

Shh signaling determines ventral cell identity in the spinal cord, but a key question is how neural diversity is generated. Here, genome-wide analysis identifies a set of Gli1 activator/Sox2 transcription factor-binding regions that direct factors central to neural tube formation. Functional analysis of Gli motifs reveals a critical role for Gli-binding affinity in ventral patterning. This study thus provides mechanistic insight into how Shh signaling achieves tissue-specific outcomes in the neural tube.
Keywords: Shh, Gli, neural tube patterning, morphogen
Abstract
In the vertebrate neural tube, regional Sonic hedgehog (Shh) signaling invokes a time- and concentration-dependent induction of six different cell populations mediated through Gli transcriptional regulators. Elsewhere in the embryo, Shh/Gli responses invoke different tissue-appropriate regulatory programs. A genome-scale analysis of DNA binding by Gli1 and Sox2, a pan-neural determinant, identified a set of shared regulatory regions associated with key factors central to cell fate determination and neural tube patterning. Functional analysis in transgenic mice validates core enhancers for each of these factors and demonstrates the dual requirement for Gli1 and Sox2 inputs for neural enhancer activity. Furthermore, through an unbiased determination of Gli-binding site preferences and analysis of binding site variants in the developing mammalian CNS, we demonstrate that differential Gli-binding affinity underlies threshold-level activator responses to Shh input. In summary, our results highlight Sox2 input as a context-specific determinant of the neural-specific Shh response and differential Gli-binding site affinity as an important cis-regulatory property critical for interpreting Shh morphogen action in the mammalian neural tube.
The template for the mammalian CNS is established during embryonic life. Initially, Sox2 and other SoxB1 family members control neural fate commitment (Pevny and Placzek 2005), while subsequent regional signals control the time and position of neural progenitor specification, ensuring position-specific elaboration of distinct neural cell types as a critical step in the formation of functional neural circuitry. In this, Sonic hedgehog (Shh) encodes a highly conserved signaling factor essential for patterning the ventral neural tube of all vertebrates (Jessell 2000; Dessaud et al. 2008).
Shh is secreted by the notochord beneath the neural epithelial anlagen of the CNS; notochordal Shh induces a secondary center of Shh production in floor plate cells, positioned at the ventral midline of the neural tissue. Five mitotically active neural progenitor domains are established dorsal to the Hh-dependent floor plate: from dorsal to ventral, pV0, pV1, pV2, pMN, and pV3 (Fig. 1A). Each progenitor pool gives rise to molecularly and functionally distinct classes of interneurons (V0, V1, V2, and V3) or motor neurons (MNs). Analysis of mutants lacking Shh, or Smoothened (Smo), a seven-pass membrane protein essential for transmission of all Hedgehog (Hh) family signals, demonstrates that Hh signaling is essential for the specification of all ventral neuronal progenitor identities and the floor plate (Chiang et al. 1996; Wijgerde et al. 2002).
Figure 1.

Genome-wide detection of Gli1 binding in neural progenitors. (A) Schematic of neural tube development at the spinal cord level. (B) Overview of directed differentiation of ESCs into neural progenitors. (C) Large-scale clustering of GBRs within 10-Mb domains comparing observed (red) distribution versus random expectation (blue). (D) Summary of de novo motifs enriched in GBRs focusing on peak regions (CisGenome) or short sequence motifs close to the peak center (DREME). (E) Histogram analysis highlighting the Gli motif centering in peak regions. (F) Neural tube marker analysis along the rostral–caudal axis at E8.5. Note that Sox2 expression precedes the emergence of Shh-dependent cell types marked by Nkx6.1. Bar, 50 μm.
Visualization of Shh movement and the transcription of general targets of Hh pathway action are consistent with Shh playing a direct role in specifying each progenitor domain (Goodrich et al. 1996; Gritli-Linde et al. 2001; Chamberlain et al. 2008). Analysis of threshold responses to Shh in inductive assays shows a striking relationship to the arrangement of neural progenitor domains in the neural tube with increasing threshold requirements for the induction of increasingly more ventral cell types, consistent with Shh acting as a morphogen in the patterning process (Marti et al. 1995; Roelink et al. 1995). More recent studies stress the importance of integrating the level of Shh signaling over time to achieve the sequential emergence of ventral progenitor domains in a dorsal-to-ventral progression and, ultimately, the final patterned structure (Dessaud et al. 2007, 2010; Balaskas et al. 2012).
The specification of progenitor populations is linked to the Shh-dependent, position-specific expression of several transcriptional regulatory factors (Fig. 1A; Dessaud et al. 2008). Broadly, these fall into two classes: those activated (class II genes) or repressed (class I genes) by Shh signaling (Briscoe et al. 2000). Loss-of-function and ectopic expression studies indicate that many of these factors play a primary role in the establishment of neural progenitor identity. Cross-repressive interactions between class I and class II targets are thought to sharpen boundaries between distinct progenitor domains (Briscoe et al. 2000; Dessaud et al. 2008).
Whereas a broad framework for Shh's actions has emerged from experimental manipulation of Shh and signaling pathway components, a significant gap remains in understanding how Shh signals direct the neural-specific transcriptional programs generating neural diversity.
The Hh signaling pathway converges on the Gli family of transcriptional regulators: Gli1, Gli2, and Gli3 in mammals. In the absence of a signal, proteasome-dependent processing of Gli3 generates a transcriptional repressor form (Gli3R) (Wang et al. 2000; Wang and Li 2006). Gli3R production is attenuated by low levels of Shh signaling; furthermore, alleviation of Gli3-mediated repression is sufficient for the activation of a subset of regulatory factors such as Nkx6.1 and Olig2 and specification of pV0, pV1, pV2, and pMN cell fates. In contrast, higher levels of Shh signaling stimulate production of a Gli2 activator form (Gli2A) (Pan et al. 2006); Gli2A is essential for activation of Nkx2.2 and Foxa2 and specification of the ventral-most pV3 and FP fates, respectively (Ding et al. 1998; Matise et al. 1998). Weak Gli2R and Gli3A activities can also be identified in specific genetic contexts (Bai et al. 2004). The third family member, Gli1, acts exclusively as an activator that is induced by, and dependent on, Hh signaling (Bai et al. 2004). Direct Gli regulatory input has been identified in proximal cis-regulatory modules (CRMs) around Nkx2.2 and Foxa2 (Sasaki et al. 1997; Lei et al. 2006; Vokes et al. 2007), but the mechanisms distinguishing their distinct responses and the regulatory controls of other class II neural progenitor determinants are unknown.
To obtain a comprehensive understanding of the gene regulatory network elicited downstream from Shh morphogen signaling in neural patterning, we performed a genome-scale identification, intersection, verification, and analysis of Gli1 and Sox2 targets. A subset of Gli1 input is integrated with Sox2 input at the level of CRMs, and such Gli1/Sox2-bound CRMs direct all class II Shh target gene expression. Comprehensive in vitro DNA-binding assay suggests a role for differential Gli-binding affinity in GliA-dependent target gene regulation. These data support a model that Sox2 integration and Gli-binding affinity are key aspects of the Shh pathway in generating dose-dependent, neural-specific transcriptional outputs.
Results
Genome-wide detection of Gli1 input to neural progenitor cells
Specification of Shh-dependent neural progenitor populations within the ventral neural tube occurs in a time- and dosage-dependent manner (Fig. 1A). To further elucidate the transcriptional regulatory program downstream from Shh signaling in neural progenitors, we used an in vitro model of Shh-dependent patterning (Fig. 1B; Wichterle et al. 2002). Shh pathway activation was achieved by exposing in vitro-derived neural progenitors to Smo agonist (SAG), a small molecule that acts as a potent activator of the pathway (Chen et al. 2002). Immunofluorescence characterization of SAG-treated neural progenitors highlighted both the time- and concentration-dependent responses of class II target genes, consistent with the in vivo patterning response (Supplemental Fig. S1A–D). Furthermore, we detected Shh-producing cells, suggesting that these culturing conditions also favor floor plate specification (Supplemental Fig. S1D).
Genome-wide location analysis of Gli1 DNA association in embryonic stem cell (ESC)-derived neural progenitors was performed using a Gli1Flag transgene (Vokes et al. 2007). We defined a prioritized set of 841 enriched Gli1-binding regions (GBRs) by intersecting chromatin immunoprecipitation (ChIP) combined with deep sequencing (ChIP-seq) data independently verified in biological replicates (false discovery rate [FDR] = 0.01) (Supplemental Fig. S2A; Supplemental Table S1).
GBRs were distributed at considerable distances from the transcriptional start site (TSS) of nearest neighboring genes; only 12% of GBRs occurred within 10 kb of a TSS, suggesting that long-range interactions are a common feature of the Gli1 regulatory program (Supplemental Fig. S2B). Whereas most putative target genes associated with a single Gli1-bound region, large-scale clustering of GBRs occurred within some key areas of the genome: flanking the pan-neural progenitor determinant Sox2; specific neural cell type determinants Nkx2.2, Nkx2.9, and Foxa2; and surrounding genes encoding Shh pathway feedback regulators Ptch1, Gli1, and Hhip (Fig. 1C; Supplemental Table S2). Ptch1, the primary negative feedback component of the Hh pathway, resides within the most GBR-dense domain in the genome (Fig. 1C; Supplemental Fig. S2E).
Within this set of 841 GBRs, a Gli-binding site (GBS) was the most highly enriched sequence recovered through de novo motif enrichment analysis (Fig. 1D), present in 768 of 841 (91%) GBRs (P < 4.6 × 10−467). As expected for a direct DNA/protein interaction, the predicted GBS centered within the GBRs (Fig. 1E) and displayed strong evolutionary constraint when compared with flanking regions (Supplemental Fig. S2C). Interestingly, only 25% of GBRs contained more than one predicted GBS, suggesting that most CRMs have limited Gli1 inputs (Supplemental Fig. S2D).
In addition to a Gli motif, de novo motif analysis recovered additional DNA sequences enriched among the Gli1 ChIP data set: nuclear hormone receptor (NHR)- and Pbx-, Fox-, Rfx-, Sox-, and Evx-binding motifs (Fig. 1D; Supplemental Tables S1, S3). These findings point to additional regulatory interactions in subsets of GBRs consistent with reports of Sox2 activity in neural programming (Thomson et al. 2011), retinoic acid signaling in several phases of neural patterning (Novitch et al. 2003), and recent reports of Foxa2 modulation of the Shh response (Ribes et al. 2010; Metzakopian et al. 2012).
The Sox motif was of particular interest given that SoxB1 family members are known to play an important role in neural progenitor specification and maintenance (Bylund et al. 2003; Graham et al. 2003; Bergsland et al. 2011; Thomson et al. 2011). Furthermore, recent analysis of differentiating ESCs suggests that the redeployment of Sox2 from maintenance of pluripotency is the key event in specifying neural fate (Thomson et al. 2011). Sox2 is expressed in the neural lineage in vivo preceding a detectable Shh response (Fig. 1F) and broadly along both the anterior–posterior and dorsal–ventral axes, consistent with Sox2 prefiguring a neural-specific output to Shh/Gli-directed regulation of target genes.
Integration of Sox2 and Gli1 input to neural CRMs
To examine the potential regulatory role for SoxB1 family input to Gli1-bound CRMs, we performed Sox2 ChIP-seq analysis in neural progenitors (Supplemental Table S4). Our data were in good agreement with recent Sox2- and Sox3-binding data from a similar neural progenitor derivation protocol, suggesting considerable overlap among SoxB1 targets once a given member is activated (Bergsland et al. 2011; data not shown). The majority of Gli1-bound regions did not intersect with Sox2 target regions in ESCs or their neural derivatives, suggesting that prior engagement of Sox2 is not essential for Gli1 binding to DNA (Supplemental Fig. S3A). However, ∼12% of GBRs did overlap specifically with neural-associated Sox2-bound regions (P < 1.3 × 10−50) (Fig. 2A), with only a small number of these also bound by Sox2 in ESCs (Supplemental Fig. S3A). Within these intersected regions, we detected both GBSs and Sox-binding sites with no strong positioning constraints, suggesting that each factor binds independently to DNA rather than through secondary recruitment (Supplemental Fig. S4A–C). Additionally, there was no significant enrichment of the de novo recovered motifs in Gli1+ Sox2+ regions compared with Gli1+ Sox2− regions (Supplemental Fig. S4A). Remarkably, this group of neural-specific Sox2 binding (Gli1+ Sox2+) includes putative CRMs around all of the known class II regulatory targets of ventral neural patterning (Figs. 2A, 3A–E) and several other genes up-regulated in neural progenitors in response to Shh pathway activation (Fig. 2A; Supplemental Table S5). Overall, the intersection of Gli1 and Sox2 cobinding was a stronger predictor that a neighboring gene shows a Shh pathway dependence than Gli1 binding alone (P < 1.2 × 10−5) (Fig. 2B).
Figure 2.

Intersection and genomic analysis of Gli1 and Sox2 binding in neural progenitors. (A, top) Venn diagram for intersection of GBRs and Sox2-binding regions in neural progenitors. (Bottom) Heat map representation of genes that are differentially expressed in response to Shh pathway stimulation and also associated with Gli1- and Sox2-cobinding regions. Class II targets are marked by an asterisk (red). (B) Normalized expression values from the microarray are shown as box plots. (C) Region-based analysis comparing changes in H3K4me2 and H3K27ac levels with and without Shh pathway activation for regions bound by Gli1, Sox2, or both Gli1 and Sox2. Class II target regions are denoted by a black bar.
Figure 3.

Gli1 and Sox2 binding at class II target genes. (A–E) Gli1 (purple) and Sox2 (red) ChIP-seq signals are shown. Blue underline denotes the Gli1-binding signal that passed the peak detection threshold, with relative distances to TSSs shown below.
To address the functional significance of cobinding, we analyzed active histone modification marks H3K4 dimethylation (H3K4me2) and H3K27 acetylation (H3K27ac) at Gli1- and Sox2-bound regions in neural progenitors with or without Shh pathway stimulation (Fig. 2C; Supplemental Fig. S3; Rada-Iglesias et al. 2011). Regions bound by Gli1 but not Sox2 (Gli1+ Sox2−) showed moderate levels of H3K4me2 and H3K27ac independent of Shh pathway stimulation (Fig. 2C; Supplemental Fig. S3). Regions bound by Sox2 but not Gli1 (Gli1− Sox2+) showed significant levels for both H3K4me2 and H3K27ac marks in a largely Shh-independent manner. On the other hand, regions bound by both Gli1 and Sox2 (Gli1+ Sox2+) showed overall high levels of H3K4me2 and H3K27ac signals that were further enhanced upon Shh pathway activation (Supplemental Fig. S3B–O). Metasite analysis of H3K4me2 and H3K72ac profiles highlighted a characteristic dip positioned at the Sox2-binding peak center that was absent from regions bound by Gli1 alone, suggesting nucleosome displacement by Sox2 but not Gli1 (Supplemental Fig. S3B–O). We identified a subgroup of Gli1+ Sox2+ regions that displayed a stark difference in H3K27ac levels with and without Shh pathway stimulation (Fig. 2C; Supplemental Fig. S4D). These regions exhibited a significant increase in H3K27ac levels upon Shh pathway stimulation and include peaks associated with class II target genes (Fig. 2A; Supplemental Fig. S4D). Other genes associated with Gli1+ Sox2+ regions show moderate levels of H3K27ac without SAG treatment, and, consistently, their expression is enhanced by, but not dependent on, SAG treatment (e.g., Ptch1 and Ccnd2) (Fig. 2A). Thus, Sox2 binding appears to contribute to an active chromatin signature in the absence of Shh pathway stimulation at a subset of regions. Cobinding defines a subpopulation of GBRs that have a strong potential to be active as CRMs functioning in a Shh-dependent manner around key class II neural targets. No significant intersection of GBRs was observed with class I neural targets. The mechanism of Shh signaling-dependent repression of class I targets is unclear. However, given that Shh signaling results in a loss of GliR and binding by GliA, these results are consistent with an indirect mechanism of gene repression in response to Shh signaling (data not shown). In contrast, a strong association was observed with Sox2 and class I targets, consistent with broad neural activity of SoxB1 family members.
All class II Shh target genes are regulated by Gli1/Sox2-bound CRMs
To examine the cis-regulatory logic governing Gli1 target gene regulation, we focused on class II target genes. Analysis of Gli1/Sox2 cobinding to DNA confirmed known regulatory sites associated with Foxa2(+10kb) (Sasaki et al. 1997), Nkx2.2(−2kb) (Lei et al. 2006; Vokes et al. 2007), Nkx2.9(−2kb) (Santagati et al. 2003), and Olig2(−33kb) (Wang et al. 2011) and further identified potential novel regulatory inputs to Foxa2(+6kb, +160kb, and +170kb), Nkx2.9(−8kb), Olig2(−16kb), Nkx6.1 (+140kb, +367kb, and +540kb), and Nkx6.2(+54kb) (Fig. 3; Supplemental Tables S1, S4).
All identified Gli1/Sox2-cobound regions coincided with discrete blocks of conservation, and mouse transgenic reporter analysis revealed neural-specific enhancer activity in all tested regions (Fig. 4; Supplemental Figs. S5, S6). Thus, all class II Shh targets identified by their transcriptional responses to pathway activation are directly engaged by Gli1 and Sox2 regulatory factors at active enhancer elements. Importantly, each enhancer module showed a precise dorsal boundary of activity that correlated well with its predicted target gene. Several CRMs around a class II target displayed similar but not identical activity; for example, the +540-kb and +140-kb enhancers about Nkx6.1 show elevated levels in the ventral and dorsal halves of the Nkx6.1 domain, respectively (Fig. 4B,C). Inappropriate reporter activity was detected within specific ventral cell populations (Nkx6.2 and Olig2), suggesting that inhibitory inputs reflected by cross-repressive interactions between class II genes (e.g., Nkx2.2 on Olig2 and Nkx6.1 on Nkx6.2) (Briscoe et al. 2000) are mediated through additional regulatory modules. Activity in post-mitotic populations (Nkx6.1 and Olig2) may reflect perdurance of the transgenic reporter or an absence of post-mitotic silencing inputs.
Figure 4.

Functional characterization of Gli1/Sox2-cobound CRMs. Transient transgenic analysis of class II regulatory regions performed at the forelimb level in E10.5 embryos. Numbers in the top right corner of each panel show expressing embryos/total transgenic embryos. (A) Nkx6.2(+54kb). Note Nkx6.2 expression flanked by Dbx1 and Nkx6.1 (Briscoe et al. 2000). (B,C,I,J) Analysis of Nkx6.1(+140kb and +540kb) Gli1-bound regions. (D,K) Characterization of Gli input to Olig2(−33kb). (E,L) Sox-binding sites are required for Nkx2.2(−2kb) enhancer activity. (F,G,H) Foxa2(+6kb, +160kb, and +170kb) enhancer activity. Bar, 50 μm.
To determine the requirement for Gli input to the Nkx6.1(+540kb) and Olig2(−33kb) regulatory elements, we performed site-directed mutagenesis to disrupt the GBS (Supplemental Fig. S6A,E). Previous genetic analyses suggested that Nkx6.1 and Olig2 are regulated primarily through derepression of Gli3 (Persson et al. 2002; Wijgerde et al. 2002). Mutation of two predicted GBSs within the Olig2(−33kb) enhancer [Olig2(−33kb)dGBS1 + 2] diminished transgene activity, indicating that GliA input contributes to Olig2 expression levels (Fig. 4D,K; Supplemental Fig. S6C,D). In contrast, Nkx6.1(+540kb)dGBS1 mutants showed a similar level of transgene expression compared with wild type but displayed a significant reduction in the number of expressing embryos, suggesting that Gli activator may contribute to the robustness of Nkx6.1(+540kb) enhancer activity (Fig. 4C,I; Supplemental Fig. S6G,H). Similar results were obtained from chick enhancer assays for Nkx6.1(+540k)dGBS1+dGBS2 (Supplemental Fig. S6E,I,J). In neither case, Olig2(-33kb) nor Nkx6.1(+540kb), did the GBS mutations abolish enhancer activity, consistent with GliR removal as the predominant Gli regulatory modality for these Shh target genes (Persson et al. 2002; Wijgerde et al. 2002; Bai et al. 2004). However, we cannot exclude the possibility that undetected GBSs may contribute to a GliA response.
Next, we assessed the significance of Sox binding to Gli1-bound CRMs by mutating all predicted Sox-binding sites in Nkx6.1(+540kb) and Nkx2.2(−2kb) enhancers (Supplemental Fig. S6K,L). The loss of Sox binding to the Nkx6.1 element [Nkx6.1(+540k)dSox] resulted in the silencing of reporter activity in nine out of 10 transgenic embryos, whereas 14 out of 16 showed activity with Sox2-bound regions intact (Fig. 4C,J). We conclude that Sox2 engagement is likely critical for Nkx6.1 enhancer activity, and the anomaly likely reflects a transgene integration event that favors neural expression (data not shown). All Nkx2.2(−2kb)dSox transgenic embryos failed to express the reporter, demonstrating an absolute dependence on Sox activity (Fig. 4E,L). Given previous evidence of Gli-dependent activity from Gli-directed mutagenesis (Lei et al. 2006), it is evident that neither Sox2 nor GliA input is sufficient to activate the Nkx2.2 CRM. Collectively, these results support a positive role for Sox2 (and likely other SoxB1 family members) in the regulation of ventrally restricted Gli-mediated CRMs.
Gli proteins show differential binding affinity at class II gene CRMs
Class II target genes respond to Shh stimulation in a graded manner and display different requirements for GliA versus GliR input. Given the prominent role of transcription factor-binding affinity in determining threshold-level responses in other model systems (Jiang and Levine 1993; Gaudet and Mango 2002; Rowan et al. 2010), we determined the individual binding site preference for each zinc finger DNA-binding domain of Gli family proteins (Gli1–3) using universal protein-binding microarrays (PBMs) (Berger et al. 2006; Berger and Bulyk 2009). Hierarchical clustering of the highest scoring 8-mers from this analysis indicated that all Gli family members share similar sequence preferences (Fig. 5A,B; Supplemental Table S6). The highest-scoring 8-mer sequence, GGGTGGTC, agrees with the core of the optimal consensus determined by several independent in vitro (e.g., SELEX) and in vivo (ChIP) methods (Kinzler and Vogelstein 1990; Hallikas et al. 2006; Vokes et al. 2007, 2008). This high-affinity variant was also the most frequently occurring site within the recovered GBRs in this study.
Figure 5.

Determination and functional characterization of intrinsic Gli-binding affinity. (A) Hierarchical clustering of bound probes in PBM data for Gli1–3. (B) Gli1–3-binding specificity motifs deduced from PBMs. (C) Reporter constructs with associated point mutations to the GBS are highlighted in red. (D–G) Transient transgenic reporter analysis performed at the forelimb level of E10.5 embryos. (D,E) Nkx2.2(−2kb) element with a high-affinity Gli site displays strong reporter activity compared with a low-affinity variant. (F,G) Foxa2(+10kb) element with a wild-type Gli site shows weak expression restricted to the most ventral cell population. The high-affinity variant shows a significant increase in GFP and extends to the dorsal limit of Nkx2.2. Bar, 50 μm.
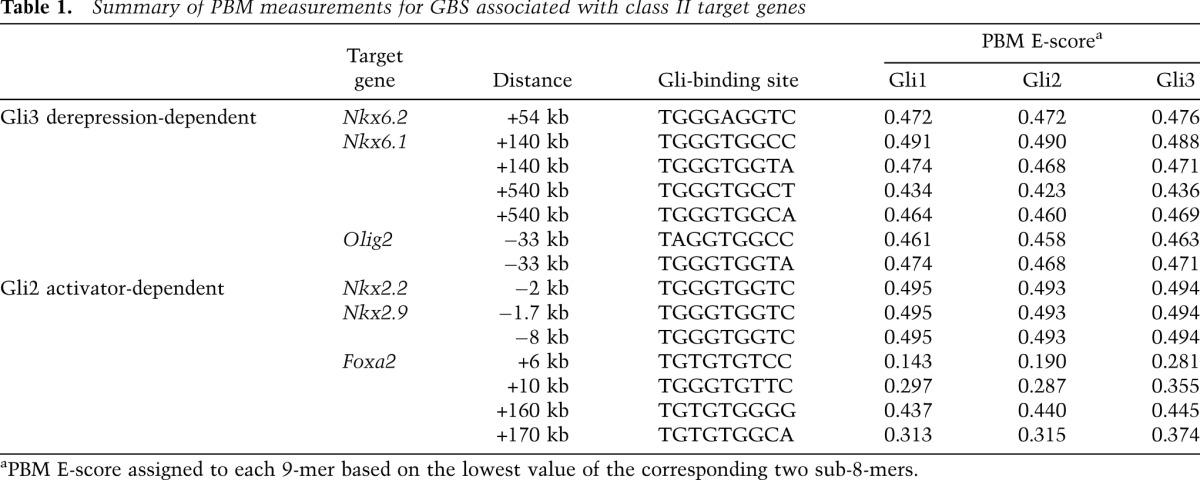
Importantly, analysis of the PBMs enabled the relative preferences of Gli factor binding to be measured for all GBSs associated with class II gene activity in neural progenitors (Table 1). Phylogenetic analysis of these GBSs indicated strong selective constraints for these sequence variants with limited nucleotide substitutions that did not impact relative binding site affinity (Supplemental Fig. S7A). Foxa2 is regulated by multiple CRMs (Fig. 3E); interestingly, all identified GBRs contain GBS variants that markedly reduced the relative affinity compared with the optimal target sequence (Table 1). In contrast, three GBRs near Nkx2.2 and Nkx2.9 all contain high-affinity variants (Santagati et al. 2003; Lei et al. 2006; Vokes et al. 2007). Foxa2 and Nkx2.2 expression depends on Gli2 activator input (Ding et al. 1998; Matise et al. 1998). Interestingly, Nkx6.1, Nkx6.2, and Olig2 are regulated by a Gli3-dependent derepression mechanism and contain CRMs with Gli motif variants that deviate from the optimal sequence (Table 1). The stark contrast between Foxa2 and Nkx2.2/Nkx2.9 GBS variants suggests that differential GBS affinity may play an important role in determining threshold-level responses to a GliA response.
Table 1.
Summary of PBM measurements for GBS associated with class II target genes
Differences in DNA-binding affinity are critical in distinguishing Nkx2.2 and Foxa2 responses to Gli activator input
To examine the potential role of GBS affinity in GliA-directed regulation of class II target genes, we introduced single-base-pair modifications that effectively swap high- and low-affinity GBSs of Nkx2.2(−2kb) and Foxa2(+10kb) CRMs, respectively (Fig. 5C; Supplemental Fig. S7B) in their enhancers. Each enhancer is dependent on a direct GliA input (Sasaki et al. 1997; Lei et al. 2006; Vokes et al. 2007). As expected, the wild-type Nkx2.2 enhancer (Nkx2.2high) drove GFP reporter expression in the pV3 progenitors and floor plate regions (Fig. 5D; Supplemental Fig. S7C); however, no activity was detected at embryonic day 10.5 (E10.5) with the low-affinity GBS variant (Nkx2.2low) (Fig. 5E; Supplemental Fig. S7D). Thus, whereas a low-affinity GBS is sufficient for Foxa2(+10kb) enhancer activity in the floor plate, a high-affinity GBS is essential for normal Nkx2.2(−2kb) enhancer activity in more dorsal pV3 progenitors.
In contrast, while the wild-type Foxa2(+10kb) element (Foxa2low) showed weak but appropriately localized floor plate activity at E10.5 (Fig. 5F), substitution of the native low-affinity GBS with the high-affinity variant (Foxa2high) resulted in a marked increase in reporter expression level as well as a dorsal expansion of the expression domain (Fig. 5G; Supplemental Fig. S7E). The dorsal boundary of reporter expression driven by the Foxa2high CRM variant extended to the limit of pV3 as indicated by Nkx2.2 (Fig. 5G). Collectively, these results are consistent with GBS affinity playing a critical role in mediating the appropriate GliA-dependent response of Foxa2(+10kb) and Nkx2.2(−2kb) CRMs. Furthermore, these results suggest that differences in Shh signaling levels in vivo may play into the timing and position of each gene's expression.
A suboptimal GBS in the Foxa2(+10kb) CRM is required for normal neural patterning
To determine whether enhancing Gli affinity within the CRM governing Foxa2 expression alters the patterning activity of Foxa2, we generated an AU1 epitope-tagged version of Foxa2 along with an independent nuclear GFP marker and generated transgenic embryos driving Foxa2 expression from wild-type (Foxa2low) or GBS variant (Foxa2high) CRMs (Fig. 6A).
Figure 6.

Critical role for Gli-binding affinity in patterning the ventral neural tube. (A) Schematic illustration of DNA constructs used in transgenic assay. (B–G) Transgenic embryos were analyzed at E10.5 at the forelimb level. (B,D,F) Expressing Foxa2 under the wild-type +10-kb element does not alter neural tube patterning. (C,E,G) Modifying the wild-type GBS in the Foxa2(+10kb) (Foxa2low) to a high-affinity variant (Foxa2high) increases reporter activity, alters floor plate morphology, and perturbs ventral patterning. (D,E) Increased levels of Foxa2 expression result in an expanded Shh domain. (F,G) Ventral neural patterning defects observed in Foxa2high embryos include a reduced number of Nkx2.2+ cells and the emergence of Foxa2/Olig2 double-positive cells. Bar, 50 μm.
As expected, the Foxa2low enhancer drove GFP reporter expression within the predicted floor plate domain at E10.5 with no detectable abnormalities (Fig. 6B,D,F). In contrast, the floor plate was expanded in embryos expressing Foxa2 from the Foxa2high enhancer, resulting in a lateral extension of Shh-expressing cells (Fig. 6D,E), elevated levels of Foxa2-AU1 and GFP levels within the Nkx2.2+ pV3 progenitor domain (Fig. 6B–E), and ectopic activity overlapping Olig2+ pMN progenitors (Fig. 6F,G). The elevated levels of Foxa2 in the Nkx2.2+ pV3 domain correlated with reduced levels of Nkx2.2, suggesting that Foxa2 attenuates Nkx2.2 expression (Fig. 6F,G). Interestingly, GFP was detected in both the Shh+ floor plate and more dorsal regions, whereas AU1 was largely excluded from the Shh+ domain (Fig. 6B–E). These observations are consistent with both an increased sensitivity of the Foxa2(+10kb) element to GliA input and the down-regulation of Hh signaling components in the floor plate (Fig. 7; Ribes et al. 2010). The observed difference between AU1 and GFP expression most likely reflects an increase in GFP protein stability compared with AU1-tagged Foxa2 and the absence of elements within the CRM that enable Foxa2-mediated autoregulation. Together, these results support a model in which GBS affinity plays a critical role in restricting Foxa2 activity, preventing a dorsal progression of Foxa2-mediated floor plate expansion.
Figure 7.

Shh signaling dynamics in developing neural tube. (A) Temporal progression of Shh signaling activity within the neural tube analyzed by smRNA FISH profiling of Hh pathway components. Position along the dorso–ventral (DV) axis is shown as relative position in each embryo (percentage to the entire length along the dorso–ventral axis). (B–G) Heat map representation of transcript densities indicating down-regulation of Ptch1 within the floor plate region followed by activation of Shh. Ventral (V) to dorsal (D), from left to right.
In vivo dynamics of Shh signaling response within the neural tube
To gain further insights into the dynamics of the Shh response, we used single-molecule RNA fluorescent in situ hybridization (smRNA FISH) as a digital readout of the Shh response (Raj et al. 2008). Both Ptch1 and Gli1 are direct targets of GliA, and their transcript levels provide an internal measure of Hh pathway activity. Strikingly, Ptch1 and Gli1 plots consistently show a maximum spike of signaling activity in the ventral-most quadrant of the neural tube at the eight- to 10-somite stages (E8.5) followed by a rapid decline in transcript level (Fig. 7A). Transcriptional activity is sufficiently high at E8.5, but not later stages, to detect two nuclear sites of intense FISH signal—presumably nascent transcripts generated at each Ptch1 allele (Supplemental Fig. S8A,B). Similar conclusions have been drawn using an artificial reporter comprising tandem repeats of the low-affinity GBS from Foxa2(+10kb), although the magnitude of the response is far greater and the decline in the response is markedly retarded when measured with the synthetic reporter of signaling levels compared with that measured in vivo through direct smRNA FISH of Shh targets (Balaskas et al. 2012).
The transient pulse of maximal signaling correlates well with the period of Shh/Gli-mediated specification of the Foxa2-dependent floor plate program and the onset of Shh transcription (Fig. 7B–G; Ribes et al. 2010). Furthermore, the rapid loss of Shh responsiveness that accompanies this spike is followed by a marked reduction in Gli2 transcription (Fig. 7A), consistent with Foxa2-directed inhibition of Hh pathway action as floor plate regulation switches from Gli-dependent to Foxa2 autoregulatory modes (Ribes et al. 2010). Indeed, Foxa2 binds to a conserved region within Gli2 that displays broad neural activity but strong silencing in the Foxa2+ floor plate domain (Supplemental Fig. S8C). In general, Gli2 RNA levels showed more time-dependent fluctuations compared with Gli3. The highest Gli2 levels were present in the intermediate neural tube, while Gli3 levels showed an inverse gradient to the Shh response, consistent with Shh-dependent inhibition of Gli3 (Fig. 7A). These data combined highlight a highly dynamic GliA-mediated signaling response in the ventral neural tube compared with a relatively stable dorsal GliR gradient.
Discussion
Analysis of the gene regulatory networks underlying developmental processes involves elucidating several layers of regulatory information that relate transcription factor occupancy to chromatin state and, ultimately, gene expression. Large-scale analyses pioneered by the ENCODE Consortium have recently defined a significant fraction of the human genome as harboring regulatory potential (Bernstein et al. 2012), yet a large gap remains between the identification of putative regulatory elements and the resolution of the mechanisms governing the tissue-specific programs of their actions. The vast amount of regulatory space highlights the potential for many disease-associated single-nucleotide polymorphisms (SNPs) to reside within regulatory elements controlling the timing, position, or level of a gene's expression, rather than the protein-coding potential of that gene. Indeed, mutations within regulatory regions are linked to altered developmental patterning responses—notably, a mutation in the 1-Mb distant enhancer of SHH associated with preaxial polydactyly (Lettice et al. 2003). Our analysis of the Shh-directed gene regulatory network in neural patterning highlights a group of conserved regulatory elements associated with key determinants of neural progenitor identity. We explored the mechanisms directing tissue-appropriate responses to Shh signaling and demonstrate that a single nucleotide substitution within a known GBS can significantly alter the activity of Gli targets and patterning of the mammalian neural tube.
Integration of Gli1-defined neural CRMs with Sox2 input
Developmental studies have highlighted a small number of signaling pathways mediating a great diversity of tissue patterning, raising the question of how tissue-specific outcomes arise from common signaling inputs. In this study, we uncovered a novel intersection between Gli and Sox factors. Sox2 is one of the earliest delineators of the neural lineage and is uniformly expressed along the dorso–ventral axis along with other SoxB1 family members. We demonstrated that Gli1 acts in conjunction with Sox activity to switch on distinct ventral neural progenitor determinants. In this Shh-directed patterning response, Sox2 likely acts permissively, binding to all CRMs directing class II gene activity independent of Shh input and class II gene activity. Shh regulation of Gli binding provides the instructive input to direct specific ventral cell fates, and Sox binding provides the neural context. neural context.
The SoxB1 family is critical for specification of neural cell fates and the maintenance of neural progenitors (Pevny and Placzek 2005). Genetic evidence indicates that SoxB1 members regulate the proliferation and timing of differentiation of neural progenitors, and recent evidence also links their actions to diversification of the progenitor pool (Bergsland et al. 2011). In a recent model, Sox factors are proposed to bind sequentially to neural target genes, with replacement of Sox2 in ESCs followed by Sox3 and then finally Sox11 in mature neurons (Bergsland et al. 2011). Although several neural-specific genes are associated with Sox2-bound regions in ESC studies, the putative CRMs highlighted by Sox2 binding do not display a permissive chromatin mark until commitment to the neural lineage, suggesting that Sox2 engagement is not sufficient to modify the chromatin signature (Bergsland et al. 2011).
In contrast, our analysis of class II targets shows that all associated GBRs, with the exception of the Nkx2.2 GBR, are not targeted by Sox2 in ESCs (Supplemental Fig. S3). The early neural Sox2 program is quite distinct from Sox2 target recognition in ESCs; however, the regulatory mechanisms that direct this shift in target recognition remain to be determined. Whereas the temporal progression of the neural patterning response is consistent with the requirement for Sox2 pre-engagement promoting Gli binding at enhancers of class II regulatory factors, this model is difficult to test. Sox2 activity is essential in both ESC and neural progenitor maintenance. In neural progenitors, loss of SoxB1 family activity leads to the rapid commitment of progenitors to post-mitotic neuron fates (Bylund et al. 2003; Graham et al. 2003). Interestingly, we show that regulatory regions near Sox2 are themselves prominent targets of Gli1 binding. Furthermore, transgenic analysis suggests that a Gli1 regulatory input into Sox2 enhancers contributes to the maintenance of Sox2 levels (data not shown). Thus, although the expression of Sox2 is initially independent of Shh, these regulatory regions may contribute to the maintenance of Sox2 levels in the ventral neural tube in order to ensure an ongoing Sox2 and Gli input over the extended period of neural progenitor patterning.
Floor plate fate specification through direct Gli regulation of Foxa2 expression
An important component of Shh-directed neural patterning is specification of the floor plate, a secondary Shh signaling center regulating ventral patterning and axon guidance (Placzek and Briscoe 2005). In the nascent floor plate, Gli2 activates Foxa2, and Foxa2 functions as a direct transcriptional activator of both Shh (Epstein et al. 1999) and its own expression, establishing an autoregulatory loop that rapidly supplants the direct Gli2 input. Foxa2 also attenuates further Shh signaling by down-regulating transcription of Gli2 and, potentially, other Hh pathway components through what is likely a direct repressive mechanism (Ribes et al. 2010; Mavromatakis et al. 2011; Metzakopian et al. 2012; this study) of the Foxa2-bound Gli2 CRM. The developmental logic to this regulatory circuit is the rapid induction and stable segregation of a new Shh signaling center at the ventral midline of the growing neural tube.
Our analysis demonstrates that GBRs associated with Foxa2 contain suboptimal GBSs. Furthermore, we show that the substitution of a high-affinity variant into a Foxa2 enhancer, previously shown in lineage-tracing experiments to only be active in the floor plate (Wang et al. 2011), displayed expanded reporter activity accompanied by an increased floor plate zone at the expense of the adjacent Nkx2.2+ pV3 population. This single nucleotide substitution is predicted to increase the sensitivity of the GBR to GliA input, extending the early period of direct Shh/Gli regulation of Foxa2. As with the same GBS in the Nkx2.2 enhancer, the modified Foxa2 enhancer supports reporter gene expression extending to the Nkx2.2/Olig2 boundary with occasional activity observed within the Olig2+, pMN zone. The dorsal limit of the Nkx2.2 population is not directly set by Gli inputs, but rather by cross-repressive interactions that likely include direct Olig2 input, as ectopic Olig2 expression represses Nkx2.2 expression (Novitch et al. 2001). The dorsal limit of the modified Foxa2 enhancer at the Nkx2.2/Olig2∷pV3/pMN boundary suggests that potential cross-repressive inputs also feed into Foxa2 CRMs.
Interestingly, ectopic Foxa2 was only observed in cells dorsal to Shh-producing cells, indicating that the Foxa2(+10kb) regulatory region lacks information required for autoregulatory function. Our studies identified additional enhancer elements supporting Foxa2 activity, including a +170-kb element that is bound by Gli1 and Foxa2 at near-adjacent sites within this CRM. Thus, while there is overlapping activity among Foxa2 enhancers, there is also likely a parsing of different regulatory modalities.
GBS affinity and initiation of target gene activity
A higher-affinity GBS is essential for the extended domain of Nkx2.2-producing cells, while a low-affinity GBS is required for transient ventral specification of the Foxa2-dependent floor plate. The floor plate is one of the first lineages to be specified, and its lineage is nonoverlapping with Nkx2.2+ cells that emerge progressively from cells that initiated earlier activity of the MN determinant Olig2 (Dessaud et al. 2007). These findings raise several interesting questions about the temporal and spatial inputs that shape the Shh response.
The first response to Shh signaling is the predicted attenuation of GliR activity. Variant GBSs with suboptimal binding are predicted to favor a rapid loss of GliR relative to GBSs with an optimal binding sequence. This likely contributes to the rapid activation of Nkx6.1 and Olig2, where loss of Gli3R is the central regulatory event, and may facilitate activation of Foxa2 prior to Nkx2.2 in ventral midline cells. The period of Shh-dependent control of Foxa2 is quite transient (Ribes et al. 2010) and corresponds to a time of maximal Shh signaling levels, as observed from direct in vivo transcript measurements of the generic Hh target genes Ptch1 and Gli1. This early spike in the Shh-directed transcriptional response may create a burst of GliA sufficient to engage the low-affinity GBSs for Foxa2 enhancers turning on Foxa2; multiple Gli-dependent CRMs for Foxa2 may also facilitate an early onset. However, the poor binding properties are predicted to favor the attenuation of Gli input as Shh signaling levels rapidly fall and a Foxa2 autoregulatory circuit completes the program of floor plate specification.
The affinity of Ci/Gli-binding sites is also postulated to control different spatial responses in Hh-dependent patterning of the Drosophila wing imaginal disc. In contrast to our data here, high-affinity sites governing ptc activity correlated with high expression restricted close to the source of Hh, while broad dpp expression was regulated through low-affinity interactions (Parker et al. 2011). In our analysis of GBS affinity, we focused on GliA-dependent outputs to control for potential differential binding responses between GliA and GliR forms encoded by different mammalian Gli genes (Sasaki et al. 1997; Ding et al. 1998; Lei et al. 2006; Vokes et al. 2007). The Drosophila Ci-regulated dpp CRM is most likely regulated through Ci-mediated derepression because Ci-binding site mutations result in broader reporter activity compared with wild type (Parker et al. 2011). Thus, weaker binding sites may favor a derepression-based mechanism for the regulation of the dpp CRM and, potentially, Olig2-, Nkx6.1-, and Nkx6.2-associated GBRs that display broader activity relative to the Shh source in the mammalian neural tube. In this view, the mechanisms governing GliA-dependent regulation of Nkx2.2 and Foxa2 CRMs are more akin to the regulation of dorsal target genes in Drosophila (Papatsenko and Levine 2005).
Importantly, while GBS affinity is one important determinant of the temporal and spatial readout of Shh morphogen signaling, there are clearly other inputs that sharpen and restrict the patterning response. No dorsal expansion of reporter gene activity was observed when GBSs in class II-regulated CRMs were mutated to disable Gli interaction (Fig. 4C,D,I,K). Thus, the dorsal limit of class II target gene expression is not set by the level of GliR input. Furthermore, loss of Gli3 does not lead to ectopic dorsal derepression of Shh targets such as Nkx6.1 or Olig2 even at early stages (Balaskas et al. 2012; data not shown). Here, class I-directed inhibitory inputs set dorsal boundaries, and our analysis predicts that these inputs are also feeding into the CRMs identified in this study. Recent evidence also points to a gene regulatory circuitry involving class I and class II factors: Cross-repressive interactions dependent on Pax6 (class I) and Olig2 (class II) can predict time-dependent responses to Shh signaling in the activation of Nkx2.2. Pax6 and Olig2 inhibition of Nkx2.2 is invoked as a barrier to Shh-mediated activation of Nkx2.2 that requires sustained, high-level Shh signaling to overcome (Balaskas et al. 2012). Although this regulatory network confers robustness to fluctuations in signaling levels over time, it does not account for the earlier onset of Olig2 activation that correlates with differential regulation by GliR and GliA forms. Other signaling inputs also appear to play a role in boundary setting: Evidence suggests that Tcf/Lef interactions indicative of canonical Wnt signaling regulate the dorsal boundary of Nkx2.2 (Lei et al. 2006; Wang et al. 2011).
Regulation of positive and negative feedback signaling components during neural patterning
Given data that argue for the regulatory significance of an early period of maximal Shh signaling for generating diversity among the most ventral Shh-dependent cell types, how are Shh signaling levels controlled by the ongoing gene regulatory networks? Cdo, Boc, and Gas1 encode coreceptors for Shh, promoting Shh engagement with its receptor, Ptch1 (Tenzen et al. 2006; Allen et al. 2007, 2011; Martinelli and Fan 2007). Triple-mutant studies indicate that activity of at least one member is essential for Shh-directed patterning (Allen et al. 2011). Notably, these genes are inhibited by Shh signaling in many contexts, and Boc and Gas1 are strongly down-regulated upon Shh pathway stimulation of neural cells, consistent with these genes being inhibited by class II factor-directed repression. Shh-responding cells also induce Ptch1 and Hhip, and their binding of Shh reduces signaling levels in a target cell (Jeong and McMahon 2005). Interestingly, Ptch1 and Hhip are direct targets of Gli1 action, and Ptch1 in particular displays an unusually complex cis-regulatory landscape with several Gli1 inputs. Thus, the absence of Shh pathway-dependent inhibitory inputs at the outset of ligand engagement of target cells is predicted to result in an early, maximal signaling response that is attenuated over time.
In summary, the studies herein demonstrate the predictive and analytical power of in vitro models in the elucidation of regulatory principles governing Shh-directed morphogen patterning of the mammalian CNS. The modality of Gli input, the affinity of GBSs, the engagement of feedback processes, and the neural context all contribute to the appropriate temporal and spatial emergence of Shh-dependent neural cell types.
Materials and methods
Neural progenitor culture, microarray analysis, and ChIP
Embryoid body culture was performed as previously described with minor modifications (Vokes et al. 2007). After 3 d of induction with 0.5 μM all-trans RA (Sigma) and 50–800 nM SAG (Alexis Biochemicals), RNA or chromatin samples were collected. Microarray analysis was performed on the Affymetrix Mouse 430 2.0 platform with paired biological triplicates. Gli1Flag ChIP was performed as described previously (Vokes et al. 2007). The antibodies used were anti-Flag (M2; Sigma), histone H3K4me2 (Abcam, ab7766), histone H3K27ac (Abcam, ab4729), and Sox2 (R&D Systems, AF2018). ChIP libraries were sequenced on the Genome Analyzer II or HiSeq platform (Illumina). To identify binding regions, an iterative conditional binomial model was applied to identify peaks enriched in experimental samples compared with a Flag-negative control ChIP on a parental cell line (YFP3.1) (Ma and Wong 2011). Region-based histone-profiling plots were generated from normalized sequence tag counts (reads per million) for a 3-kb region surrounding the peak center with overlapping regions merged. See the Supplemental Material for a complete description of analyses. All expression-profiling and ChIP-seq data associated with this study have been deposited to the Gene Expression Omnibus (GEO) database (GSE42594).
Immunofluorescence and smRNA in situ hybridization
Immunofluorescence was performed as previously described (Wijgerde et al. 2002) and imaged with Zeiss 700 or 710 confocal microscopes. Single-molecule in situ hybridization was performed as previously described (Raj et al. 2008). See the Supplemental Material for complete information.
Transgenic analysis of enhancer regions
G0 transgenic analysis was performed as previously described (Vokes et al. 2007) using a modified Hsp68-lacZ∷nGFP reporter construct (Tsanov et al. 2012). See the Supplemental Material for genomic coordinates of tested regions and a detailed description of binding site mutations. All studies involving vertebrate animals were performed with institutional approval in compliance with institutional guidelines.
Protein purification and PBM
The zinc finger domain of Gli1 (NP_034426; 223–410 amino acids), Gli2 (NP_001074594; 402–589 amino acids), or Gli3 (NP_032156; 465–652 amino acids) was cloned into pDEST15 (Invitrogen) and purified from Escherichia coli as a GST fusion protein. PBM experiments, analysis of PBM data, and determination of DNA-binding specificities were performed as described in Berger et al. (2006) and Berger and Bulyk (2009). Duplicate PBM experiments were performed on separate “all 10-mer” universal array designs in 4x44K Agilent array format (AMADID #015681 and #016060, Agilent Technologies, Inc.), and the data were averaged as described previously (Berger et al. 2006). Refer to the Supplemental Material for additional details.
Acknowledgments
We thank Zhenjuan Wang, Ying Chen, and Manfred Baetscher (Harvard Genome Manipulation Facility) for pronuclear injection; Steve Vokes (University of Texas, Austin) for advice on ChIP-seq; Yasushi Nakagawa (University of Minnesota) for Dbx1 antibody; Hiroshi Sasaki (RIKEN) for sharing the Foxa2 enhancer construct; James Briscoe (MRC) for Foxa2 cDNA; Johan Ericson (Karolinska) for sharing unpublished data; Diane Faria for technical assistance; and Alex Schier and Susan Mango for critical comments and helpful discussions. This work was supported by NIH grants R01 HG003985 (to M.L.B.), R01 RR021692-05 (to T.L.B.), R01 HG003903 (to W.H.W.), R01 HG005717 (to W.H.W.), and R37 NS033642 (to A.P.M); NRSA post-doctoral fellowships F32 GM090645 (to L.S.) and F32 GM087939 (to K.A.P); and training grant HSCI T32 HL087735 (to K.A.P.).
Note added in proof
A recent study by Oosterveen et al. (2012) provides additional support for the work herein.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.207142.112.
References
- Allen BL, Tenzen T, McMahon AP 2007. The Hedgehog-binding proteins Gas1 and Cdo cooperate to positively regulate Shh signaling during mouse development. Genes Dev 21: 1244–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen BL, Song JY, Izzi L, Althaus IW, Kang J-S, Charron F, Krauss RS, McMahon AP 2011. Overlapping roles and collective requirement for the coreceptors GAS1, CDO, and BOC in SHH pathway function. Dev Cell 20: 775–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai CB, Stephen D, Joyner AL 2004. All mouse ventral spinal cord patterning by hedgehog is Gli dependent and involves an activator function of Gli3. Dev Cell 6: 103–115 [DOI] [PubMed] [Google Scholar]
- Balaskas N, Ribeiro A, Panovska J, Dessaud E, Sasai N, Page KM, Briscoe J, Ribes V 2012. Gene regulatory logic for reading the sonic hedgehog signaling gradient in the vertebrate neural tube. Cell 148: 273–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger MF, Bulyk ML 2009. Universal protein-binding microarrays for the comprehensive characterization of the DNA-binding specificities of transcription factors. Nat Protoc 4: 393–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger MF, Philippakis AA, Qureshi AM, He FS, Estep PW III, Bulyk ML 2006. Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nat Biotechnol 24: 1429–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsland M, Ramskold D, Zaouter C, Klum S, Sandberg R, Muhr J 2011. Sequentially acting Sox transcription factors in neural lineage development. Genes Dev 25: 2453–2464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, Snyder M 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briscoe J, Pierani A, Jessell TM, Ericson J 2000. A homeodomain protein code specifies progenitor cell identity and neuronal fate in the ventral neural tube. Cell 101: 435–445 [DOI] [PubMed] [Google Scholar]
- Bylund M, Andersson E, Novitch BG, Muhr J 2003. Vertebrate neurogenesis is counteracted by Sox1-3 activity. Nat Neurosci 6: 1162–1168 [DOI] [PubMed] [Google Scholar]
- Chamberlain CE, Jeong J, Guo C, Allen BL, McMahon AP 2008. Notochord-derived Shh concentrates in close association with the apically positioned basal body in neural target cells and forms a dynamic gradient during neural patterning. Development 135: 1097–1106 [DOI] [PubMed] [Google Scholar]
- Chen JK, Taipale J, Young KE, Maiti T, Beachy PA 2002. Small molecule modulation of Smoothened activity. Proc Natl Acad Sci 99: 14071–14076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C, Litingtung Y, Lee E, Young KE, Corden JL, Westphal H, Beachy PA 1996. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature 383: 407–413 [DOI] [PubMed] [Google Scholar]
- Dessaud E, Yang LL, Hill K, Cox B, Ulloa F, Ribeiro A, Mynett A, Novitch BG, Briscoe J 2007. Interpretation of the sonic hedgehog morphogen gradient by a temporal adaptation mechanism. Nature 450: 717–720 [DOI] [PubMed] [Google Scholar]
- Dessaud E, McMahon AP, Briscoe J 2008. Pattern formation in the vertebrate neural tube: A sonic hedgehog morphogen-regulated transcriptional network. Development 135: 2489–2503 [DOI] [PubMed] [Google Scholar]
- Dessaud E, Ribes V, Balaskas N, Yang LL, Pierani A, Kicheva A, Novitch BG, Briscoe J, Sasai N 2010. Dynamic assignment and maintenance of positional identity in the ventral neural tube by the morphogen Sonic Hedgehog. PLoS Biol 8: e1000382 doi: 10.1371/journal.pbio.1000382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q, Motoyama J, Gasca S, Mo R, Sasaki H, Rossant J, Hui CC 1998. Diminished Sonic hedgehog signaling and lack of floor plate differentiation in Gli2 mutant mice. Development 125: 2533–2543 [DOI] [PubMed] [Google Scholar]
- Epstein DJ, McMahon AP, Joyner AL 1999. Regionalization of Sonic hedgehog transcription along the anteroposterior axis of the mouse central nervous system is regulated by Hnf3-dependent and -independent mechanisms. Development 126: 281–292 [DOI] [PubMed] [Google Scholar]
- Gaudet J, Mango SE 2002. Regulation of organogenesis by the Caenorhabditis elegans FoxA protein PHA-4. Science 295: 821–825 [DOI] [PubMed] [Google Scholar]
- Goodrich LV, Johnson RL, Milenkovic L, McMahon JA, Scott MP 1996. Conservation of the hedgehog/patched signaling pathway from flies to mice: Induction of a mouse patched gene by Hedgehog. Genes Dev 10: 301–312 [DOI] [PubMed] [Google Scholar]
- Graham V, Khudyakov J, Ellis P, Pevny L 2003. SOX2 functions to maintain neural progenitor identity. Neuron 39: 749–765 [DOI] [PubMed] [Google Scholar]
- Gritli-Linde A, Lewis P, McMahon AP, Linde A 2001. The whereabouts of a morphogen: Direct evidence for short- and graded long-range activity of hedgehog signaling peptides. Dev Biol 236: 364–386 [DOI] [PubMed] [Google Scholar]
- Hallikas O, Palin K, Sinjushina N, Rautiainen R, Partanen J, Ukkonen E, Taipale J 2006. Genome-wide prediction of mammalian enhancers based on analysis of transcription-factor binding affinity. Cell 124: 47–59 [DOI] [PubMed] [Google Scholar]
- Jeong J, McMahon AP 2005. Growth and pattern of the mammalian neural tube are governed by partially overlapping feedback activities of the hedgehog antagonists patched 1 and Hhip1. Development 132: 143–154 [DOI] [PubMed] [Google Scholar]
- Jessell TM 2000. Neuronal specification in the spinal cord: Inductive signals and transcriptional codes. Nat Rev Genet 1: 20–29 [DOI] [PubMed] [Google Scholar]
- Jiang J, Levine M 1993. Binding affinities and cooperative interactions with bHLH activators delimit threshold responses to the dorsal gradient morphogen. Cell 72: 741–752 [DOI] [PubMed] [Google Scholar]
- Kinzler KW, Vogelstein B 1990. The GLI gene encodes a nuclear protein which binds specific sequences in the human genome. Mol Cell Biol 10: 634–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei Q, Jeong Y, Misra K, Li S, Zelman AK, Epstein DJ, Matise MP 2006. Wnt signaling inhibitors regulate the transcriptional response to morphogenetic Shh–Gli signaling in the neural tube. Dev Cell 11: 325–337 [DOI] [PubMed] [Google Scholar]
- Lettice LA, Heaney SJ, Purdie LA, Li L, de Beer P, Oostra BA, Goode D, Elgar G, Hill RE, de Graaff E 2003. A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum Mol Genet 12: 1725–1735 [DOI] [PubMed] [Google Scholar]
- Ma W, Wong WH 2011. The analysis of ChIP-seq data. Methods Enzymol 497: 51–73 [DOI] [PubMed] [Google Scholar]
- Marti E, Bumcrot DA, Takada R, McMahon AP 1995. Requirement of 19K form of Sonic hedgehog for induction of distinct ventral cell types in CNS explants. Nature 375: 322–325 [DOI] [PubMed] [Google Scholar]
- Martinelli DC, Fan CM 2007. Gas1 extends the range of Hedgehog action by facilitating its signaling. Genes Dev 21: 1231–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matise MP, Epstein DJ, Park HL, Platt KA, Joyner AL 1998. Gli2 is required for induction of floor plate and adjacent cells, but not most ventral neurons in the mouse central nervous system. Development 125: 2759–2770 [DOI] [PubMed] [Google Scholar]
- Mavromatakis YE, Lin W, Metzakopian E, Ferri AL, Yan CH, Sasaki H, Whisett J, Ang SL 2011. Foxa1 and Foxa2 positively and negatively regulate Shh signalling to specify ventral midbrain progenitor identity. Mech Dev 128: 90–103 [DOI] [PubMed] [Google Scholar]
- Metzakopian E, Lin W, Salmon-Divon M, Dvinge H, Andersson E, Ericson J, Perlmann T, Whitsett JA, Bertone P, Ang SL 2012. Genome-wide characterization of Foxa2 targets reveals upregulation of floor plate genes and repression of ventrolateral genes in midbrain dopaminergic progenitors. Development 139: 2625–2634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novitch BG, Chen AI, Jessell TM 2001. Coordinate regulation of motor neuron subtype identity and pan-neuronal properties by the bHLH repressor Olig2. Neuron 31: 773–789 [DOI] [PubMed] [Google Scholar]
- Novitch BG, Wichterle H, Jessell TM, Sockanathan S 2003. A requirement for retinoic acid-mediated transcriptional activation in ventral neural patterning and motor neuron specification. Neuron 40: 81–95 [DOI] [PubMed] [Google Scholar]
- Oosterveen T, Kurdija S, Alekseenko Z, Uhde CW, Bergsland M, Sandberg M, Andersson E, Dias JM, Muhr J, Ericson J 2012. Mechanistic differences in the transcriptional interpretation of local and long-range shh morphogen signaling. Dev Cell 23: 1006–1019 [DOI] [PubMed] [Google Scholar]
- Pan Y, Bai CB, Joyner AL, Wang B 2006. Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol Cell Biol 26: 3365–3377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papatsenko D, Levine M 2005. Quantitative analysis of binding motifs mediating diverse spatial readouts of the Dorsal gradient in the Drosophila embryo. Proc Natl Acad Sci 102: 4966–4971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker DS, White MA, Ramos AI, Cohen BA, Barolo S 2011. The cis-regulatory logic of Hedgehog gradient responses: Key roles for gli binding affinity, competition, and cooperativity. Sci Signal 4: ra38 doi: 10.1126/scisignal.2002077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson M, Stamataki D, te Welscher P, Andersson E, Bose J, Ruther U, Ericson J, Briscoe J 2002. Dorsal–ventral patterning of the spinal cord requires Gli3 transcriptional repressor activity. Genes Dev 16: 2865–2878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pevny L, Placzek M 2005. SOX genes and neural progenitor identity. Curr Opin Neurobiol 15: 7–13 [DOI] [PubMed] [Google Scholar]
- Placzek M, Briscoe J 2005. The floor plate: Multiple cells, multiple signals. Nat Rev Neurosci 6: 230–240 [DOI] [PubMed] [Google Scholar]
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J 2011. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470: 279–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, Tyagi S 2008. Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods 5: 877–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribes V, Balaskas N, Sasai N, Cruz C, Dessaud E, Cayuso J, Tozer S, Yang LL, Novitch B, Marti E, et al. 2010. Distinct Sonic Hedgehog signaling dynamics specify floor plate and ventral neuronal progenitors in the vertebrate neural tube. Genes Dev 24: 1186–1200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roelink H, Porter JA, Chiang C, Tanabe Y, Chang DT, Beachy PA, Jessell TM 1995. Floor plate and motor neuron induction by different concentrations of the amino-terminal cleavage product of sonic hedgehog autoproteolysis. Cell 81: 445–455 [DOI] [PubMed] [Google Scholar]
- Rowan S, Siggers T, Lachke SA, Yue Y, Bulyk ML, Maas RL 2010. Precise temporal control of the eye regulatory gene Pax6 via enhancer-binding site affinity. Genes Dev 24: 980–985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santagati F, Abe K, Schmidt V, Schmitt-John T, Suzuki M, Yamamura K, Imai K 2003. Identification of cis-regulatory elements in the mouse Pax9/Nkx2-9 genomic region: Implication for evolutionary conserved synteny. Genetics 165: 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki H, Hui C, Nakafuku M, Kondoh H 1997. A binding site for Gli proteins is essential for HNF-3β floor plate enhancer activity in transgenics and can respond to Shh in vitro. Development 124: 1313–1322 [DOI] [PubMed] [Google Scholar]
- Tenzen T, Allen BL, Cole F, Kang JS, Krauss RS, McMahon AP 2006. The cell surface membrane proteins Cdo and Boc are components and targets of the Hedgehog signaling pathway and feedback network in mice. Dev Cell 10: 647–656 [DOI] [PubMed] [Google Scholar]
- Thomson M, Liu SJ, Zou LN, Smith Z, Meissner A, Ramanathan S 2011. Pluripotency factors in embryonic stem cells regulate differentiation into germ layers. Cell 145: 875–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsanov KM, Nishi Y, Peterson KA, Liu J, Baetscher M, McMahon AP 2012. An embryonic stem cell-based system for rapid analysis of transcriptional enhancers. Genesis 50: 443–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vokes SA, Ji H, McCuine S, Tenzen T, Giles S, Zhong S, Longabaugh WJ, Davidson EH, Wong WH, McMahon AP 2007. Genomic characterization of Gli-activator targets in sonic hedgehog-mediated neural patterning. Development 134: 1977–1989 [DOI] [PubMed] [Google Scholar]
- Vokes SA, Ji H, Wong WH, McMahon AP 2008. A genome-scale analysis of the cis-regulatory circuitry underlying sonic hedgehog-mediated patterning of the mammalian limb. Genes Dev 22: 2651–2663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Li Y 2006. Evidence for the direct involvement of βTrCP in Gli3 protein processing. Proc Natl Acad Sci 103: 33–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Fallon JF, Beachy PA 2000. Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell 100: 423–434 [DOI] [PubMed] [Google Scholar]
- Wang H, Lei Q, Oosterveen T, Ericson J, Matise MP 2011. Tcf/Lef repressors differentially regulate Shh–Gli target gene activation thresholds to generate progenitor patterning in the developing CNS. Development 138: 3711–3721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichterle H, Lieberam I, Porter JA, Jessell TM 2002. Directed differentiation of embryonic stem cells into motor neurons. Cell 110: 385–397 [DOI] [PubMed] [Google Scholar]
- Wijgerde M, McMahon JA, Rule M, McMahon AP 2002. A direct requirement for Hedgehog signaling for normal specification of all ventral progenitor domains in the presumptive mammalian spinal cord. Genes Dev 16: 2849–2864 [DOI] [PMC free article] [PubMed] [Google Scholar]