

Abstract
The anaphase promoting complex/cyclosome (APC/C) is an E3 ligase regulated by Cdh1. Beyond its role in controlling cell cycle progression, APC/C–Cdh1 has been detected in neurons and plays a role in long-lasting synaptic plasticity and long-term memory. Herein, we further examined the role of Cdh1 in synaptic plasticity and memory by generating knockout mice where Cdh1 was conditionally eliminated from the forebrain post-developmentally. Although spatial learning and memory in the Morris water maze (MWM) was normal, the Cdh1 conditional knockout (cKO) mice displayed enhanced reversal learning in the MWM and in a water-based Y maze. In addition, we found that the Cdh1 cKO mice had impaired associative fear memory and exhibited impaired long-term potentiation (LTP) in amygdala slices. Finally, we observed increased expression of Shank1 and NR2A expression in amygdalar slices from the Cdh1 cKO mice following the induction of LTP, suggesting a possible molecular mechanism underlying the behavioral and synaptic plasticity impairments displayed in these mice. Our findings are consistent with a role for the APC/C–Cdh1 in fear memory and synaptic plasticity in the amygdala.
In addition to de novo protein synthesis, protein degradation via the ubiquitin proteasome system (UPS) has emerged as a crucial component of synaptic plasticity and memory (Lopez-Salon et al. 2001; Ehlers 2003; Bingol and Schuman 2006; Fonseca et al. 2006; Karpova et al. 2006; Lee et al. 2008). Activity of three types of enzymes is coordinated in order to covalently ligate a chain of ubiquitin molecules onto a target protein, which is subsequently detected and degraded by the proteasome. This unique two-step mechanism of tagging followed by degradation enables the UPS to play a critical role in cellular processes requiring precise and distinct degradation of substrates, such as cell cycle regulation, DNA repair, and learning and memory (Weissman 2001). Though evidence for the UPS in learning and memory is emerging, little is known about the identity of the E3 ligases involved.
The anaphase promoting complex/cyclosome (APC/C) is an E3 ligase that has been well characterized for its role in driving cells through the completion of mitosis and maintaining them in interphase (Harper et al. 2002). Cdh1, a regulatory protein of the APC/C, along with several APC/C subunits, has been detected in postmitotic neurons, suggesting a novel role for APC/C–Cdh1 in neurons (Gieffers et al. 1999). Subsequently, several studies have demonstrated a range of roles for APC/C–Cdh1 in neurons, from preventing cell cycle progression (Almeida et al. 2005) to regulating axonal growth and patterning (Konishi et al. 2004), as well as contributing to synaptic plasticity (Juo and Kaplan 2004; van Roessel et al. 2004) and memory (Li et al. 2008; Kuczera et al. 2011). In addition, a number of novel substrates of APC/C–Cdh1 have been detected in neurons such as SnoN (Stegmüller et al. 2006) and Id2 (Lasorella et al. 2006). Because of the evidence showing the involvement of APC/C–Cdh1 in synaptic plasticity and memory, we sought to better characterize and determine the precise contribution of Cdh1 to learning and memory in a mouse model.
Using the cre-lox system, we generated Cdh1 conditional knockout mice (cKO) where Cdh1 was eliminated from excitatory neurons in the forebrain of adult mice. Although hippocampal synaptic plasticity and hippocampus-dependent memory remained intact, the Cdh1 cKO mice demonstrated enhanced behavioral flexibility in both the Morris water maze (MWM) and a water-based Y maze. Furthermore, we identified a role for Cdh1 in the amygdala as long-term potentiation (LTP) in amygdala slices, as well as both contextual and cued fear memory was impaired in the Cdh1 cKO mice. At the molecular level, we detected increased expression of a synaptic scaffolding protein, Shank1, and an NMDAR subunit, NR2A, after LTP-inducing stimulation in amygdala slices from the Cdh1 cKO mice, suggesting a possible molecular mechanism underlying the LTP impairments and memory deficits in these mice. To the best of our knowledge, this is the first time that a specific E3 ligase has been shown to play a role in synaptic plasticity and memory in the amygdala.
Results
Localization of Cdh1 in PSD fractions in neurons
Cdh1 was found to be expressed in postmitotic neurons (Gieffers et al. 1999) and has been detected in the nucleus in cultured cerebellar neurons (Konishi et al. 2004). Because there is increasing evidence that Cdh1 is involved in synaptic plasticity and memory (Juo and Kaplan 2004; van Roessel et al. 2004; Li et al. 2008), we hypothesized that it also could be localized in dendrites and postsynaptic densities (PSD). We first examined whether we could detect Cdh1 in dendrites using cultured rat hippocampal neurons. We employed immunofluorescence and were able to detect Cdh1 in dendrites (Fig. 1A). In order to detect synaptic Cdh1 in mammalian brain tissue, we fractionated hippocampal lysates from adult mouse brains and probed the fractions for the presence of Cdh1. We detected Cdh1 in both synaptoneurosome and PSD preparations, where Cdh1 appeared to be enriched (Fig. 1B). Furthermore, we confirmed previous findings that reported the presence of Cdh1 in the nucleus of cerebellar neurons (Konishi et al. 2004) by fractioning hippocampal lysates and detecting Cdh1 in all fractions examined, including the nuclear fraction (Fig. 1C). These findings suggest that the APC/C–Cdh1 is present in locations where it could play a role in dendritic and synaptic function, including synaptic plasticity.
Figure 1.
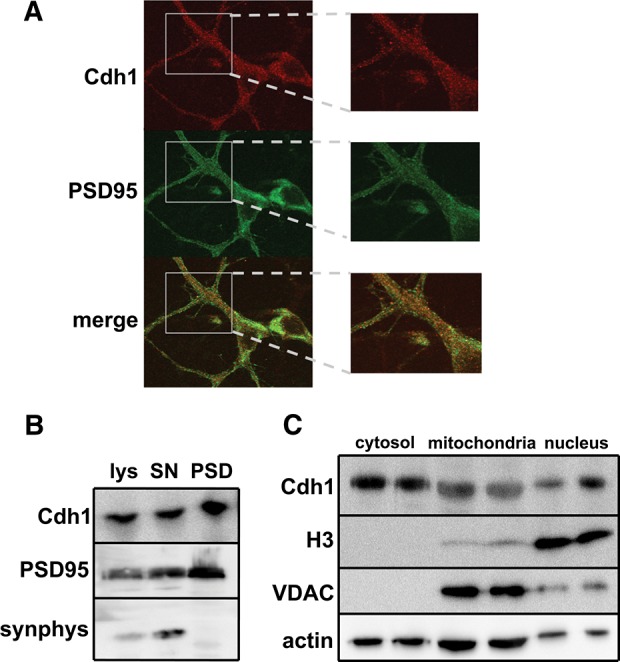
Subcellular localization of Cdh1 in neurons. (A) Immunofluorescent images of Cdh1 and PSD95 in dendrites of cultured rat hippocampal neurons. (Left) Images acquired with a 63× objective, (right) digital zoom highlights localization. (B) Western blots showing expression of Cdh1, PSD95, and synaptophysin (synphys) from mouse hippocampal brain lysates (lys), synaptoneurosome preparations (SN), and PSD preparations (PSD). (C) Western blots showing expression of Cdh1, H3, VDAC, and actin in mouse brains fractionated into cytosol, mitochondria, and nucleus preparations.
Generation of Cdh1 conditional knockout mice
A previous study from our laboratory demonstrated the involvement of Cdh1 in synaptic plasticity and memory by detecting impaired hippocampal LTP and contextual fear memory in constitutive Cdh1 heterozygous knockout mice (Li et al. 2008). However, because Cdh1 was globally reduced from the beginning of brain development, the precise contribution of Cdh1 to the synaptic plasticity and memory deficits is unclear. In order to assess the role of Cdh1 in the adult mammalian brain in a more precise manner, we generated conditional knockout (cKO) mice where Cdh1 was deleted specifically in excitatory neurons in the adult hippocampus and forebrain late in brain development.
In order to specifically eliminate Cdh1 from adult pyramidal neurons, we utilized the cre-lox system (Tsien et al. 1996). The Cdh1 protein is encoded by the Fzr1 gene. Mice expressing a loxP tag flanking exons 2 and 3 in the Fzr1 allele (García-Higuera et al. 2008) were crossed with mice expressing cre recombinase under a CaMKII promoter (Fig. 2A, T-29 line; Tsien et al. 1996). Under this promoter, cre recombinase is both regionally and temporally limited to expression in the hippocampus and forebrain, and expression begins ∼3 wk after birth (Tsien et al. 1996; Hoeffer et al. 2008).
Figure 2.

Generation of Cdh1 conditional knockout mice. (A) Schematic of the conditional Fzr1 allele. (Top) Triangles represent two loxP sites flanking exons 2 and 3 (E2–E3) in the Fzr1(lox) mice. (Bottom) After recombination with CamKIIα-Cre, exons 2 and 3 are excised thus preventing the functional expression of the Fzr1 allele in the Fzr1(−) mice. (B) PCR identification of alleles of Fzr1 and CaMKIIα-Cre. (C) Nissl stained sagittal sections of the hippocampus (HIP) (4× and 40×) and amygdala (AMY) (4×) for Cdh1 conditional knockout (cKO) and their wild-type (WT) littermates. (D) (Top) Representative Western blot showing CaMKIIα-driven cre disruption of Cdh1 in the prefrontal cortex (PFC), striatum (STR), amygdala (AMY), hippocampus (HIP), and cerebellum (CER) in Cdh1 cKO mice. GADPH was the loading control. (Bottom) Quantification of Cdh1 expression in various brain regions of WT and Cdh1 cKO mice. Mice were between 12 and 16 wk old, WT, n = 4 mice; cKO, n = 4 mice. (*) P < 0.05, Student's t-test; (**) P ≤ 0.01, Student's t-test.
Expression of the cre allele and the floxed Fzr1 gene was confirmed using PCR specific primers (Fig. 2B). Gross neuroanatomical structure remained intact in these mice (Fig. 2C). To confirm the knockdown of Cdh1, we examined tissue from prefrontal cortex, striatum, amygdala, hippocampus, and cerebellum of adult knockout mice (12–16 wk old). Robust reduction of Cdh1 expression was detected in the hippocampus and forebrain regions in the Cdh1 cKO mice when compared with their wild-type (WT) littermates (Fig. 2D).
Cdh1 cKO mice exhibit normal hippocampal long-term potentiation
Because it previously was demonstrated that constitutive Cdh1 heterozygous knockout mice had impairments in late phase LTP (L-LTP), but not early LTP (E-LTP) (Li et al. 2008), we first explored whether the Cdh1 cKO mice exhibited similar LTP phenotypes. E-LTP typically is induced with one train of high-frequency stimulation (HFS) (100 Hz) and requires posttranslational modifications of existing proteins whereas L-LTP typically is induced with four trains of HFS and, in addition to posttranslational modifications, requires new protein synthesis and protein degradation. Consistent with studies of the Cdh1 heterozygous knockout mice (Li et al. 2008), we detected no significant difference in E-LTP between the Cdh1 cKO mice and their wild-type littermates (Fig. 3A). However, in contrast with the Cdh1 heterozygous knockout mice, we found that L-LTP was indistinguishable between the Cdh1 cKO mice and their wild-type littermates (Fig. 3B). These findings suggest that previously described L-LTP impairments in the Cdh1 heterozygous knockout mice likely are due to developmental abnormalities rather than a requirement for Cdh1 function in the adult hippocampus.
Figure 3.

E-LTP and L-LTP are normal in hippocampal slices from Cdh1 cKO mice. (A) E-LTP induced with one train of high-frequency stimulation (HFS) was similar in slices from WT and Cdh1 cKO mice. WT, n = 7 slices; cKO, n = 8 slices. P > 0.05, ANOVA. (B) L-LTP induced with four trains of HFS remained potentiated after 180 min of recording in hippocampal slices from Cdh1 cKO mice, which did not differ from WT slices. WT, n = 10 slices; cKO, n = 11 slices. P > 0.05, ANOVA.
Cdh1 cKO mice exhibit impaired exploration on an open field arena task but normal behavior in sensory and motor behavioral tasks
In order to characterize the Cdh1 cKO mice and assess their sensory and motor capabilities, we tested the mice on a battery of behavioral tasks. Exploratory behavior was examined using an open field arena task. Mice were exposed to a brightly lit arena for fifteen minutes, and the amount of time the mice spent exploring the center of the arena vs. the periphery was recorded. Cdh1 cKO mice spent significantly less time in the center of the arena (Fig. 4A) and conversely more time in the periphery (Fig. 4B) compared with their wild-type littermates, suggesting that the tendency for the Cdh1 cKO mice to explore is impaired.
Figure 4.

Cdh1 cKO mice are impaired in their exploration in the open field task, but exhibit normal phenotypes in other behavioral tasks. (A,B) Cdh1 cKO mice spend a significantly lower percentage of time in the center and a higher percentage of time in the periphery than WT mice in an open field arena task. (C) There is no significant difference in the percentage of time Cdh1 cKO mice spend in either the closed or open arms of an elevated plus maze when compared with their WT littermates. (D) Cdh1 cKO mice and their WT littermates travel at a similar velocity in the elevated plus maze. (E) Cdh1 cKO mice and their WT littermates express a similar percentage of inhibition at varying decibel (db) levels of prepulse intensity in the prepulse inhibition task. (F) Both Cdh1 cKO mice and their WT littermates stayed on an accelerating rotarod for similar amounts of time over the course of 2 d on a rotarod task. WT, n = 8; cKO, N = 10. (**) P < 0.01, Student's t-test.
In order to examine whether the reluctance of the Cdh1 cKO mice to explore is reflective of enhanced anxiety-like behavior in these mice, we tested the Cdh1 cKO mice on an elevated plus maze (EPM). Mice were placed on a plus shaped elevated platform in which two of the four arms had high protective walls (closed arms), while the other two were open (open arms). Cdh1 cKO mice spent a similar amount of time in the closed arms and open arms as compared with their wild-type littermates (Fig. 4C). In addition, there was no difference in the velocity with which the Cdh1 cKO mice moved around the maze compared with their wild-type littermates (Fig. 4D). Thus, in contrast to the open field arena, the Cdh1 cKO mice exhibited normal anxiety-like behavior in the EPM.
In order to examine sensorimotor gating we tested the Cdh1 cKO mice on a prepulse inhibition (PPI) task. An auditory startle response can be inhibited when a strong startle stimulus is immediately preceded by a weaker auditory stimulus. Cdh1 cKO mice exhibited similar levels of PPI as their wild-type littermates (Fig. 4E), indicating that their sensorimotor gating capabilities are intact.
In order to assess the motor learning capabilities of the Cdh1 cKO mice, we used an accelerating rotarod task. Mice were placed on an accelerating rotarod for four trials over 2 d, and their latency to fall off was recorded. Cdh1 cKO mice were able to learn and perform this task as well as their wild-type littermates (Fig. 4F). These results indicate that the motor coordination and motor learning capabilities of Cdh1 cKO mice are intact.
Cdh1 cKO mice exhibit normal hippocampus-dependent memory but enhanced behavioral flexibility
We subsequently explored the spatial learning and memory of the Cdh1 cKO mice with the Morris water maze (MWM) and a water-based Y maze, tasks known to require intact hippocampal function (Morris et al. 1982; Hoeffer et al. 2008). In the MWM task, mice were placed in a pool of opaque water and were trained for 5 d to learn the location of a hidden platform by using spatial cues. After the final day of training, the hidden platform was removed and a probe test was performed. The latency for the Cdh1 cKO mice to find the hidden platform did not differ from wild-type mice during the training period (Fig. 5A). In addition, we detected no significant difference between Cdh1 cKO mice and their wild-type littermates during the probe test in either the amount of time spent in the target quadrant (Fig. 5B) or the number of platform crossings (Fig. 5C). Similarly, in a water-based Y maze task where mice are trained to learn which of the two arms contains a hidden escape platform, there was no difference between Cdh1 cKO mice and their wild-type littermates either in the number of trials required to achieve criteria or in their memory of which arm contained the hidden platform (Fig. 6A,B). Taken together, these findings indicate that Cdh1 cKO exhibit normal hippocampus-dependent spatial learning and memory.
Figure 5.

Cdh1 cKO mice exhibit normal spatial learning and memory, but enhanced reversal learning in the Morris water maze (MWM). (A) Average latency of Cdh1 cKO mice and their WT littermates to find the hidden platform in the MWM during a 5-d training protocol (B) Percentage of time that Cdh1 cKO and WT mice spent in each zone during the probe test. Platform is hidden in target zone. (C) Average number of times that Cdh1 cKO mice and their WT littermates crossed the location of the previously hidden platform during probe test. (D) Average latency of Cdh1 cKO mice and their WT littermates to find the hidden platform in the MWM over a 2-d reversal task. (E) Average latency of Cdh1 cKO and WT mice to find the new location of the hidden platform over the first three trials on day 1 of the reversal task. (F) Average latency of Cdh1 cKO mice and their WT littermates to find the hidden platform in the MWM over a 2-d visible platform task. WT, n = 8 mice; cKO, n = 10 mice. (*) P < 0.05; F(1,16) = 4.19 ANOVA.
Figure 6.

Cdh1 cKO mice exhibit enhanced reversal learning in the Y maze. (A) Average percentage of correct trials of Cdh1 cKO mice and their WT littermates on a Y maze task. (B) Average number of trials required for Cdh1 cKO mice and their WT littermates to achieve a criterion of 10 out of 20 correct arm choices during training. (C) Average number of trials required for Cdh1 cKO mice and their WT littermates to achieve a criterion of nine out of 10 correct arm choices during the reversal task. WT, n = 12 mice; cKO, n = 15 mice. (**) P < 0.001; F(10,165) = 3.37, ANOVA.
Although the Cdh1 cKO mice demonstrated normal learning and memory on the hippocampus-dependent tasks, their behavioral flexibility was enhanced when they subsequently had to find a new location of the hidden platform. In the MWM, after the initial 5-d training period, the hidden platform was moved to a different quadrant, and the latency of the mice to find the new location of the hidden platform was recorded. Over the first three trials on the first day of reversal learning, the knockout mice found the hidden platform significantly faster than their wild-type littermates (Fig. 5D,E). Similarly, when the hidden platform was moved to the other arm in the Y maze, the Cdh1 cKO mice consistently found the new location of the hidden platform significantly faster than their wild-type littermates (Fig. 6A,C). When the hidden platform was raised to a visible level above the water, Cdh1 cKO mice performed similarly to their wild-type littermates (Fig. 5F), confirming that their vision was normal. These findings suggest that, although reducing Cdh1 in neurons of adult mice had no impact on hippocampus-dependent learning and memory, their behavioral flexibility was enhanced when the mice needed to adjust their prior knowledge and find the new location of the hidden platform.
Cdh1 cKO mice exhibit impairments in both contextual and cued fear conditioning
We subsequently examined the associative memory of the Cdh1 cKO mice using a standard fear conditioning protocol. Mice were placed in a unique training environment and were conditioned to associate the novel context and a neutral auditory tone (CS) with a mild footshock (US). Wild-type mice learn to make these associations and will demonstrate freezing behavior when later reintroduced to the same training environment or re-exposed to the auditory cue. Although the same training protocol is employed for both cued and contextual fear conditioning and both paradigms require an intact amygdala, only contextual fear conditioning requires the involvement of the hippocampus (Phillips and LeDoux 1992).
It previously was demonstrated that constitutive Cdh1 heterozygous knockout mice are impaired in contextual, but not cued, fear conditioning (Li et al. 2008). Consistent with Cdh1 heterozygous knockout mice, the Cdh1 cKO mice exhibited a significant reduction in their freezing behavior in contextual fear conditioning when their freezing behavior was measured 24 h after training (Fig. 7A,B). However, in contrast to the constitutive Cdh1 heterozygous knockout mice, the freezing behavior of the Cdh1 cKO mice also was significantly impaired in cued fear conditioning tested 24 h after training (Fig. 7C,D). This impairment in cued fear conditioning was not due to the mice recognizing the original context because the baseline freezing prior to the onset of the first test tone was comparable to that of the wild-type mice (Fig. 7C,D). All together these findings indicate that Cdh1 cKO mice have impaired associative fear memory, both contextual and cued, compared with their wild-type littermates.
Figure 7.

Cdh1 cKO mice exhibit impaired associative fear memory. (A) Average percentage of time that Cdh1 cKO mice and their WT littermates spent freezing over each minute for the 5-min training context re-exposure 24 h after training. (B) Average percentage of time over the entire 5 min that Cdh1 cKO mice and their WT littermates spent freezing when re-exposed to the training context 24 h after training. WT, n = 8; cKO, n = 10, (*) P < 0.05; F(1,16) = 5.5, one-way ANOVA. (C) Average percentage of time that Cdh1 cKO mice and their WT littermates spent freezing during each conditioned stimulus (CS1 and CS2) presentation in a novel context, 25 h after training. (D) Average percentage of time that Cdh1 cKO mice and their WT littermates spent freezing in a novel context during the entire auditory cue (tone) presentation as well as 30 sec prior to onset of auditory cue (pre-tone), 25 h after training. WT, n = 8; cKO, n = 10, (**) P = 0.01; F(1,16) = 8.29, one-way ANOVA.
Cdh1 cKO mice exhibit impairments in LTP in the amygdala
Because the amygdala is known to be crucial for both cued and contextual fear memory, and because we were unable to detect any impairments in hippocampal plasticity and memory in the Cdh1 cKO mice, we asked whether the impairment in their associative fear memory was due to a requirement for Cdh1 in synaptic plasticity in the amygdala. To test this prediction, we first examined whether a presynaptic form of synaptic plasticity, paired pulse facilitation (PPF), was altered in the Cdh1 cKO mice. We detected no difference in PPF between the Cdh1 cKO mice and their wild-type littermates (Fig. 8A). We proceeded to use a slightly modified protocol known to induce L-LTP by stimulating thalamic afferents and recording in the amygdala (Huang and Kandel 1998) in coronal slices from Cdh1 cKO mice and their wild-type littermates. Upon stimulating the slices with five trains of HFS, we detected an impairment in L-LTP in amygdala slices from Cdh1 cKO mice beginning ∼2 h after the last train of HFS (Fig. 8B). These findings suggest that, although reduction of Cdh1 does not alter synaptic plasticity or memory in the adult mouse hippocampus, post-developmental removal of Cdh1 results in impaired synaptic plasticity and memory deficits in the adult amygdala.
Figure 8.

L-LTP in the amygdala is impaired in Cdh1 cKO mice. (A) Paired-pulse facilitation (PPF) in Cdh1 cKO mice and their wild-type (WT) littermates is shown for interpulse intervals of 10–300 msec. PPF in Cdh1 cKO mice was not different from that in WT littermates. WT, n = 16 slices; cKO n = 16 slices. (B) L-LTP induced with five trains of HFS is impaired in amygdala slices. WT, n = 8 slices; cKO, n = 10 slices. P < 0.05, Student's t-test for the average of the final 30 min of recording.
The expression of Shank1 and NR2A are increased in the amygdala of Cdh1 cKO mice after LTP-inducing stimulation
In order to identify a potential molecular mechanism that could underlie the impairments that we detected in associative fear memory and amygdala LTP, we reasoned that since Cdh1 is found in abundance in the PSD (Fig. 1B), perhaps impairing Cdh1 leads to abnormal increases in PSD proteins that are involved in synaptic plasticity. Shank1 is a member of the family of Shank proteins found at the PSD and a major scaffolding protein that links together several different protein complexes containing different types of glutamate receptors, including NMDA receptors (Lim et al. 1999; Naisbitt et al. 1999; Sheng and Kim 2000). Shank1 has been shown to be ubiquitinated and degraded by the UPS, and is involved in activity-dependent restructuring of the PSD (Ehlers 2003; Lee et al. 2008; Hung et al. 2010). Shank1 also contains a D box, one of the sequences recognized by APC/C–Cdh1, and it has been reported in cultured hippocampal neurons that eliminating APC2, a subunit of the APC/C, results in increased expression of Shank1 (Hung et al. 2010). We initially examined the basal expression of Shank1 in amygdala tissue from Cdh1 cKO and wild-type mice but did not observe any differences between the genotypes. We then applied the LTP-inducing protocol of five trains of HFS to amygdala slices and examined the expression levels of Shank1 45 min after the stimulation. We detected a robust LTP-associated increase in the levels of Shank1 in amygdala slices from the Cdh1 cKO mice compared with slices from their wild-type littermates (Fig. 9A,B). These findings indicate that Shank1 expression is abnormally increased during LTP in the Cdh1 cKO mice.
Figure 9.

L-LTP inducing stimulation results in increased expression of Shank1 and NR2A in Cdh1 cKO mice. (A) Representative Western blot of Shank1 in amygdala tissue from WT and Cdh1 cKO mice after no stimulation (control) and five trains of HFS. Actin was used as a loading control. The Shank1 antibody recognizes multiple isoforms (Lim et al. 1999; Lee et al. 2008). (B) Quantification of Shank1 expression in WT and Cdh1 cKO mice after no stimulation (control) and five trains of HFS. WT, n = 5 slices; cKO, n = 4 slices. (**) P < 0.01, Student's t-test. (C) Representative Western blot of NR2A in amygdala tissue from WT and Cdh1 cKO mice after no stimulation (control) and five trains of HFS stimulation. (D) Quantification of NR2A expression in WT and Cdh1 cKO mice after no stimulation (control) and five trains of HFS. WT, n = 5 slices; cKO, n = 4 slices. (**) P < 0.01, Student's t-test.
Because Shank1 is a synaptic scaffolding protein that is known to bind proteins that anchor receptors in the PSD, we further explored other PSD proteins, including various glutamate receptor subunits, to determine whether the LTP-induced increase in Shank1 expression in the Cdh1 cKO mice was correlated with increased expression of other PSD proteins. Although we did not detect any differences in the basal level of protein expression between Cdh1 cKO and wild-type mice, expression of NR2A was increased in amygdala slices in Cdh1 cKO mice following the induction of L-LTP (Fig. 9C,D). The increases in Shank1 and NR2A expression are consistent with the idea that Shank1 could be a substrate of Cdh1 and point to a possible mechanism that may underlie the fear memory and LTP phenotypes we have detected in the Cdh1 cKO mice.
The expression of NR2A is unchanged in hippocampal slices after LTP-inducing stimulation and blocking NR2A activity does not rescue the LTP impairment in amygdala slices
Because Shank1 and NR2A expression was increased in the amygdala following the induction of LTP, we next examined whether the expression of Shank1 and NR2A was increased in hippocampal slices from Cdh1 cKO mice following induction of L-LTP. Because we did not detect any differences in hippocampal LTP in the Cdh1 cKO mice (Fig. 3), we expected there to be no change in expression of either Shank1 or NR2A. However, we did detect enhanced Shank1 expression following the induction of L-LTP in hippocampal slices from Cdh1 cKO mice when compared with their wild-type littermates (Fig. 10A). In contrast, we did not detect any change in expression of NR2A in hippocampal slices from Cdh1 cKO mice following induction of LTP (Fig. 10A). These findings indicate that in the hippocampus, the induction of L-LTP results in abnormal expression of Shank1 but not NR2A.
Figure 10.

NR2A expression is unaltered in hippocampal slices after the induction L-LTP, and inhibition of NR2A does not rescue L-LTP impairments in the amygdala of Cdh1 cKO mice. (A) (Left) Representative Western blots of Shank1 and NR2A in hippocampal tissue from WT and Cdh1 cKO mice after either no stimulation (control) or four trains of HFS. Actin was used as a loading control. (Right) Quantification of Shank1 and NR2A expression in WT and Cdh1 cKO mice after either no stimulation (control) or four trains of HFS. WT, n = 8 slices; cKO, n = 9 slices. (**) P < 0.01, Student's t-test. (B) L-LTP induced with five trains of HFS is impaired in amygdala slices from Cdh1 cKO mice in control slices and slices treated with 400 nM PEAQX. Control, n = 4 slices; PEAQX, n = 3 slices.
Because NR2A expression was enhanced following induction of L-LTP in amygdala but not hippocampal slices, we determined whether pharmacologically impairing the activity of NR2A-containing NMDARs following induction of LTP could rescue the L-LTP impairment in the Cdh1 cKO mice. Amygdala slices from the Cdh1 cKO mice were incubated with either vehicle or 400 nM of PEAQX, which preferentially antagonizes NR2A-containing NMDARs (Berberich et al. 2005), beginning 20 min after the final train of HFS. We found that L-LTP was impaired in slices from Cdh1 cKO mice regardless of whether they were treated with either vehicle or PEAQX (Fig. 10B). These findings suggest that enhanced NR2A activity is not responsible for the impaired L-LTP in amygdalar slices from Cdh1 cKO mice.
Discussion
Although evidence of the involvement of the UPS in synaptic plasticity and memory is growing (Lopez-Salon et al. 2001; Ehlers 2003; Bingol and Schuman 2006; Bingol and Sheng 2011), there are only a handful of studies describing the involvement of specific E3 ligases in vertebrates (Jiang et al. 1998; Yao et al. 2011). Accumulating evidence demonstrating the expression of APC/C–Cdh1 in neurons indicates that APC/C–Cdh1 plays a role in regulating synaptic plasticity (Juo and Kaplan 2004; Konishi et al. 2004; van Roessel et al. 2004; Fu et al. 2011) and memory (García-Higuera et al. 2008; Li et al. 2008; Kuczera et al. 2011). Here, we confirmed that Cdh1 is expressed at synapses in adult mice by examining the specific subcellular localization of Cdh1 in neurons. In addition, using Cdh1 cKO mice where Cdh1 is removed post-developmentally in the forebrain, we found evidence for the involvement of Cdh1 in behavioral flexibility, amygdala-dependent synaptic plasticity and memory, and plasticity-associated changes in molecular targets that may underlie these differences.
Previous work from our laboratory demonstrated the involvement of Cdh1 in synaptic plasticity and memory that was based on the detection of impairments in contextual fear conditioning and hippocampal L-LTP in constitutive Cdh1 heterozygous knockout mice (Li et al. 2008). Because Cdh1 expression was reduced globally in the mice, we wanted to examine if the synaptic plasticity impairments and memory deficits were maintained when Cdh1 was eliminated only in excitatory neurons of adult mice. Utilizing the cre-lox system, we generated mice with reduced post-developmental expression of Cdh1 in the forebrain. Although Cdh1 cKO mice displayed normal hippocampal synaptic plasticity and memory, they did display enhanced behavioral flexibility, as they reversed faster than their wild-type littermates during reversal tasks in the MWM and the water-based Y maze.
We considered two potential explanations for the enhanced behavioral flexibility in the Cdh1 cKO mice. The medial PFC (mPFC) is known to mediate behavioral flexibility (Sotres-Bayon et al. 2006), and consistent with the observation that there was a robust decrease in Cdh1 expression in the PFC of Cdh1 cKO mice (Fig. 2D), it is possible that impaired Cdh1 activity in the mPFC may underlie the enhanced behavioral flexibility in the Cdh1 cKO mice. An alternative explanation is that the mechanisms underlying behavioral flexibility may be intact in the Cdh1 cKO mice, but the confidence in their memory may be impaired. Thus, although we were unable to detect any impairment in the hippocampus-dependent memory tasks we examined, if the Cdh1 cKO mice were not as confident in their memory, they might be more flexible and willing to explore new locations of the hidden platform.
There are several ways to account for the fact that hippocampal synaptic plasticity and memory impairments were detected in constitutive Cdh1 heterozygous knockout mice, but not in Cdh1 cKO mice. First, in the Cdh1 heterozygous knockout mice, Cdh1 was reduced in all cells, whereas Cdh1 is reduced only in excitatory neurons in the Cdh1 cKO mice. Because Cdh1 is known to have important functions in glial cells (Silies and Klämbt 2010), it is possible that eliminating Cdh1 from glial cells has a detrimental effect on hippocampal synaptic plasticity and memory. Alternatively, because the reduction of Cdh1 in constitutive heterozygous knockout mice begins at the earliest stages of embryonic development whereas Cdh1 is eliminated in the cKO mice 2 to 3 wk after birth, it is possible that the contribution of Cdh1 in the hippocampus is developmental and does not play a role in synaptic plasticity and memory in adult mice.
Most studies exploring the involvement of the UPS and E3 ligases in synaptic plasticity and memory have been limited to the hippocampus (Jiang et al. 1998; Fonseca et al. 2006; Karpova et al. 2006; Lee et al. 2008; Li et al. 2008; Yao et al. 2011). Although there are several studies that are consistent with the notion of a role for the proteasome in the amygdala (Yeh et al. 2006; Mao et al. 2008; Jarome et al. 2011; Rodriguez-Ortiz et al. 2011), to the best of our knowledge there are no studies that have identified a role for a specific E3 ligase in amygdala-dependent synaptic plasticity and memory. In this study we have shown that Cdh1 is required for amygdala-dependent memory using Cdh1 cKO mice. The Cdh1 cKO mice exhibited deficits in both contextual and cued fear memory, both of which require a functioning amygdala (Phillips and LeDoux 1992), as well as impaired L-LTP in amygdala slices. Consistent with these impairments, Cdh1 expression was reduced in the amygdala of these mice. In addition, we have detected increased expression of two PSD proteins, Shank1 and NR2A, after the induction of LTP in amygdala slices from Cdh1 cKO mice. These findings suggest that Cdh1 normally is involved in maintaining the appropriate protein complexes in the PSD following neuronal activity.
Together, these findings suggest that Cdh1 has distinctive roles in different brain regions either at various developmental times or in different types of cells. Because Cdh1 is part of the APC/C, which is known to cause the degradation of numerous substrates, it is possible that the divergent effects of Cdh1 activity in the hippocampus and amygdala are due to a diverse expression of the various Cdh1 substrates in these brain regions. Alternatively it is possible that different temporal expression of Cdh1 substrates may underlie the differences in the contribution of Cdh1 in the amygdala and hippocampus.
Our finding of increased expression of Shank1 following the induction of LTP in amygdala slices of Cdh1 cKO mice is consistent with several studies that demonstrate degradation of Shank1 in an activity-dependent manner via the UPS (Ehlers 2003; Lee et al. 2008; Hung et al. 2010), and lends further support to the idea that Shank1 may be a substrate of APC/C–Cdh1. Thus, Cdh1 may normally degrade Shank1 following synaptic stimulation, and eliminating Cdh1 results in increases in Shank1 and the NR2A subunit of the NMDAR.
Because Shank1 expression was enhanced in both amygdala and hippocampal slices following the induction of L-LTP but NR2A expression was only enhanced following induction of L-LTP in the amygdala, it is possible that there is an interaction between Shank1 and NR2A in the amygdala, but not in the hippocampus. Further studies are needed to determine whether there are amygdala-specific interactions of Shank1 and NR2A and how they are altered by the lack of Cdh1.
Although inhibiting NR2A activity did not rescue the impaired L-LTP in amygdala slices from Cdh1 cKO mice (Fig. 10), it is possible that after induction of L-LTP normal NR2A removal from the synapse and degradation is prevented in the Cdh1 cKO mice. Thus, overexpression of NR2A would limit the amount of space available for the normal insertion of AMPARs following the induction of L-LTP. Thus, the physical overexpression of NR2A rather than increased NR2A activity may result in impaired L-LTP. This possibility remains to be determined.
It has been shown that increases in NR2A in the amygdala and the hippocampus can have detrimental behavioral effects. For example, NR2A levels in the rat amygdala are normally reduced after fear conditioning (Zinebi et al. 2003), suggesting that increased NR2A levels could lead to altered fear memory. In addition, increased levels of NR2A in the amygdala are associated with depression (Karolewicz et al. 2009). Furthermore, mouse models of Down syndrome overexpressing the Dyrk1A gene have impaired hippocampal synaptic plasticity and memory (Altafaj et al. 2008), and the increased expression of the Dyrk1A gene is associated with increased expression of NR2A (Ahn et al. 2006). These studies support the notion that aberrant increases in NR2A expression can have adverse effects on normal amygdala function.
Although earlier studies of the molecular targets of Cdh1 in neurons focused on nuclear substrates, specifically transcription factors, those studies examined the function of Cdh1 in a basal state (Konishi et al. 2004; Lasorella et al. 2006; Stegmüller et al. 2006). A recent study demonstrated that Cdh1 may play a role in synaptic function, mediating the degradation of GluR1 during homeostatic plasticity (Fu et al. 2011). Our finding of Cdh1 localization in the PSD in addition to the increases in expression of the PSD proteins Shank1 and NR2A after induction of synaptic plasticity is consistent with this expanded role of Cdh1 in regulating synaptic proteins in response to neuronal activity.
A recent study was conducted on APC2 cKO mice in which the same CaMKII promoter was used to eliminate APC2, a subunit from the APC/C (Kuczera et al. 2011). The APC2 cKO mice displayed impairments in spatial memory in the MWM and associative fear memory that differ from our findings. Although both studies used the same promoter to impair the same E3 ligase, they diverged in the particular subunit that was eliminated. APC2 is an essential protein of the catalytic region of the APC/C (Thornton et al. 2006), and eliminating it would impair general APC/C activity. However, eliminating Cdh1 does not necessarily impair general APC/C activity because the APC/C can function through a second adaptor protein, Cdc20, that has been found in postmitotic neurons as well (Yang et al. 2009; Puram et al. 2011). Thus, it is possible that the memory impairments displayed by the APC2 cKO mice are due to the elimination of all APC/C activity, whereas the memory impairments displayed by the Cdh1 cKO mice are the result of elimination of only one branch of APC/C activity in excitatory neurons.
There is an alternative explanation for the different behavioral phenotypes displayed by the APC2 cKO and Cdh1 cKO mice. It is known that Cdh1 not only regulates APC/C activity, but it also is a target of the APC/C and helps mediate its own degradation (Listovsky et al. 2004). Consistent with this idea, although the APC/C is impaired in the APC2 cKO mice, the expression of Cdh1 itself is increased, presumably due to the lack of degradation via the APC/C (Kuczera et al. 2011). Thus, it is possible that the increased expression of Cdh1 in the APC2 cKO mice either has an alternative function independent of the APC/C or competitively binds to and isolates its targets from normal function (Wan et al. 2011).
In closing, we have shown a role for Cdh1 in amygdala-dependent synaptic plasticity and memory, demonstrating a role for a specific E3 ligase in these processes in a brain region other than the hippocampus. In addition, we have presented evidence that Cdh1 may play a role regulating the expression of PSD proteins in response to synaptic activity. Further studies are required to determine whether deregulation of Shank1 and NR2A expression in the amygdala are responsible for the L-LTP impairments and memory deficits displayed by the Cdh1 cKO mice.
Materials and Methods
Conditional knockout mice
Mice with floxed Fzr1 (Fzr1fl) alleles were generated as described previously (García-Higuera et al. 2008). The mouse Fzr1 locus encodes the Cdh1 gene. Mice expressing cre recombinase (T-29) under the CaMKII promoter were kindly provided by Dr. Susumu Tonegawa from MIT (Tsien et al. 1996). Mice were genotyped using cre-specific primers and primers that identify floxed alleles of the Fzr1 locus. Mice for these studies were generated using the following breeding strategy: Male cre(−/−) Fzr1fl(+/−) Fzr1(+/−) were crossed to cre(+/+) Fzr1fl(+/−) Fzr1(+/−) females. The WT mice in this study were cre(+/−) Fzr1fl(−/−) Fzr1(+/+), whereas the knockout mice were cre(+/−) Fzr1fl(+/+) Fzr1(−/−).
Immunohistochemistry
Cultured rat hippocampal neurons were kindly provided by Dr. Moses Chao from the NYU Langone Medical Center. Neurons were treated and processed for fluorescence microscopy as described previously (Deinhardt et al. 2011). Briefly, neurons were fixed for 10 min in phosphate-buffered solution (PBS) with 4% paraformaldehyde and 20% sucrose, followed by 5 min in 50 mM NH4Cl in PBS. Cells then were permeabilized by incubating for 1–3 min in 0.1% Triton X-100 in PBS. Cells were blocked in Tris-buffered solution (TBS) containing 10% normal donkey serum, 2% bovine serum albumin, and 0.25% fish skin gelatin for 30 min, incubated with primary antibodies in blocking solution for 30 min, washed three times in TBS, followed by another 30-min incubation with secondary antibodies bound to fluorophores in blocking solution, and washed three times with TBS before mounting onto slides for imaging.
Cellular fractionation
Mouse brains were pooled and homogenized in buffer (4 mM HEPES, 320 mM sucrose, 1 mM DTT, 1 mM orthovanadate, 1× complete protease inhibitor cocktail) on ice with a 2-mL Kontes Dounce tissue grinder (A and B pestle, 20 strokes each). All centrifugations were performed at 4°C. The homogenate was centrifuged at 800g for 3 min, and the enriched nuclear pellet was carefully washed with buffer and centrifuged five to six times 800g for 3 min. The supernatant was subsequently centrifuged at 17,000g for 55 min. The resulting pellet, enriched with mitochondria, was washed twice at 17,000g for 15 min, and the remaining supernatant contained the cytoplasm enriched fraction. The resulting pellets were resuspended in buffer for Western blot analysis.
PSD preparations
PSD preps were performed as described previously (Villasana et al. 2006). Briefly, mouse brains were homogenized in buffer (10 mM HEPES, 1 mM EDTA, 2 mM EGTA, 0.5 mM DTT, 1 mM orthovanadate, 1× complete protease inhibitor cocktail) on ice with a 2-mL Kontes Dounce tissue grinder (A and B pestle, 20 strokes each). The sample was filtered twice through three layers of a pre-wetted 100-μm pore nylon filter. The resulting filtrate was filtered through a pre-wetted 5-μm pore hydrophilic filter. The filtrate was centrifuged at 1000g for 10 min. The pellet containing synaptoneurosomes was resuspended in buffer. The synaptoneurosomes then were diluted with buffer containing 1% Triton X-100, 32 mM sucrose, and 12 mM Tris-HCl and centrifuged at 33,000g for 20 min. The pellet was resuspended in buffer and layered onto a sucrose gradient containing 1 M sucrose and 1 mM NaHCO3, as well as 1.5 M sucrose and 1 mM NaHCO3, and centrifuged for 2 h at 200,000g. The pellet was resuspended in buffer containing 1% Triton X-100 and 150 mM KCl and centrifuged for 30 min at 167,000g. The resulting pellet was resuspended for Western blot analysis.
Western blots
Total protein levels were quantified using a BCA assay (Bio-Rad Protein Assay) and 15 µg of protein per lane was loaded on a 10% SDS-polyacrylamide gel and separated by electrophoresis. Protein bands were transferred onto a nitrocellulose membrane and blots were blocked in 5% nonfat dry milk +0.1% Tween-20 for 60 min. Membranes were incubated with antibodies to either Cdh1 (MBL, 1:500), GAPDH (Cell Signaling, 1:10,000), actin (Sigma, 1:10,000), PSD95 (Chemicon, 1:2000), H3 (Millipore, 1:5000), VDAC (Abcam, 1:1000), Shank1 (Novus Biologicals, 1:250), NR2A (Imgenex, 1:2000), or synaptophysin (Chemicon, 1:1000) overnight at 4°C. Excess antibody was removed by three washes in T-TBS for 5 min each. Membranes then were incubated in secondary HRP-conjugated anti mouse or anti-rabbit IgG (Promega, 1:5000) for 60 min and excess antibody was again removed by three washes in T-TBS for 5 min each. Blots were visualized using a Kodak imager. Protein bands were quantified using the ImageJ software.
Nissl staining
Mice were perfused with 4% PFA in PBS, brains were removed and stored in 4% PFA. Forty-micrometer thick slices were sectioned using a vibratome, mounted on slides, and dried overnight. Slices were stained using Cresyl violet, coverslipped with Permount, and examined using light microscopy.
Open field arena
Mice were placed in the center of a brightly lit, open field arena (40 × 40 × 30 cm) and permitted to explore for 15 min on a single day. Location and movements of the mice were determined by beam breaks in horizontal emissions of infrared light from two adjacent sides of the arena. Movements were recorded and analyzed using the Activity Monitor program (Med Associates Inc). The time spent in the periphery and the center was calculated as a percentage of the total exploration time. The results were analyzed by Student's t-test with P < 0.05 as significance criteria.
Elevated plus maze
A plus shaped maze was elevated 40 cm off the floor with two closed arms (30 × 5 × 15 cm) and two open arms (30 × 5 cm) facing each other. The animal was placed in a central region (5 × 5 cm) to which each of the four arms attach and allowed to explore for 5 min. Movements were recorded and analyzed by Ethovision XT (Noldus) and the time spent in the closed arms and open arms was calculated as a percentage of the total exploration time. The results were analyzed by Student's t-test with P < 0.05 as significance criteria.
Prepulse inhibition
Mice were individually placed in a plexiglass cylinder attached to a startle detector (San Diego Instruments) and a range of prepulse stimuli at varying intensities from 74 to 90 db were semi-randomly presented while paired with an acoustic startle stimulus (120 db). Startle responses were measured using the Startle Response program (San Diego Instruments). The percentage of PPI was calculated by normalizing prepulse intensity responses to the response to the startle stimulus alone. The results were analyzed by Student's t-test with P < 0.05 as significance criteria.
Rotarod
Mice were placed on a rotarod (UGO Basile) and their latency to fall off was measured in seconds. Each mouse was tested four times over the course of 2 d. The rotarod accelerated from a speed of 4–40 rpm over a 5-min period. The results were analyzed by Student's t-test with P < 0.05 as significance criteria.
Morris water maze
A hidden platform was placed in a pool of opaque water. Mice were placed into the pool and permitted to swim for 60 sec. If they found the platform, they were immediately removed and returned to their home cage. If they did not find the platform after 60 sec, they were guided to the platform and were repeatedly placed on the platform until they remained on the platform, unassisted, for a total of 15 sec. Mice were trained with four trials each day with at least a 10-minute intertrial interval. On the final day, the platform was removed and a probe test was performed. The escape latency was measured on each training trial. During the probe test, the number of times the mice crossed the location of the hidden platform, along with the amount of time they spent in the target quadrant was recorded. Two days after the probe test, the reversal task was started. The hidden platform was placed in the opposite quadrant, and the latency to find the new location of the platform was recorded. The reversal task continued for 3 d exactly as the initial training protocol. A 2-d visible platform task ensued after the reversal task where a visible platform and flag were placed in the water and moved to a different zone for each of the four trials on both days. Latency of the mice to climb onto the visible platform was recorded. For analysis of escape latencies, a repeated-measures analysis of variance (ANOVA) was used to analyze the escape latencies, while a one-way ANOVA was used for analyzing the probe test with P < 0.05 as significance criteria.
Water-based Y maze
A submerged Y maze was placed in a pool of opaque water. On day 1, the mice were habituated to the maze and allowed to swim in the maze for 60 sec. On day 2, a hidden platform was placed into one of the two arms and mice were positioned in the starting arm and swam into one of the two arms. If the mice selected the correct arm and climbed onto the hidden platform, they immediately were removed and returned to their cage. If the mice selected the wrong arm, they were trapped in the arm for 20 sec before being removed and returned to the cage. If the mice selected the wrong arm on the first trial, after being trapped for 20 sec they were shown the hidden platform and were placed repeatedly on it until it remained on the platform, unassisted, for a total of 15 sec. Mice were tested with an intertrial interval of at least 10 min until they correctly selected the arm with the hidden platform on 10 out of 20 trials. On day 3, the platform was removed and the memory of the mice was assessed by observing whether they swam into the arm containing the hidden platform. Mice that remembered the location of the hidden platform on at least four of five trials were subsequently used for the reversal task where the hidden platform was moved to the other arm. In the reversal task, mice were repeatedly tested until they found the new location of the hidden platform on nine of 10 trials. A one-way ANOVA was used for analyzing the average number of trials required to achieve criteria with P < 0.05 as significance criteria.
Fear conditioning
Mice were introduced to a novel plexiglass cage (white overhead light, metallic grid floor, peppermint odor) and were allowed to explore for 150 sec before exposure to two tone-footshock pairings separated by one minute (tone, 85 db white noise, 30 sec duration; footshock intensity, 0.6 mA, 2 sec duration). Contextual memory tests were carried out 24 h later in the same environment where the mice were allowed to explore for 5 min. Cued memory tests were carried out 25 h after training in an altered environment (red overhead light, plastic floor, vanilla odor) where mice were allowed to explore for 150 sec before re-exposure to the same auditory cues. Behavior was videotaped and scored blind. One-way ANOVA was used for statistical analysis with P < 0.05 as significance criteria.
Electrophysiological recordings
Eight- to twelve-week-old mice were killed by cervical dislocation and the brains were sectioned with a vibratome into 400-μm thick slices in an oxygenated sucrose solution. Brains were sectioned either horizontally for hippocampal slices or coronally for amygdala slices. The slices were equilibrated with oxygenated ACSF at 32°C for at least 1 h before recording. For hippocampal recordings, extracellular recordings of field EPSPs (fEPSPs) were obtained from the stratum radiatum using microelectrodes filled with ACSF, and a bipolar Teflon-coated platinum electrode was placed in stratum radiatum to stimulate Schaffer collateral-commissural afferents. For the amygdala recordings, fEPSPs were obtained from the lateral amygdala while a bipolar Teflon-coated platinum electrode was placed in the internal capsule to stimulate thalamic afferents. The stimulation strength was set to elicit a response equivalent to 50% of the maximal fEPSPs. The slope of the fEPSP for hippocampal slices and the amplitude of the fEPSP for amygdala slices were expressed as a percentage of the baseline average prior to stimulation. PEAQX (Sigma) was dissolved in H2O and a final concentration of 400 nM was used to treat slices.
Acknowledgments
This work was supported by National Institutes of Health grants NS034007, NS047384, and NS078708 (E.K.) and an NRSA Predoctoral Award AG034005 (J.E.P.).
References
- Ahn K-J, Jeong HK, Choi H-S, Ryoo S-R, Kim YJ, Goo J-S, Choi S-Y, Han J-S, Ha I, Song W-J 2006. DYRK1A BAC transgenic mice show altered synaptic plasticity with learning and memory defects. Neurobiol Dis 22: 463–472 [DOI] [PubMed] [Google Scholar]
- Almeida A, Bolaños JP, Moreno S 2005. Cdh1/Hct1-APC is essential for the survival of postmitotic neurons. J Neurosci 25: 8115–8121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altafaj X, Ortiz-Abalia J, Fernández M, Potier MC, Laffaire J, Andreu N, Dierssen M, González-García C, Ceña V, Martí E, et al. 2008. Increased NR2A expression and prolonged decay of NMDA-induced calcium transient in cerebellum of TgDyrk1A mice, a mouse model of Down syndrome. Neurobiol Dis 32: 377–384 [DOI] [PubMed] [Google Scholar]
- Berberich S, Punnakkal P, Jensen V, Pawlak V, Seeburg PH, Hvalby Ø, Köhr G 2005. Lack of NMDA receptor subtype selectivity for hippocampal long-term potentiation. J Neurosci 25: 6907–6910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingol B, Schuman EM 2006. Activity-dependent dynamics and sequestration of proteasomes in dendritic spines. Nature 441: 1144–1148 [DOI] [PubMed] [Google Scholar]
- Bingol B, Sheng M 2011. Deconstruction for reconstruction: The role of proteolysis in neural plasticity and disease. Neuron 69: 22–32 [DOI] [PubMed] [Google Scholar]
- Deinhardt K, Kim T, Spellman DS, Mains RE, Eipper BA, Neubert TA, Chao MV, Hempstead BL 2011. Neuronal growth cone retraction relies on proneurotrophin receptor signaling through Rac. Sci Signal 4: ra82 10.1126/scisignal.2002060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD 2003. Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci 6: 231–242 [DOI] [PubMed] [Google Scholar]
- Fonseca R, Vabulas RM, Hartl FU, Bonhoeffer T, Nagerl UV 2006. A balance of protein synthesis and proteasome-dependent degradation determines the maintenance of LTP. Neuron 52: 239–245 [DOI] [PubMed] [Google Scholar]
- Fu AKY, Hung K-W, Fu W-Y, Shen C, Chen Y, Xia J, Lai K-O, Ip NY 2011. APC(Cdh1) mediates EphA4-dependent downregulation of AMPA receptors in homeostatic plasticity. Nat Neurosci 14: 181–189 [DOI] [PubMed] [Google Scholar]
- García-Higuera I, Manchado E, Dubus P, Cañamero M, Méndez J, Moreno S, Malumbres M 2008. Genomic stability and tumour suppression by the APC/C cofactor Cdh1. Nat Cell Biol 10: 802–811 [DOI] [PubMed] [Google Scholar]
- Gieffers C, Peters BH, Kramer ER, Dotti CG, Peters JM 1999. Expression of the CDH1-associated form of the anaphase-promoting complex in postmitotic neurons. Proc Natl Acad Sci 96: 11317–11322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JW, Burton JL, Solomon MJ 2002. The anaphase-promoting complex: It's not just for mitosis any more. Genes Dev 16: 2179–2206 [DOI] [PubMed] [Google Scholar]
- Hoeffer CA, Tang W, Wong H, Santillan A, Patterson RJ, Martinez LA, Tejada-Simon MV, Paylor R, Hamilton SL, Klann E 2008. Removal of FKBP12 enhances mTOR-Raptor interactions, LTP, memory, and perseverative/repetitive behavior. Neuron 60: 832–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YY, Kandel ER 1998. Postsynaptic induction and PKA-dependent expression of LTP in the lateral amygdala. Neuron 21: 169–178 [DOI] [PubMed] [Google Scholar]
- Hung AY, Sung CC, Brito IL, Sheng M 2010. Degradation of postsynaptic scaffold GKAP and regulation of dendritic spine morphology by the TRIM3 ubiquitin ligase in rat hippocampal neurons. PLoS One 5: e9842 10.1371/journal.pone.0009842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarome TJ, Werner CT, Kwapis JL, Helmstetter FJ 2011. Activity dependent protein degradation is critical for the formation and stability of fear memory in the amygdala. PLoS One 6: e24349 10.1371/journal.pone.0024349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang YH, Armstrong D, Albrecht U, Atkins CM, Noebels JL, Eichele G, Sweatt JD, Beaudet AL 1998. Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron 21: 799–811 [DOI] [PubMed] [Google Scholar]
- Juo P, Kaplan JM 2004. The anaphase-promoting complex regulates the abundance of GLR-1 glutamate receptors in the ventral nerve cord of C. elegans. Curr Biol 14: 2057–2062 [DOI] [PubMed] [Google Scholar]
- Karolewicz B, Szebeni K, Gilmore T, Maciag D, Stockmeier CA, Ordway GA 2009. Elevated levels of NR2A and PSD-95 in the lateral amygdala in depression. Int J Neuropsychopharmacol 12: 143–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpova A, Mikhaylova M, Thomas U, Knöpfel T, Behnisch T 2006. Involvement of protein synthesis and degradation in long-term potentiation of Schaffer collateral CA1 synapses. J Neurosci 26: 4949–4955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi Y, Stegmüller J, Matsuda T, Bonni S, Bonni A 2004. Cdh1-APC controls axonal growth and patterning in the mammalian brain. Science 303: 1026–1030 [DOI] [PubMed] [Google Scholar]
- Kuczera T, Stilling RM, Hsia H-E, Bahari-Javan S, Irniger S, Nasmyth K, Sananbenesi F, Fischer A 2011. The anaphase promoting complex is required for memory function in mice. Learn Mem 18: 49–57 [DOI] [PubMed] [Google Scholar]
- Lasorella A, Stegmüller J, Guardavaccaro D, Liu G, Carro MS, Rothschild G, La Torre-Ubieta De L, Pagano M, Bonni A, Iavarone A 2006. Degradation of Id2 by the anaphase-promoting complex couples cell cycle exit and axonal growth. Nature 442: 471–474 [DOI] [PubMed] [Google Scholar]
- Lee S-H, Choi J-H, Lee N, Lee H-R, Kim J-I, Yu N-K, Choi S-L, Lee S-H, Kim H, Kaang B-K 2008. Synaptic protein degradation underlies destabilization of retrieved fear memory. Science 319: 1253–1256 [DOI] [PubMed] [Google Scholar]
- Li M, Shin Y-H, Hou L, Huang X, Wei Z, Klann E, Zhang P 2008. The adaptor protein of the anaphase promoting complex Cdh1 is essential in maintaining replicative lifespan and in learning and memory. Nat Cell Biol 10: 1083–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S, Naisbitt S, Yoon J, Hwang JI, Suh PG, Sheng M, Kim E 1999. Characterization of the Shank family of synaptic proteins. Multiple genes, alternative splicing, and differential expression in brain and development. J Biol Chem 274: 29510–29518 [DOI] [PubMed] [Google Scholar]
- Listovsky T, Oren YS, Yudkovsky Y, Mahbubani HM, Weiss AM, Lebendiker M, Brandeis M 2004. Mammalian Cdh1/Fzr mediates its own degradation. EMBO J 23: 1619–1626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Salon M, Alonso M, Vianna MR, Viola H, Mello e Souza T, Izquierdo I, Pasquini JM, Medina JH 2001. The ubiquitin-proteasome cascade is required for mammalian long-term memory formation. Eur J Neurosci 14: 1820–1826 [DOI] [PubMed] [Google Scholar]
- Mao S-C, Lin H-C, Gean P-W 2008. Augmentation of fear extinction by D-cycloserine is blocked by proteasome inhibitors. Neuropsychopharmacology 33: 3085–3095 [DOI] [PubMed] [Google Scholar]
- Morris RG, Garrud P, Rawlins JN, O'Keefe J 1982. Place navigation impaired in rats with hippocampal lesions. Nature 297: 681–683 [DOI] [PubMed] [Google Scholar]
- Naisbitt S, Kim E, Tu JC, Xiao B, Sala C, Valtschanoff J, Weinberg RJ, Worley PF, Sheng M 1999. Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron 23: 569–582 [DOI] [PubMed] [Google Scholar]
- Phillips RG, LeDoux JE 1992. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci 106: 274–285 [DOI] [PubMed] [Google Scholar]
- Puram SV, Kim AH, Ikeuchi Y, Wilson-Grady JT, Merdes A, Gygi SP, Bonni A 2011. A CaMKIIβ signaling pathway at the centrosome regulates dendrite patterning in the brain. Nat Neurosci 14: 973–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Ortiz CJ, Balderas I, Saucedo-Alquicira F, Cruz-Castañeda P, Bermudez-Rattoni F 2011. Long-term aversive taste memory requires insular and amygdala protein degradation. Neurobiol Learn Mem 95: 311–315 [DOI] [PubMed] [Google Scholar]
- Sheng M, Kim E 2000. The Shank family of scaffold proteins. J Cell Sci 113: 1851–1856 [DOI] [PubMed] [Google Scholar]
- Silies M, Klämbt C 2010. APC/C(Fzr/Cdh1)-dependent regulation of cell adhesion controls glial migration in the Drosophila PNS. Nat Neurosci 13: 1357–1364 [DOI] [PubMed] [Google Scholar]
- Sotres-Bayon F, Cain CK, Ledoux JE 2006. Brain mechanisms of fear extinction: Historical perspectives on the contribution of prefrontal cortex. Biol Psychiatry 60: 329–336 [DOI] [PubMed] [Google Scholar]
- Stegmüller J, Konishi Y, Huynh MA, Yuan Z, Dibacco S, Bonni A 2006. Cell-intrinsic regulation of axonal morphogenesis by the Cdh1-APC target SnoN. Neuron 50: 389–400 [DOI] [PubMed] [Google Scholar]
- Thornton BR, Ng TM, Matyskiela ME, Carroll CW, Morgan DO, Toczyski DP 2006. An architectural map of the anaphase-promoting complex. Genes Dev 20: 449–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, Mayford M, Kandel ER, Tonegawa S 1996. Subregion- and cell type-restricted gene knockout in mouse brain. Cell 87: 1317–1326 [DOI] [PubMed] [Google Scholar]
- van Roessel P, Elliott DA, Robinson IM, Prokop A, Brand AH 2004. Independent regulation of synaptic size and activity by the anaphase-promoting complex. Cell 119: 707–718 [DOI] [PubMed] [Google Scholar]
- Villasana LE, Klann E, Tejada-Simon MV 2006. Rapid isolation of synaptoneurosomes and postsynaptic densities from adult mouse hippocampus. J Neurosci Methods 158: 30–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan L, Zou W, Gao D, Inuzuka H, Fukushima H, Berg AH, Drapp R, Shaik S, Hu D, Lester C, et al. 2011. Cdh1 regulates osteoblast function through an APC/C-independent modulation of Smurf1. Mol Cell 44: 721–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman AM 2001. Themes and variations on ubiquitylation. Nat Rev Mol Cell Biol 2: 169–178 [DOI] [PubMed] [Google Scholar]
- Yang Y, Kim AH, Yamada T, Wu B, Bilimoria PM, Ikeuchi Y, la Iglesia de N, Shen J, Bonni A 2009. A Cdc20-APC ubiquitin signaling pathway regulates presynaptic differentiation. Science 326: 575–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao I, Takao K, Miyakawa T, Ito S, Setou M 2011. Synaptic E3 ligase SCRAPPER in contextual fear conditioning: Extensive behavioral phenotyping of Scrapper heterozygote and overexpressing mutant mice. PLoS One 6: e17317 10.1371/journal.pone.0017317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh S-H, Mao S-C, Lin H-C, Gean P-W 2006. Synaptic expression of glutamate receptor after encoding of fear memory in the rat amygdala. Mol Pharmacol 69: 299–308 [DOI] [PubMed] [Google Scholar]
- Zinebi F, Xie J, Liu J, Russell RT, Gallagher JP, McKernan MG, Shinnick-Gallagher P 2003. NMDA currents and receptor protein are downregulated in the amygdala during maintenance of fear memory. J Neurosci 23: 10283–10291 [DOI] [PMC free article] [PubMed] [Google Scholar]