

Abstract
During exercise, oxygen delivery to skeletal muscle is elevated to meet the increased oxygen demand. The increase in blood flow to skeletal muscle is achieved by vasodilators formed locally in the muscle tissue, either on the intraluminal or on the extraluminal side of the blood vessels. A number of vasodilators have been shown to bring about this increase in blood flow and, importantly, interactions between these compounds seem to be essential for the precise regulation of blood flow. Two compounds stand out as central in these vasodilator interactions: nitric oxide (NO) and prostacyclin. These two vasodilators are both stimulated by several compounds, e.g. adenosine, ATP, acetylcholine and bradykinin, and are affected by mechanically induced signals, such as shear stress. NO and prostacyclin have also been shown to interact in a redundant manner where one system can take over when formation of the other is compromised. Although numerous studies have examined the role of single and multiple pharmacological inhibition of different vasodilator systems, and important vasodilators and interactions have been identified, a large part of the exercise hyperaemic response remains unexplained. It is plausible that this remaining hyperaemia may be explained by cAMP- and cGMP-independent smooth muscle relaxation, such as effects of endothelial derived hyperpolarization factors (EDHFs) or through metabolic modulation of sympathetic effects. The nature and role of EDHF as well as potential novel mechanisms in muscle blood flow regulation remain to be further explored to fully elucidate the regulation of exercise hyperaemia.
 |
Ylva Hellsten is head of the cardiovascular research group at the Department of Exercise and Sport Sciences, Section for Integrated Physiology, University of Copenhagen. The research group investigates the regulation of skeletal muscle blood flow and skeletal muscle angiogenesis in health and cardiovascular disease. Senior researcher Stefan P. Mortensen, DMSci, is leader of the cardiovascular group at the Centre of Inflammation and Metabolism at Rigshospitalet. He earned his master's degree from the University of Copenhagen and received post-doctoral training with Professor Bengt Saltin in Copenhagen. His main research interest is cardiovascular regulation during exercise and alterations in disease states.
Introduction
Blood flow to skeletal muscle is highly dynamic and increases markedly with exercise at a rate closely related to the oxygen demand of the muscle (Andersen & Saltin, 1985). Overall, muscle blood flow is regulated through a balance between, on the one hand, sympathetic activity and vasoconstrictors and, on the other hand, vasodilators and compounds modulating the effect of sympathetic activity. These vasodilating compounds are formed locally in the skeletal muscle tissue and are released from endothelial cells, red blood cells and skeletal muscle cells as a result of signals primarily related to the balance between oxygen delivery and demand. Several vasodilators, including nitric oxide (NO), prostacyclin, ATP, adenosine, potassium and compounds associated with the endothelium derived hyperpolarizing factor (EDHF) concept, such as 11,12-eicosatrienoic acid (11,12-EET), have been proposed to be of importance for muscle blood flow regulation. For review on this topic see Clifford & Hellsten (2004) and Sarelius & Pohl (2010). Evidence for the role of these vasodilators in exercise hyperaemia stems from studies showing that the vasodilators are formed in exercising muscle and from studies using pharmacological interventions to either inhibit or promote the vasodilator systems. None of the proposed vasodilators seem to operate independently or to be essential for reaching adequate blood flow during exercise, but they show a close interaction with other vasodilator systems. Vasodilator interactions may serve two purposes, where one is a redundancy mechanism whereby one vasodilator can take over when the formation of another vasodilator is impaired, and the other is activation of other vasodilator systems. The redundancy interaction may occur either chemically, by direct interactions between the vasodilator systems, or be functional and coupled to the demand for oxygen. Redundancy is a physiologically important concept as it can secure adequate oxygen supply despite impairments in vasodilator function. It is important to keep in mind that functional redundancy only becomes apparent in experimental settings when there is a demand for oxygen in the tissue, such as during exercise or hypoxia, whereas it is lacking in in vitro set-ups and experiments utilizing infusion of vasodilators. The other kind of vasodilator interaction serves to promote the formation of one or several other vasodilating systems, thereby potentially enhancing the vasodilator effect. NO and prostacyclin appear to be central in both of these interactions as they share a redundancy interaction and as they both are activated by multiple compounds and mechanical signals.
It should be emphasized that several other compounds than mentioned in this review have been proposed to contribute to exercise hyperaemia, e.g. potassium and lactate. Due to restrictions in length of this review we have chosen to discuss only selected compounds and their interactions and with a focus on human studies.
Functional role of NO, prostanoids and EDHF in exercise hyperaemia
In 1969 prostanoids were proposed to be involved in muscle blood flow regulation based on the findings that infusion of prostanoids into the brachial artery increased blood flow (Bevegård & Orö, 1969). A role for prostanoids in exercise hyperaemia was later supported by the findings that both plasma (Wilson & Kapoor, 1993) and interstitial (Frandsen et al. 2000) prostacyclin and prostaglandin E2 concentrations were increased during muscle contractions in the forearm and leg, respectively. However, infusion of cyclooxygenase (COX) inhibitors to inhibit the formation of prostanoids, has shown no effect on blood flow at rest or during exercise in the human forarm (Shoemaker et al. 1996) or leg (Fig. 1) (Mortensen et al. 2007; Schrage et al. 2010).
Figure 1. Change in exercise hyperaemia in the leg during inhibition of nitric oxide, prostaglandins, EDHF and the purinergic P1 receptor in young healthy male subjects.
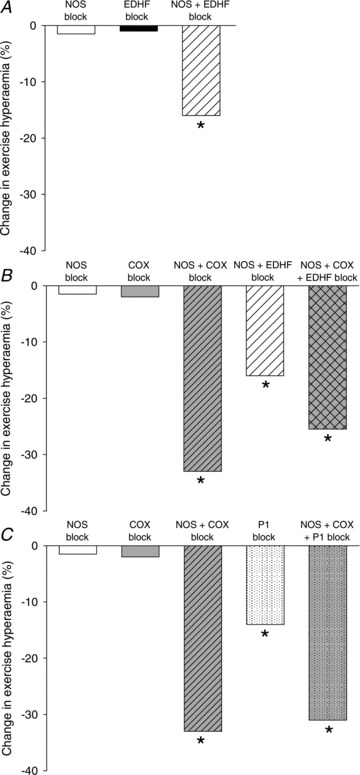
A, relative change in leg exercise hyperaemia with single or combined inhibition of nitric oxide synthase (NOS) and the endothelial derived hyperpolarizing factor (EDHF) cytochrome P450 2C9. B, relative change in leg exercise hyperaemia with single, double or triple blockade of NOS, EDHF and cyclooxygenase (COX). C, relative change in leg exercise hyperaemia with single, double or triple blockade of NOS, COX and the adenosine P1 receptor. In all experiments the exercise performed was single leg knee extensor exercise. Inhibition was achieved by arterial or venous infusion: NOS inhibition by NG-monomethyl-l-arginine (l-NMMA) or NG-nitroarginine methyl ester (l-NAME), COX inhibition by indomethacin, CYP 2 C9 by sulfaphenazole, EDHF by tetraammoniumchloride, and Pi receptor blockade by theophylline. Blood flow was determined by either thermodilution or ultrasound Doppler technique. Adapted from Frandsen et al. (2000), Hillig et al. (2002), Mortensen et al. (2007) and Mortensen et al. (2009).
In the late 1980s, Vallance and co-workers blocked NO synthase (NOS) by infusion of NG-monomethyl-l-arginine (l-NMMA) and showed a 50% reduction in resting forearm blood flow (Vallance et al. 1989). The importance of NO for resting blood flow, blood flow in recovery from exercise, and blood flow during passive movement has since been widely confirmed in the leg (Rådegran & Saltin, 1999; Mortensen et al. 2009b; Heinonen et al. 2011) and in the forearm (Panza et al. 1993; Gilligan et al. 1994; Dyke et al. 1995). During exercise, however, inhibition of NO formation has been shown not to reduce blood flow to the leg (Rådegran & Saltin, 1999; Bradley et al. 1999; Frandsen et al. 2001; Kingwell et al. 2002; Heinonen et al. 2011), whereas a transient effect has been observed in the forearm (Schrage, 2004). Thus, at least in the leg, neither NO nor prostanoids appear to be obligatory for exercise hyperaemia (Fig. 1). However, experiments in which the synthesis of NO and prostanoids have been inhibited simultaneously have demonstrated a clear reduction in leg blood flow during exercise (Boushel et al. 2002; Mortensen et al. 2007; 2009b; Heinonen et al. 2011). The observation that single inhibition of a system has no effect on exercise hyperaemia whereas combined inhibition markedly lowers blood flow suggests that there is a compensatory formation of the other vasodilator so that adequate blood flow is achieved. Direct interactions between the vasodilator systems may explain this redundancy, as described below.
EDHF is a concept derived from the observation that acetylcholine induces hyperpolarization of smooth muscle cells in the presence of NOS and COX inhibitors (Busse et al. 2002). The EDHF concept, which has not yet been fully elucidated, entails multiple compounds and the identity of these compounds varies according to tissue and blood vessel type (Busse et al. 2002). In coronary and skeletal muscle resistance arteries, the product of CYP 2C9, 11,12-eicosatrienoic acid (11,12-EET), as well as other eicosatrienoic acids (EETs), such as 8,9- and 14,15-EETs, have been identified as EDHFs (Fisslthaler et al. 1999; Bolz et al. 2000). In the human forearm, bradykinin has been shown to elevate blood flow during NOS and COX blockade, indicating the presence of an EDHF mediated mechanism (Halcox et al. 2001). In the human leg, single blockade of CYP 2C9 by arterial infusion of sulphaphenazole has no effect on exercise hyperaemia but combined inhibition of NO synthesis and CYP 2C9 lowers blood flow by ∼15% (Fig. 1) (Hillig et al. 2003). This finding suggests an interaction between these two vasodilator systems, similar to the redundancy interaction observed between NO synthase and COX. Interestingly, when EDHF blockade by infusion of the non-specific potassium channel blocker tetraammonium chloride (TEA) is added to combined inhibition of NO synthase and COX, exercise hyperaemia is not reduced beyond that of NO synthase and COX inhibition combined (Mortensen et al. 2007). The immediate interpretation could be that EDHF induced vasodilatation cannot compensate for the impaired NO and prostanoid systems, but an equally possible explanation is that TEA infusion is not sufficiently specific to examine an EDHF effect.
Stimulators of nitric oxide and prostanoid formation in endothelial cells
NO produced from eNOS and prostacyclin produced by the COX pathway play critical roles in normal vascular biology and pathophysiology and regulate vascular conductance, platelet aggregation, angiogenesis and vascular smooth muscle proliferation (Dudzinski & Michel, 2007; Félétou et al. 2011). eNOS is activated by increases in intracellular Ca2+ with concurrent binding of calmodulin (CaM) to the enzyme (Nathan & Xie, 1994) and by protein phosphorylation at several sites (Fleming & Busse, 2003; Mount et al. 2007). The formation of prostacyclin is stimulated by increases in intracellular Ca2+ in endothelial cells that lead to liberation of arachidonic acid and activation of the COX pathway (Fig. 2) (Carter et al. 1988; Ray & Marshall, 2006; Domeier & Segal, 2007). Numerous chemical and mechanical stimuli including ATP, adenosine, ACh, insulin, bradykinin, histamine, thrombin, stretch and shear stress have been shown to activate eNOS through increases in Ca2+ and/or phosphorylation status (de Wit et al. 1997; Fleming & Busse, 2003; Ray & Marshall, 2006; Domeier & Segal, 2007; Dudzinski & Michel, 2007; da Silva et al. 2009; Nyberg et al. 2010; Raqeeb et al. 2011). Similarly, prostacyclin formation has been shown to be increased by several of the same stimuli, e.g. ACh, ATP, adenosine, bradykinin, histamine and shear stress (Baenziger et al. 1980; Grabowski et al. 1985; Carter et al. 1988; Koller et al. 1994; Ray & Marshall, 2006; Domeier & Segal, 2007; Nyberg et al. 2010). The multiple stimulators of eNOS and the COX pathway highlight the importance of these vasoactive systems for vascular function and illustrate a dynamic control of NO and prostanoid bioactivity. The advantage of this design is that activation can occur even if some activation pathways are weak. On the other hand, impairments in these central vasodlator systems, as can occur in cardiovascular disease (Vanhoutte et al. 2009) can have a large impact on vascular function.
Figure 2. Schematic illustration of vasodilator interactions in skeletal muscle arterioles.

A, illustration of a simplified view of endothelium-dependent induction of vasodilatation. Vasodilator agonist, such as adenosine or acetylcholine (ACh), acts on specific receptors on endothelial cells lining the luminal side of an arteriole. The receptor activation leads to the formation of compounds which cause relaxation of smooth muscle cells situated adjacent to the endothelial cells resulting in vasodilatation of the arteriole. B, detailed illustration of how vasodilator systems in the vascular wall can be activated and of proposed interactions between vasodilator systems. In vascular endothelial cells, several compounds, including acetylcholine, ATP, adenosine and bradykinin as well as mechanical signals including shear stress, activate both endothelial NO synthase (eNOS) and the arachidonic acid pathway leading to formation of prostacyclin (PGI2) and eicosatrienoic acids (EETs). Activation of NOS and the arachidonic acid pathway occurs via an increase in intracellular calcium but eNOS activity is also regulated by protein phosphorylation at different sites. Redundancy exists between the NO and the prostacyclin systems where one described mechanism is inhibition of NOS by prostacyclin. Redundancy also exists between the NO system and cytochrome P450 2C9 (CYP 2C9), which produces 11,12-eicosatrienoic acid (11,12-EET) that can induce smooth muscle cell (SMC) relaxation by hyperpolarization. During normal conditions, NO exerts an inhibitory effect on CYP 2C9 but when NO formation is inhibited, the activity of CYP 2C9 increases. In the smooth muscle cell cyclic guanosine monophosphate (cGMP) can promote cyclic adenosine monophosphate (cAMP) levels by inhibiting phosphodiesterase III, which degrades cAMP. Abbreviations: AChR: acetylcholine receptor; AA: arachidonic acid; ATP: adenosine 5′-triphosphate; BKR: bradykinin receptor; 11, 12 EETs: 11, 12 eicosatrienoic acid; CaM: calmodulin; CaMK: calmodulin kinase; cAMP: cyclic adenosine monophosphate; cGMP: cyclic guanosine monophosphate; COX: cyclooxygenase; CYP 2C9: cytochrome P450 2C9; HR: histamine receptor; PGI2: prostacyclin; PKA, protein kinase A; PKC: protein kinase C; P1: purinergic receptor 1; P2: purinergic receptor 2; SMC: smooth muscle cell.
Interactions of nitric oxide and prostanoids in endothelial cells
In addition to their effects on smooth muscle cells, NO and prostanoids may also influence autacoid production in the endothelial layer. Inhibition of prostanoid synthesis leads to an increased NO formation in endothelial cells, an effect brought about by a cAMP induced reduction in the intracellular calcium level (Fig. 2) (Bolz & Pohl, 1997). The inhibitory effect of prostanoids on NO formation (Bolz & Pohl, 1997) could explain why exercise hyperaemia is not reduced when COX is inhibited in humans (Mortensen et al. 2007; 2009b). In this setting, inhibition of prostanoid formation would increase intracellular calcium, leading to a compensating increase in NO formation. In contrast, inhibition of the NO system may not severely affect NO function as this system is already suppressed by the contraction-induced formation of prostanoids (Frandsen et al. 2000; Karamouzis et al. 2001). This latter finding is supported by a lack of effect of NO synthase inhibition on the exercise induced increase in prostacyclin levels in the skeletal muscle interstitium (Frandsen et al. 2000). A redundancy interaction between the prostacyclin and the NO system may also explain the synergistic effect of combined NOS and COX inhibition in reducing ATP induced vasodilatation (Mortensen et al. 2009b).
As described above, the NOS system also shows an interaction with CYP 2C9 in that inhibition of CYP 2C9 or NOS alone does not lower exercise hyperaemia, whereas the combined inhibition does (Hillig et al. 2003). The explanation may lie in the observation that NO can inhibit the activity of CYP 2C so that when NO is inhibited the activity of CYP2C is enhanced and more of the vasodilator 11,12-EET is formed (Bauersachs et al. 1996).
Interactions of cAMP and cGMP in smooth muscle cells
The intracellular second messengers involved in smooth muscle cell (SMC) relaxation are the cyclic nucleotides, cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP), generated by the activity of adenylate and guanylate cyclase, respectively. cGMP is considered to be the main mediator of the cellular effects produced by NO (Bina et al. 1994), although at least a part of the NO-induced SMC relaxation is cGMP independent (Carvajal et al. 2000), whereas the vasodilator effects of prostanoids are mediated via elevations in cAMP (Vanhoutte & Mombouli, 1996). Interestingly, cGMP has been shown to have an inhibitory effect on the degradation of cAMP (Maurice & Haslam, 1990), which may explain the synergistic effect of NO and prostanoids on SMC relaxation (Fig. 2) (de Wit et al. 1994). This synergism is physiologically interesting as it increases the sensitivity of the system, but on the other hand it also indicates that even a small attenuation in the formation of cGMP could have a large effect on the resulting dilatation. The latter suggestion could explain why the different vasodilator systems that stimulate cGMP and cAMP can be stimulated by so many different compounds. In this context, simultaneous inhibition of NO and prostanoid formation could depress SMC levels of cAMP and cGMP to such an extent that additional inhibition of vasodilator systems coupled to these cyclic nucleotides may not have a further effect.
Vasodilator activation by ATP and adenosine
Adenosine binds to P1 purinergic receptors whereas ATP binds to P2 receptors (Ralevic & Burnstock, 1998). The vasodilator effect of both adenosine (Ray et al. 2002; Mortensen et al. 2009b; Nyberg et al. 2010) and ATP (McCullough et al. 1997; Hammer et al. 2001; Mortensen et al. 2009a; Crecelius et al. 2011) have been shown to be mediated in part via formation of NO and prostanoids (Fig. 3). As ATP is degraded rapidly by membrane-bound and soluble nucleotidases within the vasculature (Gordon, 1986; Yegutkin, 2008), one explanation for the convergence of downstream signalling could be that the vasodilator effect of ATP is mediated via adenosine. However, inhibition of P1 receptors does not reduce the vasodilator response to intra-arterial ATP infusion in humans (Rongen et al. 1994; Mortensen et al. 2009b; Kirby et al. 2010), suggesting that the vasodilator effect of intravascular ATP is independent of adenosine. This suggestion is also in congruence with observations in isolated endothelial cells demonstrating a similar potency of ATP and the P2Y receptor specific agonist UTP (da Silva et al. 2009; Raqeeb et al. 2011). Interestingly, in contrast to the adenosine-independent vasodilator effect of intravascular ATP, extraluminal application of ATP to blood-perfused arterioles has been suggested to be dependent on the action of adenosine on P1 receptors (Duza & Sarelius, 2003). This discrepancy between mechanisms underlying interstitial and intravascular ATP-induced vasodilatation is likely to reflect differences in receptor expression and/or the capacity for nucleotide degradation in the two compartments, but more evidence is needed to clarify the interaction of adenosine and ATP in the intravascular and interstitial space.
Figure 3. Adenosine induced stimulation of NO and prostacyclin formation.

A, muscle interstitial nitrite and nitrate (NOx) and 6-PGF1α concentrations during baseline conditions and interstitial adenosine infusion through microdialysis probes. *Significantly different from baseline (P < 0.05). B, effect of adenosine on release of NO from skeletal muscle and microvascular endothelial cells. *Significant formation (P < 0.05). C, effect of adenosine on release of 6-PGF1α from skeletal muscle and microvascular endothelial cells. *Significant formation (P < 0.05). Adapted from Nyberg et al. (2010).
Forearm and leg: do both models reflect skeletal muscle vasculature?
Experimental considerations in the forearm versus leg model
Evaluation of vascular function and blood flow regulation relies on the determination of blood flow and/or changes in arterial vessel diameter which can be evaluated by various methods in the forearm and leg model (Casey et al. 2008). Because the forearm volume is low, smaller doses of first-pass pharmacological drugs can be infused into the arterial circulation, which reduces the risk of confounding systemic effects such as an increase in blood pressure. A drawback of the forearm model, however, is that arterio-venous differences across the forearm cannot be obtained, because there is no single vein draining the forearm (Wahren 1966). Consequently, release and uptake of substances across the experimental limb cannot be determined and forearm oxygen uptake ( ) cannot be estimated. Without forearm
) cannot be estimated. Without forearm 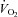 , the physiological importance of differences in blood flow is undisclosed because it is not known if it is secondary to changes in vascular function or a change in metabolic demand. In regards to the latter, it is well known that some infused substances can alter local metabolism (Mortensen et al. 2007; Boushel et al. 2012).
, the physiological importance of differences in blood flow is undisclosed because it is not known if it is secondary to changes in vascular function or a change in metabolic demand. In regards to the latter, it is well known that some infused substances can alter local metabolism (Mortensen et al. 2007; Boushel et al. 2012).
Limb specific vascular function
Due to the upright posture, the human legs are much more exposed to hydrostatic pressure when compared to the forearm (Rowell, 1993). In addition, the use of the leg muscles in locomotion holds the skeletal muscle tissue more active than the arm muscles, even in sedentary individuals. It therefore seems likely that there are limb differences in vascular function and blood flow regulation and studies that have compared the arm and leg have also reported differences in vasodilator responsiveness to endothelium-dependant and –independent substances (Newcomer et al. 2004) and α1-adrenergic responsiveness (Pawelczyk & Levine, 2002). In the forearm, adenosine infusion shows a large difference in vasodilator effect among individuals (Martin et al. 2006) whereas the inter-subject variation is less in the leg (Mortensen et al. 2009b; Nyberg et al. 2010; Hellsten et al. 2012). Moreover, a large number of studies show that the vasodilator response to ACh is markedly reduced in the forearm in individuals with cardiovascular disease (Virdis et al. 2010); however, when measured in the leg, the vasodilator response to ACh has been found to be similar in hypertensive and normotensive individuals, and in Type II diabetics and healthy controls (Thaning et al. 2011; Hellsten et al. 2012). This lack of difference in ACh induced vasodilatation also suggests that the finding that ACh infusion induces vasoconstriction in the forearm of individuals with cardiovascular disease, primarily by binding of prostacyclin to TP receptors (Félétou et al. 2010), is more evident in the arm. Ageing also appears to affect the endothelial function of the legs more than the arms (Thijssen et al. 2011). With regards to blood flow regulation, NO appears to be obligatory for exercise hyperaemia in the forearm of young subjects (Schrage, 2004), whereas it is not in the leg (Rådegran & Saltin, 1999; Frandsen et al. 2001; Nyberg et al. 2012). Notably, the capacity to form prostacyclin has also been shown to be lower in plasma and leg skeletal muscle in individuals with hypertension compared to normotensive control subjects (Hellsten et al. 2012).
Thus, existing evidence suggest that there are differences in blood flow regulation in young healthy individuals and that ageing and cardiovascular diseases affect the arms and legs differently (Thijssen et al. 2011). Flow mediated dilatation in the forearm is widely used to evaluate endothelial function, but because of the apparent differences in vascular function and relative small mass of the forearm, it is likely that the leg or a combination between evaluation of the endothelial function of the arm and leg is a better indicator of the general cardiovascular system (Thijssen et al. 2011; Mortensen et al. 2012). More studies comparing vascular function and regulation in the arms and legs and evaluating the prognostic value of the endothelial function in the arm and leg in regards to cardiovascular risks are needed, but existing data suggest that observations in one limb cannot be extrapolated to other limbs.
Conclusion
It is clear that regulation of skeletal muscle blood flow is a complex process involving several cellular sources, and many compounds and mechanisms. Most of the vasodilators proposed to be of importance in skeletal muscle blood flow regulation do not appear to be essential as their vasodilator effect can be compensated for by the formation of others. NO and prostacyclin are two central vasodilators which are stimulated by a number of other vasodilator compounds including ACh, ATP, adenosine and mechanical signalling. The many stimulators of NO and prostacyclin can probably be explained by redundancy and fine-tuned control of blood flow that ensures the vital delivery of O2 even under compromised conditions.
Pharmacological interventions in humans designed to reveal mechanisms underlying the regulation of skeletal muscle blood flow have succeeded in identifying specific vasodilators and vasodilator interactions of importance for exercise hyperaemia. However, regardless of the pharmacological interventions used, exercise hyperaemia has only been partially reduced, suggesting that yet unknown vasodilator mechanisms, potentially independent of cAMP and cGMP formation, remain to be revealed. Future studies should focus on the functional role of EDHF as well as potential novel mechanisms in muscle blood flow regulation.
Acknowledgments
Funding for the referenced work of the authors was gratefully received from The Danish Council for Independent Research – Medical Sciences, Lundbeck Foundation, Novo Nordisk Foundation, The Danish Ministry of Culture, The Danish Heart Association and P. Carl Petersen Foundation.
Glossary
- ACh
acetylcholine
- AA
arachidonic acid
- BKR
bradykinin receptor 2
- CaM
calmodulin
- CaMK
calmodulin kinase
- cAMP
cyclic adenosine monophosphate
- cGMP
cyclic guanosine monophosphate
- COX
cyclooxygenase
- CYP 2C9
cytochrome P450 2C9
- EET
eicosatrienoic acid
- EDHF
endothelial derived hyperpolarizing factor
- HR
histamine receptor
- PGI2
prostacyclin
- PKA
protein kinase A
- PKC
protein kinase C
- P1
purinergic receptor 1
- P2
purinergic receptor 2
- SMC
smooth muscle cell
References
- Andersen P, Saltin B. Maximal perfusion of skeletal-muscle in man. J Physiol. 1985;366:233–249. doi: 10.1113/jphysiol.1985.sp015794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baenziger NL, Force LE, Becherer PR. Histamine stimulate prostacyclin synthesis in cultured human umbilical vein endothelial cells. Biochem Biophys Res Commun. 1980;92:1435–1440. doi: 10.1016/0006-291x(80)90447-7. [DOI] [PubMed] [Google Scholar]
- Bauersachs J, Popp R, Hecker M, Sauer E, Fleming I, Busse R. Nitric oxide attenuates the release of endothelium-derived hyperpolarizing factor. Circulation. 1996;94:3341–3347. doi: 10.1161/01.cir.94.12.3341. [DOI] [PubMed] [Google Scholar]
- Bevegård S, Orö L. Effect of prostaglandin E1 on forearm blood flow. Scand J Clin Lab Inevest. 1969;23:347–353. doi: 10.3109/00365516909081700. [DOI] [PubMed] [Google Scholar]
- Bina S, Hart JL, Muldoon SM. Comparative effects of exogenous nitrovasodilators on cGMP levels in different canine blood vessels. Life Sci. 1994;56:33–38. doi: 10.1016/0024-3205(94)00424-q. [DOI] [PubMed] [Google Scholar]
- Bolz SS, Pohl U. Indomethacin enhances endothelial NO release – evidence for a role of PGI2 in the autocrine control of calcium-dependent autacoid production. Cardiovasc Res. 1997;36:437–444. doi: 10.1016/s0008-6363(97)00197-1. [DOI] [PubMed] [Google Scholar]
- Bolz SS, Fisslthaler B, Pieperhoff S, Wit CD, Fleming I, Busse R, Pohl U. Antisense oligonucleotides against cytochrome P450 2C8 attenuate EDHF-mediated Ca2+ changes and dilation in isolated resistance arteries. FASEB J. 2000;14:255–260. doi: 10.1096/fasebj.14.2.255. [DOI] [PubMed] [Google Scholar]
- Boushel R, Fuentes T, Hellsten Y, Saltin B. Opposing effects of nitric oxide and prostaglandin inhibition on muscle mitochondrial VO2 during exercise. Am J Physiol Regul Integr Comp Physiol. 2012;303:R94–R100. doi: 10.1152/ajpregu.00044.2012. [DOI] [PubMed] [Google Scholar]
- Boushel R, Langberg H, Gemmer C, Olesen J, Crameri R, Scheede C, Sander M, Kjaer M. Combined inhibition of nitric oxide and prostaglandins reduces human skeletal muscle blood flow during exercise. J Physiol. 2002;543:691–698. doi: 10.1113/jphysiol.2002.021477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley SJ, Kingwell BA, McConell GK. Nitric oxide synthase inhibition reduces leg glucose uptake but not blood flow during dynamic exercise in humans. Diabetes. 1999;48:1815–1821. doi: 10.2337/diabetes.48.9.1815. [DOI] [PubMed] [Google Scholar]
- Busse R, Edwards G, Félétou M, Fleming I. EDHF: bringing the concepts together. Trends Pharmacol Sci. 2002;23:374–380. doi: 10.1016/s0165-6147(02)02050-3. [DOI] [PubMed] [Google Scholar]
- Casey DP, Curry TB, Joyner MJ. Measuring muscle blood flow: a key link between systemic and regional metabolism. Curr Opin Clin Nutr Metab Care. 2008;11(5):580–586. doi: 10.1097/MCO.0b013e32830b5b34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter TD, Hallam TJ, Cusack NJ, Pearson JD. Regulation of P2y-purinoceptor-mediated prostacyclin release from human endothelial cells by cytoplasmic calcium concentration. Br J Pharmacol. 1988;95:1181–1190. doi: 10.1111/j.1476-5381.1988.tb11754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvajal JA, Germain AM, Huidobro-Toro JP, Weiner CP. Molecular mechanism of cGMP-mediated smooth muscle relaxation. J Cell Physiol. 2000;184:409–420. doi: 10.1002/1097-4652(200009)184:3<409::AID-JCP16>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Crecelius AR, Kirby BS, Voyles WF, Dinenno FA. Augmented skeletal muscle hyperaemia during hypoxic exercise in humans is blunted by combined inhibition of nitric oxide and vasodilating prostaglandins. J Physiol. 2011;589:3671–3683. doi: 10.1113/jphysiol.2011.209486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifford PS, Hellsten Y. Vasodilatory mechanisms in contracting skeletal muscle. J Appl Physiol. 2004;97:393–403. doi: 10.1152/japplphysiol.00179.2004. [DOI] [PubMed] [Google Scholar]
- da Silva CG, Specht A, Wegiel B, Ferran C, Kaczmarek E. Mechanism of purinergic activation of endothelial nitric oxide synthase in endothelial cells. Circulation. 2009;119:871–879. doi: 10.1161/CIRCULATIONAHA.108.764571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit C, von Bismarck P, Pohl U. Synergistic action of vasodilators that increase cGMP and cAMP in the hamster cremaster microcirculation. Cardiovasc Res. 1994;28:1513–1518. doi: 10.1093/cvr/28.10.1513. [DOI] [PubMed] [Google Scholar]
- de Wit C, Schäfer C, von Bismarck P, Bolz SS, Pohl U. Elevation of plasma viscosity induces sustained NO-mediated dilation in the hamster cremaster microcirculation in vivo. Pflugers Arch. 1997;434:354–361. doi: 10.1007/s004240050408. [DOI] [PubMed] [Google Scholar]
- Domeier TL, Segal SS. Electromechanical and pharmacomechanical signalling pathways for conducted vasodilatation along endothelium of hamster feed arteries. J Physiol. 2007;579:175–186. doi: 10.1113/jphysiol.2006.124529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudzinski D, Michel T. Life history of eNOS: partners and pathways. Cardiovasc Res. 2007;75:247–260. doi: 10.1016/j.cardiores.2007.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duza T, Sarelius IH. Conducted dilations initiated by purines in arterioles are endothelium dependent and require endothelial Ca2+ Am J Physiol Heart Circ Physiol. 2003;285:H26–H37. doi: 10.1152/ajpheart.00788.2002. [DOI] [PubMed] [Google Scholar]
- Dyke CK, Proctor DN, Dietz NM, Joyner MJ. Role of nitric oxide in exercise hyperaemia during prolonged rhythmic handgripping in humans. J Physiol. 1995;488:259–265. doi: 10.1113/jphysiol.1995.sp020964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Félétou M, Huang Y, Vanhoutte PM. Vasoconstrictor prostanoids. Pflugers Arch. 2010;459:941–950. doi: 10.1007/s00424-010-0812-6. [DOI] [PubMed] [Google Scholar]
- Félétou M, Huang Y, Vanhoutte PM. Endothelium-mediated control of vascular tone: COX-1 and COX-2 products. Br J Pharmacol. 2011;164:894–912. doi: 10.1111/j.1476-5381.2011.01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisslthaler B, Popp R, Kiss L, Potente M, Harder DR, Fleming I, Busse R. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature. 1999;401:493–497. doi: 10.1038/46816. [DOI] [PubMed] [Google Scholar]
- Fleming I, Busse R. Molecular mechanisms involved in the regulation of the endothelial nitric oxide synthase. Am J Physiol Regul Integr Comp Physiol. 2003;284:R1–R12. doi: 10.1152/ajpregu.00323.2002. [DOI] [PubMed] [Google Scholar]
- Frandsen U, Bangsbo J, Langberg H, Saltin B, Hellsten Y. Inhibition of nitric oxide synthesis by systemic NG-monomethyl- L-arginine administration in humans: effects on interstitial adenosine, prostacyclin and potassium concentrations in resting and contracting skeletal muscle. J Vasc Res. 2000;37:297–302. doi: 10.1159/000025743. [DOI] [PubMed] [Google Scholar]
- Frandsen U, Bangsbo J, Sander M, Hoffner L, Betak A, Saltin B, Hellsten Y. Exercise-induced hyperaemia and leg oxygen uptake are not altered during effective inhibition of nitric oxide synthase with NG-nitro-L-arginine methyl ester in humans. J Physiol. 2001;531:257–264. doi: 10.1111/j.1469-7793.2001.0257j.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilligan DM, Panza JA, Kilcoyne CM, Waclawiw MA, Casino PR, Quyyumi AA. Contribution of endothelium-derived nitric oxide to exercise-induced vasodilation. Circulation. 1994;90:2853–2858. doi: 10.1161/01.cir.90.6.2853. [DOI] [PubMed] [Google Scholar]
- Gordon JL. Extracellular ATP: effects, sources and fate. Biochem J. 1986;233:309–319. doi: 10.1042/bj2330309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabowski EF, Jaffe EA, Weksler BB. Prostacyclin production by cultured endothelial cell monolayers exposed to step increases in shear stress. J Lab Clin Med. 1985;105:36–43. [PubMed] [Google Scholar]
- Halcox JP, Narayanan S, Cramer-Joyce L, Mincemoyer R, Quyyumi AA. Characterization of endothelium-derived hyperpolarizing factor in the human forearm microcirculation. Am J Physiol Heart Circ Physiol. 2001;280:H2470–H2477. doi: 10.1152/ajpheart.2001.280.6.H2470. [DOI] [PubMed] [Google Scholar]
- Hammer LW, Ligon AL, Hester RL. ATP-mediated release of arachidonic acid metabolites from venular endothelium causes arteriolar dilation. Am J Physiol Heart Circ Physiol. 2001;280:H2616–H2622. doi: 10.1152/ajpheart.2001.280.6.H2616. [DOI] [PubMed] [Google Scholar]
- Heinonen I, Bengt S, Jukka K, Sipilä HT, Vesa O, Pirjo N, Juhani K, Kari K, Ylva H. Skeletal muscle blood flow and oxygen uptake at rest and during exercise in humans: a pet study with nitric oxide and cyclooxygenase inhibition. Am J Physiol Heart Circ Physiol. 2011;300:1510–1517. doi: 10.1152/ajpheart.00996.2010. [DOI] [PubMed] [Google Scholar]
- Hellsten Y, Nyberg M, Mortensen SP. Contribution of intravascular versus interstitial purines and nitric oxide in the regulation of exercise hyperaemia in humans. J Physiol. 2012;590:5015–5023. doi: 10.1113/jphysiol.2012.234963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillig T, Krustrup P, Fleming I, Osada T, Saltin B, Hellsten Y. Cytochrome P450 2C9 plays an important role in the regulation of exercise-induced skeletal muscle blood flow and oxygen uptake in humans. J Physiol (lond) 2003;546:307–314. doi: 10.1113/jphysiol.2002.030833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karamouzis M, Langberg H, Skovgaard D, Bülow J, Kjaer M, Saltin B. In situ microdialysis of intramuscular prostaglandin and thromboxane in contracting skeletal muscle in humans. Acta Physiol Scand. 2001;171:71–76. doi: 10.1046/j.1365-201X.2001.00775.x. [DOI] [PubMed] [Google Scholar]
- Kingwell BA, Formosa M, Muhlmann M, Bradley SJ, McConell GK. Nitric oxide synthase inhibition reduces glucose uptake during exercise in individuals with type 2 diabetes more than in control subjects. Diabetes. 2002;51:2572–2580. doi: 10.2337/diabetes.51.8.2572. [DOI] [PubMed] [Google Scholar]
- Kirby BS, Crecelius AR, Voyles WF, Dinenno FA. Vasodilatory responsiveness to adenosine triphosphate in ageing humans. J Physiol (Lond) 2010;588:4017–4027. doi: 10.1113/jphysiol.2010.197814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koller A, Sun D, Huang A, Kaley G. Corelease of nitric oxide and prostaglandins mediates flow-dependent dilation of rat gracilis muscle arterioles. Am J Physiol Heart Circ Physiol. 1994;267:H326–H332. doi: 10.1152/ajpheart.1994.267.1.H326. [DOI] [PubMed] [Google Scholar]
- Martin EA, Nicholson WT, Eisenach JH, Charkoudian N, Joyner MJ. Influences of adenosine receptor antagonism on vasodilator responses to adenosine and exercise in adenosine responders and nonresponders. J Appl Physiol. 2006;101:1678–1684. doi: 10.1152/japplphysiol.00546.2006. [DOI] [PubMed] [Google Scholar]
- Maurice DH, Haslam RJ. Molecular basis of the synergistic inhibition of platelet function by nitrovasodilators and activators of adenylate cyclase: inhibition of cyclic AMP breakdown by cyclic GMP. Mol Pharmacol. 1990;37:671–681. [PubMed] [Google Scholar]
- McCullough W, Collins D, Ellsworth M. Arteriolar responses to extracellular ATP in striated muscle. Am J Physiol Heart Circ Physiol. 1997;272:H1886–H1891. doi: 10.1152/ajpheart.1997.272.4.H1886. [DOI] [PubMed] [Google Scholar]
- Mortensen SP, Askew CD, Walker M, Nyberg M, Hellsten Y. The hyperaemic response to passive leg movement is dependent on nitric oxide; a new tool to evaluate endothelial nitric oxide function. J Physiol. 2012;590:4391–4404. doi: 10.1113/jphysiol.2012.235952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen SP, Gonzalez-Alonso J, Damsgaard R, Saltin B, Hellsten Y. Inhibition of nitric oxide and prostaglandins, but not endothelial-derived hyperpolarizing factors, reduces blood flow and aerobic energy turnover in the exercising human leg. J Physiol. 2007;581:853–861. doi: 10.1113/jphysiol.2006.127423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen SP, González-Alonso J, Bune LT, Saltin B, Pilegaard H, Hellsten Y. ATP-induced vasodilation and purinergic receptors in the human leg: roles of nitric oxide, prostaglandins, and adenosine. Am J Physiol Regul Integr Comp Physiol. 2009a;296:R1140–R1148. doi: 10.1152/ajpregu.90822.2008. [DOI] [PubMed] [Google Scholar]
- Mortensen SP, Nyberg M, Thaning P, Saltin B, Hellsten Y. Adenosine contributes to blood flow regulation in the exercising human leg by increasing prostaglandin and nitric oxide formation. Hypertension. 2009b;53:993–999. doi: 10.1161/HYPERTENSIONAHA.109.130880. [DOI] [PubMed] [Google Scholar]
- Mount PF, Kemp BE, Power DA. Regulation of endothelial and myocardial NO synthesis by multi-site eNOS phosphorylation. J Mol Cell Cardiol. 2007;42:271–279. doi: 10.1016/j.yjmcc.2006.05.023. [DOI] [PubMed] [Google Scholar]
- Nathan C, Xie QW. Nitric oxide synthases: roles, tolls, and controls. Cell. 1994;78:915–918. doi: 10.1016/0092-8674(94)90266-6. [DOI] [PubMed] [Google Scholar]
- Newcomer SC, Leuenberger UA, Hogeman CS, Handly BD, Proctor DN. Different vasodilator responses of human arms and legs. J Physiol. 2004;556:1001–1011. doi: 10.1113/jphysiol.2003.059717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyberg M, Jensen LG, Thaning P, Hellsten Y, Mortensen SP. Role of nitric oxide and prostanoids in the regulation of leg blood flow and blood pressure in humans with essential hypertension: effect of high-intensity aerobic training. J Physiol. 2012;590:1481–1494. doi: 10.1113/jphysiol.2011.225136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyberg M, Mortensen SP, Thaning P, Saltin B, Hellsten Y. Interstitial and plasma adenosine stimulate nitric oxide and prostacyclin formation in human skeletal muscle. Hypertension. 2010;56:1102–1108. doi: 10.1161/HYPERTENSIONAHA.110.161521. [DOI] [PubMed] [Google Scholar]
- Panza JA, Casino PR, Badar DM, Quyyumi AA. Effect of increased availability of endothelium-derived nitric oxide precursor on endothelium-dependent vascular relaxation in normal subjects and in patients with essential hypertension. Circulation. 1993;87:1475–1481. doi: 10.1161/01.cir.87.5.1475. [DOI] [PubMed] [Google Scholar]
- Pawelczyk JA, Levine BD. Heterogeneous responses of human limbs to infused adrenergic agonists: a gravitational effect. J Appl Physiol. 2002;92:2105–2113. doi: 10.1152/japplphysiol.00979.2001. [DOI] [PubMed] [Google Scholar]
- Ralevic V, Burnstock G. Receptors for purines and pyrimidines. Pharmacol Rev. 1998;50(3):413–492. Review. [PubMed] [Google Scholar]
- Raqeeb A, Sheng J, Ao N, Braun AP. Purinergic P2Y2 receptors mediate rapid Ca2+ mobilization, membrane hyperpolarization and nitric oxide production in human vascular endothelial cells. Cell Calcium. 2011;49:240–248. doi: 10.1016/j.ceca.2011.02.008. [DOI] [PubMed] [Google Scholar]
- Ray C, Marshall J. The cellular mechanisms by which adenosine evokes release of nitric oxide from rat aortic endothelium. J Physiol. 2006;570:85–96. doi: 10.1113/jphysiol.2005.099390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray CJ, Abbas MR, Coney AM, Marshall JM. Interactions of adenosine, prostaglandins and nitric oxide in hypoxia-induced vasodilatation: in vivo and in vitro studies. J Physiol. 2002;544:195–209. doi: 10.1113/jphysiol.2002.023440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rådegran G, Saltin B. Nitric oxide in the regulation of vasomotor tone in human skeletal muscle. Am J Physiol Heart Circ Physiol. 1999;276:H1951–H1960. doi: 10.1152/ajpheart.1999.276.6.H1951. [DOI] [PubMed] [Google Scholar]
- Rongen GA, Smits P, Thien T. Characterization of ATP-induced vasodilation in the human forearm vascular bed. Circulation. 1994;90:1891–1898. doi: 10.1161/01.cir.90.4.1891. [DOI] [PubMed] [Google Scholar]
- Rowell LB. Human Cardiovascular Control. New York: Oxford University Press; 1993. [Google Scholar]
- Sarelius I, Pohl U. Control of muscle blood flow during exercise: local factors and integrative mechanisms. Acta Physiologica. 2010;199:349–365. doi: 10.1111/j.1748-1716.2010.02129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrage WG. Local inhibition of nitric oxide and prostaglandins independently reduces forearm exercise hyperaemia in humans. J Physiol. 2004;557:599–611. doi: 10.1113/jphysiol.2004.061283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrage WG, Wilkins BW, Johnson CP, Eisenach JH, Limberg JK, Dietz NM, Curry TB, Joyner MJ. Roles of nitric oxide synthase and cyclooxygenase in leg vasodilation and oxygen consumption during prolonged low-intensity exercise in untrained humans. J Appl Physiol. 2010;109:768–777. doi: 10.1152/japplphysiol.00326.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker JK, Naylor HL, Pozeg ZI, Hughson RL. Failure of prostaglandins to modulate the time course of blood flow during dynamic forearm exercise in humans. J Appl Physiol. 1996;81:1516–1521. doi: 10.1152/jappl.1996.81.4.1516. [DOI] [PubMed] [Google Scholar]
- Thaning P, Bune LT, Zaar M, Saltin B, Rosenmeier JB. Functional sympatholysis during exercise in patients with type 2 diabetes with intact response to acetylcholine. Diabetes Care. 2011;34:1186–1191. doi: 10.2337/dc10-2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thijssen DHJ, Rowley N, Padilla J, Simmons GH, Laughlin MH, Whyte G, Cable NT, Green DJ. Relationship between upper and lower limb conduit artery vasodilator function in humans. J Appl Physiol. 2011;111:244–250. doi: 10.1152/japplphysiol.00290.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallance P, Collier J, Moncada S. Effects of endothelium-derived nitric oxide on peripheral arteriolar tone in man. Lancet. 1989;334:997–1000. doi: 10.1016/s0140-6736(89)91013-1. [DOI] [PubMed] [Google Scholar]
- Vanhoutte PM, Mombouli JV. Vascular endothelium: vasoactive mediators. Prog Cardiovasc Dis. 1996;39:229–238. doi: 10.1016/s0033-0620(96)80003-x. [DOI] [PubMed] [Google Scholar]
- Vanhoutte PM, Shimokawa H, Tang EHC, Félétou M. Endothelial dysfunction and vascular disease. Acta Physiologica. 2009;196:193–222. doi: 10.1111/j.1748-1716.2009.01964.x. [DOI] [PubMed] [Google Scholar]
- Virdis A, Ghiadoni L, Taddei S. Human endothelial dysfunction: EDCFs. Pflugers Arch. 2010;459:1015–1023. doi: 10.1007/s00424-009-0783-7. [DOI] [PubMed] [Google Scholar]
- Wahren J. Quantitative aspects of blood flow and oxygen uptake in the human forearm during rhythmic exercise. Acta Physiol Scand Suppl. 1966;269:1–93. [PubMed] [Google Scholar]
- Wilson JR, Kapoor SC. Contribution of prostaglandins to exercise-induced vasodilation in humans. Am J Physiol Heart Circ Physiol. 1993;265:H171–H175. doi: 10.1152/ajpheart.1993.265.1.H171. [DOI] [PubMed] [Google Scholar]
- Yegutkin GG. Nucleotide- and nucleoside-converting ectoenzymes: Important modulators of purinergic signalling cascade. Biochim Biophys Acta. 2008;1783:673–694. doi: 10.1016/j.bbamcr.2008.01.024. [DOI] [PubMed] [Google Scholar]