinbothe TM, Ivarsson R, Li DQ, et al. Glutaredoxin-1 mediates NADPH-dependent stimulation of calcium-dependent insulin secretion. Molecular Endocrinology. 2009;23(6):893–900. doi: 10.1210/me.2008-0306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zhao F, Wang Q. The protective effect of peroxiredoxin II on oxidative stress induced apoptosis in pancreatic β-cells. Cell Bioscience. 2012;2(1, article 22) doi: 10.1186/2045-3701-2-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Yang KS, Kang SW, Woo HA, et al. Inactivation of human peroxiredoxin I during catalysis as the result of the oxidation of the catalytic site cysteine to cysteine-sulfinic acid. Journal of Biological Chemistry. 2002;277(41):38029–38036. doi: 10.1074/jbc.M206626200. [DOI] [PubMed] [Google Scholar]
- 146.Purves T, Middlemas A, Agthong S, et al. A role for mitogen-activated protein kinases in the etiology of diabetic neuropathy. FASEB Journal. 2001;15(13):2508–2514. doi: 10.1096/fj.01-0253hyp. [DOI] [PubMed] [Google Scholar]
- 147.Koya D, King GL. Protein kinase C activation and the development of diabetic complications. Diabetes. 1998;47(6):859–866. doi: 10.2337/diabetes.47.6.859. [DOI] [PubMed] [Google Scholar]
- 148.Kaneto H, Kajimoto Y, Miyagawa JI, et al. Beneficial effects of antioxidants in diabetes: possible protection of pancreatic β-cells against glucose toxicity. Diabetes. 1999;48(12):2398–2406. doi: 10.2337/diabetes.48.12.2398. [DOI] [PubMed] [Google Scholar]
- 149.Harmon JS, Stein R, Robertson RP. Oxidative stress-mediated, post-translational loss of MafA protein as a contributing mechanism to loss of insulin gene expression in glucotoxic beta cells. Journal of Biological Chemistry. 2005;280(12):11107–11113. doi: 10.1074/jbc.M410345200. [DOI] [PubMed] [Google Scholar]
- 150.Giniatullin AR, Darios F, Shakirzyanova A, Davletov B, Giniatullin R. SNAP25 is a pre-synaptic target for the depressant action of reactive oxygen species on transmitter release. Journal of Neurochemistry. 2006;98(6):1789–1797. doi: 10.1111/j.1471-4159.2006.03997.x. [DOI] [PubMed] [Google Scholar]
- 151.Smolková K, Ježek P. The role of mitochondrial NADPH-dependent isocitrate dehydrogenase in cancer cells. International Journal of Biochemistry & Cell Biology. 2012;2012 doi: 10.1155/2012/273947.273947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Gray JP, Alavian KN, Jonas EA, Heart EA. NAD kinase regulates the size of the NADPH pool and insulin secretion in pancreatic β-cells. American Journal of Physiology. 2012;303(2):E191–E199. doi: 10.1152/ajpendo.00465.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Tang C, Koulajian K, Schuiki I, et al. Glucose-induced beta cell dysfunction in vivo in rats: link between oxidative stress and endoplasmic reticulum stress. Diabetologia. 2012;55(5):1366–1379. doi: 10.1007/s00125-012-2474-8. [DOI] [PubMed] [Google Scholar]
- 154.Li N, Frigerio F, Maechler P. The sensitivity of pancreatic β-cells to mitochondrial injuries triggered by lipotoxicity and oxidative stress. Biochemical Society Transactions. 2008;36(5):930–934. doi: 10.1042/BST0360930. [DOI] [PubMed] [Google Scholar]
- 155.Victor VM, Rocha M, Herance R, Hernandez-Mijares A. Oxidative stress and mitochondrial dysfunction in type 2 diabetes. Current Pharmacological Design. 2011;17(36):3947–3958. doi: 10.2174/138161211798764915. [DOI] [PubMed] [Google Scholar]
- 156.Pitocco D, Zaccardi F, Di Stasio E, et al. Oxidative stress, nitric oxide, and diabetes. The Review of Diabetic Studies. 2010;7(1):15–25. doi: 10.1900/RDS.2010.7.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Newsholme P, Haber EP, Hirabara SM, et al. Diabetes associated cell stress and dysfunction: role of mitochondrial and non-mitochondrial ROS production and activity. Journal of Physiology. 2007;583(1):9–24. doi: 10.1113/jphysiol.2007.135871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Supale S, Ning L, Brun T, Maechler P. Mitochondrial dysfunction in pancreatic β cells. Trends in Endocrinology and Metabolism. 2012;23(9):477–487. doi: 10.1016/j.tem.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 159.Patti ME, Corvera S. The role of mitochondria in the pathogenesis of type 2 diabetes. Endocrine Reviews. 2010;31(3):364–395. doi: 10.1210/er.2009-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wang X, Vatamaniuk MZ, Roneker CA, et al. Knockouts of SOD1 and GPX1 exert different impacts on murine islet function and pancreatic integrity. Antioxidants and Redox Signaling. 2011;14(3):391–401. doi: 10.1089/ars.2010.3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Acharya JD, Ghaskadbi SS. Islets and their antioxidant defense. Islets. 2010;2(4):225–235. doi: 10.4161/isl.2.4.12219. [DOI] [PubMed] [Google Scholar]
- 162.Robertson RP, Harmon JS. Pancreatic islet β-cell and oxidative stress: the importance of glutathione peroxidase. FEBS Letters. 2007;581(19):3743–3748. doi: 10.1016/j.febslet.2007.03.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Harmon JS, Bogdani M, Parazzoli SD, et al. β-cell-specific overexpression of glutathione peroxidase preserves intranuclear MafA and reverses diabetes in db/db mice. Endocrinology. 2009;150(11):4855–4862. doi: 10.1210/en.2009-0708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hoehn KL, Salmon AB, Hohnen-Behrens C, et al. Insulin resistance is a cellular antioxidant defense mechanism. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(42):17787–17792. doi: 10.1073/pnas.0902380106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hur KY, Jung HS, Lee MS. Role of autophagy in β-cell function and mass. Diabetes, Obesity and Metabolism. 2010;12(Supplement 2):20–26. doi: 10.1111/j.1463-1326.2010.01278.x. [DOI] [PubMed] [Google Scholar]
- 166.Twig G, Elorza A, Molina AJA, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO Journal. 2008;27(2):433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Jung HS, Chung KW, Won Kim J, et al. Loss of autophagy diminishes pancreatic β cell mass and function with resultant hyperglycemia. Cell Metabolism. 2008;8(4):318–324. doi: 10.1016/j.cmet.2008.08.013. [DOI] [PubMed] [Google Scholar]
- 168.Roma LP, Pascal SM, Duprez J, Jonas JC. Mitochondrial oxidative stress contributes differently to rat pancreatic islet cell apoptosis and insulin secretory defects after prolonged culture in a low non-stimulating glucose concentration. Diabetologia. 2012;55(8):2226–2237. doi: 10.1007/s00125-012-2581-6. [DOI] [PubMed] [Google Scholar]
- 169.Mehmeti I, Gurgul-Convey E, Lenzen S, Lortz S. Induction of the intrinsic apoptosis pathway in insulin-secreting cells is dependent on oxidative damage of mitochondria but independent of caspase-12 activation. Biochimica et Biophysica Acta. 2011;1813(10):1827–1835. doi: 10.1016/j.bbamcr.2011.06.022. [DOI] [PubMed] [Google Scholar]
- 170.Hou N, Torii S, Saito N, Hosaka M, Takeuchi T. Reactive oxygen species-mediated pancreatic β-cell death is regulated by interactions between stress-activated protein kinases, p38 and c-jun N-terminal kinase, and mitogen-activated protein kinase phosphatases. Endocrinology. 2008;149(4):1654–1665. doi: 10.1210/en.2007-0988. [DOI] [PubMed] [Google Scholar]
- 171.Gehrmann W, Elsner M, Lenzen S. Role of metabolically generated reactive oxygen species for lipotoxicity in pancreatic β-cells. Diabetes, Obesity and Metabolism. 2010;12(Supplement 2):149–158. doi: 10.1111/j.1463-1326.2010.01265.x. [DOI] [PubMed] [Google Scholar]
- 172.Giacca A, Xiao C, Oprescu AI, Carpentier AC, Lewis GF. Lipid-induced pancreatic β-cell dysfunction: focus on in vivo studies. American Journal of Physiology. 2011;300(2):E255–E262. doi: 10.1152/ajpendo.00416.2010. [DOI] [PubMed] [Google Scholar]
- 173.Graciano MF, Valle MM, Kowluru A, Curi R, Carpinelli AR. Regulation of insulin secretion and reactive oxygen species production by free fatty acids in pancreatic islets. Islets. 2011;3(5):213–223. doi: 10.4161/isl.3.5.15935. [DOI] [PubMed] [Google Scholar]
- 174.Bensellam M, Laybutt DR, Jonas JC. The molecular mechanisms of pancreatic β-cell glucotoxicity: recent findings and future research directions. Molecular and Cellular Endocrinology. 2012;364(1-2):1–27. doi: 10.1016/j.mce.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 175.Nishikawa T, Araki E. Impact of mitochondrial ROS production in the pathogenesis of diabetes mellitus and its complications. Antioxidants and Redox Signaling. 2007;9(3):343–353. doi: 10.1089/ars.2006.1458. [DOI] [PubMed] [Google Scholar]
- 176.Sivitz WI, Yorek MA. Mitochondrial dysfunction in diabetes: from molecular mechanisms to functional significance and therapeutic opportunities. Antioxidants and Redox Signaling. 2010;12(4):537–577. doi: 10.1089/ars.2009.2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Lu H, Koshkin V, Allister EM, Gyulkhandanyan AV, Wheeler MB. Molecular and metabolic evidence for mitochondrial defects associated with β-cell dysfunction in a mouse model of type 2 diabetes. Diabetes. 2010;59(2):448–459. doi: 10.2337/db09-0129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Liou CW, Chen JB, Tiao MM, et al. Mitochondrial DNA coding and control region variants as genetic risk factors for type 2 diabetes mellitus. Diabetes. 2012;2(1):2642–2651. doi: 10.2337/db11-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Weiss H, Wester-Rosenloef L, Koch C, et al. The mitochondrial Atp8 mutation induces mitochondrial ROS generation, secretory dysfunction, and β-cell mass adaptation in conplastic B6-mtFVB mice. Endocrinology. 2012;153(10):4666–4676. doi: 10.1210/en.2012-1296. [DOI] [PubMed] [Google Scholar]
- 180.Bensch KG, Mott JL, Chang SW, et al. Selective mtDNA mutation accumulation results in β-cell apoptosis and diabetes development. American Journal of Physiology. 2009;296(4):E672–E680. doi: 10.1152/ajpendo.90839.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Hashizume O, Shimizu A, Yokota M, et al. Specific mitochondrial DNA mutation in mice regulates diabetes and lymphoma development. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(26):10528–10533. doi: 10.1073/pnas.1202367109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Wang PW, Lin TK, Weng SW, Liou CW. Mitochondrial DNA variants in the pathogenesis of type 2 diabetes—relevance of Asian population studies. The Review of Diabetic Studies. 2009;6(4):237–246. doi: 10.1900/RDS.2009.6.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Cree LM, Patel SK, Pyle A, et al. Age-related decline in mitochondrial DNA copy number in isolated human pancreatic islets. Diabetologia. 2008;51(8):1440–1443. doi: 10.1007/s00125-008-1054-4. [DOI] [PubMed] [Google Scholar]
- 184.Salles JE, Kasamatsu TS, Dib SA, Moisés RS. β-cell function in individuals carrying the mitochondrial tRNA Leu (UUR) mutation. Pancreas. 2007;34(1):133–137. doi: 10.1097/01.mpa.0000246659.38375.4d. [DOI] [PubMed] [Google Scholar]
- 185.Koeck T, Olsson AH, Nitert MD, et al. A common variant in TFB1M is associated with reduced insulin secretion and increased future risk of type 2 diabetes. Cell Metabolism. 2011;13(1):80–91. doi: 10.1016/j.cmet.2010.12.007. [DOI] [PubMed] [Google Scholar]
- 186.Silva JP, Köhler M, Graff C, et al. Impaired insulin secretion and β-cell loss in tissue-specific knockout mice with mitochondrial diabetes. Nature Genetics. 2000;26(3):336–340. doi: 10.1038/81649. [DOI] [PubMed] [Google Scholar]
- 187.Mizukami H, Wada R, Koyama M, et al. Augmented β cell loss and mitochondrial abnormalities in sucrose-fed GK rats. Virchows Archiv. 2008;452(4):383–392. doi: 10.1007/s00428-007-0508-2. [DOI] [PubMed] [Google Scholar]
- 188.Östenson CG, Efendic S. Islet gene expression and function in type 2 diabetes; studies in the Goto-Kakizaki rat and humans. Diabetes, Obesity and Metabolism. 2007;9(Supplement 2):180–186. doi: 10.1111/j.1463-1326.2007.00787.x. [DOI] [PubMed] [Google Scholar]
- 189.Serradas P, Giroix MH, Saulnier C, et al. Mitochondrial deoxyribonucleic acid content is specifically decreased in adult, but not fetal, pancreatic islets of the Goto-Kakizaki rat, a genetic model of noninsulin-dependent diabetes. Endocrinology. 1995;136(12):5623–5631. doi: 10.1210/endo.136.12.7588317. [DOI] [PubMed] [Google Scholar]
- 190.Alán L, Špaček T, Zelenka J, et al. Assessment of mitochondrial DNA as an indicator of islet quality: an example in Goto Kakizaki rats. Transplantation Proceedings. 2011;43(9):3281–3284. doi: 10.1016/j.transproceed.2011.09.055. [DOI] [PubMed] [Google Scholar]
- 191.Dlasková A, Špaček T, Šantorová J, et al. 4Pi microscopy reveals an impaired three-dimensional mitochondrial network of pancreatic islet β-cells, an experimental model of type-2 diabetes. Biochimica et Biophysica Acta. 2010;1797(6-7):1327–1341. doi: 10.1016/j.bbabio.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 192.Sethumadhavan S, Vasquez-Vivar J, Migrino RQ, Harmann L, Jacob HJ, Lazar J. Mitochondrial DNA variant for complex I reveals a role in diabetic cardiac remodeling. Journal of Biological Chemistry. 2012;287(26):22174–22182. doi: 10.1074/jbc.M111.327866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Ihara Y, Toyokuni S, Uchida K, et al. Hyperglycemia causes oxidative stress in pancreatic β-cells of GK rats, a model of type 2 diabetes. Diabetes. 1999;48(4):927–932. doi: 10.2337/diabetes.48.4.927. [DOI] [PubMed] [Google Scholar]
- 194.Del Guerra S, Lupi R, Marselli L, et al. Functional and molecular defects of pancreatic islets in human Type 2 diabetes. Diabetes. 2005;54(3):727–735. doi: 10.2337/diabetes.54.3.727. [DOI] [PubMed] [Google Scholar]
- 195.Gorogawa SI, Kajimoto Y, Umayahara Y, et al. Probucol preserves pancreatic β-cell function through reduction of oxidative stress in type 2 diabetes. Diabetes Research and Clinical Practice. 2002;57(1):1–10. doi: 10.1016/s0168-8227(02)00005-0. [DOI] [PubMed] [Google Scholar]
- 196.Norton VG, Marvin KW, Yau P, Bradbury EM. Nucleosome linking number change controlled by acetylation of histones H3 and H4. Journal of Biological Chemistry. 1990;265(32):19848–19852. [PubMed] [Google Scholar]
- 197.Boucher MJ, Selander L, Carlsson L, Edlund H. Phosphorylation marks IPF1/PDX1 protein for degradation by glycogen synthase kinase 3-dependent mechanisms. Journal of Biological Chemistry. 2006;281(10):6395–6403. doi: 10.1074/jbc.M511597200. [DOI] [PubMed] [Google Scholar]
- 198.Wu KL, Gannon M, Peshavaria M, et al. Hepatocyte nuclear factor 3β is involved in pancreatic β-cell-specific transcription of the pdx-1 gene. Molecular and Cellular Biology. 1997;17(10):6002–6013. doi: 10.1128/mcb.17.10.6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Panowski SH, Wolff S, Aguilaniu H, Durieux J, Dillin A. PHA-4/Foxa mediates diet-restriction-induced longevity of C. elegans . Nature. 2007;447(7144):550–555. doi: 10.1038/nature05837. [DOI] [PubMed] [Google Scholar]
- 200.Liu D, Pavlovic D, Chen MC, Flodström M, Sandler S, Eizirik DL. Cytokines induce apoptosis in β-cells isolated from mice lacking the inducible isoform of nitric oxide synthase (iNOS(-/-)) Diabetes. 2000;49(7):1116–1122. doi: 10.2337/diabetes.49.7.1116. [DOI] [PubMed] [Google Scholar]
- 201.Azevedo-Martins AK, Lortz S, Lenzen S, Curi R, Eizirik DL, Tiedge M. Improvement of the mitochondrial antioxidant defense status prevents cytokine-induced nuclear factor-κB activation in insulin-producing cells. Diabetes. 2003;52(1):93–101. doi: 10.2337/diabetes.52.1.93. [DOI] [PubMed] [Google Scholar]
- 202.Saxena G, Chen J, Shalev A. Intracellular shuttling and mitochondrial function of thioredoxin-interacting protein. Journal of Biological Chemistry. 2010;285(6):3997–4005. doi: 10.1074/jbc.M109.034421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Chen J, Hui ST, Couto FM, et al. Thioredoxin-interacting protein deficiency induces Akt/Bcl-xL signaling and pancreatic beta-cell mass and protects against diabetes. FASEB Journal. 2008;22(10):3581–3594. doi: 10.1096/fj.08-111690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Chen J, Fontes G, Saxena G, Poitout V, Shalev A. Lack of TXNIP protects against mitochondria-mediated apoptosis but not against fatty acid-induced ER stress-mediated β-cell death. Diabetes. 2010;59(2):440–447. doi: 10.2337/db09-0949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Spindel ON, World C, Berk BC. Thioredoxin interacting protein: redox dependent and independent regulatory mechanisms. Antioxidants and Redox Signaling. 2012;16(6):587–596. doi: 10.1089/ars.2011.4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Oslowski CM, Hara T, O'Sullivan-Murphy B, et al. Thioredoxin-interacting protein mediates ER stress-induced β Cell death through initiation of the inflammasome. Cell Metabolism. 2012;16(2):265–273. doi: 10.1016/j.cmet.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Sarre A, Gabrielli J, Vial G, Leverve XM, Assimacopoulos-Jeannet F. Reactive oxygen species are produced at low glucose and contribute to the activation of AMPK in insulin-secreting cells. Free Radical Biology and Medicine. 2012;52(1):142–150. doi: 10.1016/j.freeradbiomed.2011.10.437. [DOI] [PubMed] [Google Scholar]
- 208.Laybutt DR, Hawkins YC, Lock J, et al. Influence of diabetes on the loss of beta cell differentiation after islet transplantation in rats. Diabetologia. 2007;50(10):2117–2125. doi: 10.1007/s00125-007-0749-2. [DOI] [PubMed] [Google Scholar]
- 209.Wolf G, Aumann N, Michalska M, et al. Peroxiredoxin III protects pancreatic β cells from apoptosis. Journal of Endocrinology. 2010;207(2):163–175. doi: 10.1677/JOE-09-0455. [DOI] [PubMed] [Google Scholar]
- 210.Lortz S, Tiedge M. Sequential inactivation of reactive oxygen species by combined overexpression of SOD isoforms and catalase in insulin-producing cells. Free Radical Biology and Medicine. 2003;34(6):683–688. doi: 10.1016/s0891-5849(02)01371-0. [DOI] [PubMed] [Google Scholar]
- 211.Lortz S, Gurgul-Convey E, Lenzen S, Tiedge M. Importance of mitochondrial superoxide dismutase expression in insulin-producing cells for the toxicity of reactive oxygen species and proinflammatory cytokines. Diabetologia. 2005;48(8):1541–1548. doi: 10.1007/s00125-005-1822-3. [DOI] [PubMed] [Google Scholar]
- 212.Huebschmann AG, Regensteiner JG, Vlassara H, Reusch JEB. Diabetes and advanced glycoxidation end products. Diabetes Care. 2006;29(6):1420–1432. doi: 10.2337/dc05-2096. [DOI] [PubMed] [Google Scholar]
- 213.Hamaoka R, Fujii J, Miyagawa JI, et al. Overexpression of the aldose reductase gene induces apoptosis in pancreatic β-cells by causing a redox imbalance. Journal of Biochemistry. 1999;126(1):41–47. doi: 10.1093/oxfordjournals.jbchem.a022434. [DOI] [PubMed] [Google Scholar]
- 214.Kaneto H, Xu G, Song KH, et al. Activation of the hexosamine pathway leads to deterioration of pancreatic β-cell function through the induction of oxidative stress. Journal of Biological Chemistry. 2001;276(33):31099–31104. doi: 10.1074/jbc.M104115200. [DOI] [PubMed] [Google Scholar]
- 215.Konarkowska B, Aitken JF, Kistler J, Zhang S, Cooper GJS. Thiol reducing compounds prevent human amylin-evoked cytotoxicity. FEBS Journal. 2005;272(19):4949–4959. doi: 10.1111/j.1742-4658.2005.04903.x. [DOI] [PubMed] [Google Scholar]
- 216.Montane J, A. Klimek-Abercrombie A, Potter KJ, Westwell-Roper C, Bruce Verchere C. Metabolic stress, IAPP and islet amyloid. Diabetes, Obesity and Metabolism. 2012;14(Supplement 3):68–77. doi: 10.1111/j.1463-1326.2012.01657.x. [DOI] [PubMed] [Google Scholar]
- 217.Westermark P, Andersson A, Westermark GT. Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiological Reviews. 2011;91(3):795–826. doi: 10.1152/physrev.00042.2009. [DOI] [PubMed] [Google Scholar]