

Abstract
Hypoxia can have profound influences on the circulation. In humans, acute exposure to moderate hypoxia has been demonstrated to result in vasodilatation in the coronary, cerebral, splanchnic and skeletal muscle vascular beds. The combination of submaximal exercise and hypoxia produces a ‘compensatory’ vasodilatation and augmented blood flow in contracting skeletal muscles relative to the same level of exercise under normoxic conditions. This augmented vasodilatation exceeds that predicted by a simple sum of the individual dilator responses to hypoxia alone and normoxic exercise. Additionally, this enhanced hypoxic exercise hyperaemia is proportional to the hypoxia-induced fall in arterial oxygen (O2) content, thus preserving muscle O2 delivery and ensuring it is matched to demand. Several vasodilator pathways have been proposed and examined as likely regulators of skeletal muscle blood flow in response to changes in arterial O2 content. The purpose of this review is to put into context the present evidence regarding mechanisms responsible for the compensatory vasodilatation observed during hypoxic exercise in humans. Along these lines, this review will highlight the interactions between various local metabolic and endothelial derived substances that influence vascular tone during hypoxic exercise.
 |
Darren P. Casey is an Assistant Professor of Physiology at the Mayo Clinic. He received his undergraduate (1999) and Masters (2001) degrees from Syracuse University, and doctorate from the University of Florida (2007) and completed his research training at Mayo. In general, his research interest is in the regulation of vascular tone and peripheral blood flow during exercise. More specifically the role that local factors and the sympathetic nervous system have on blood flow during exercise under conditions of low oxygen delivery (i.e. hypoxia and hypoperfusion) and in conditions of aging and cardiovascular disease. Michael J. Joyner, M.D., is the Caywood Professor of Anesthesiology at Mayo Clinic where he was named Distinguished Investigator in 2010. His undergraduate (1981) and medical (1987) degrees are from the University of Arizona with residency and research training at Mayo. His interests include cardiovascular regulation in humans, the physiology of world records, and autonomic regulation of blood glucose. He has held leadership positions at Mayo, in the extramural research community, and with leading journals. His lab has been funded by the NIH since 1993, and former fellows have established independent research programs at leading institutions throughout the world.
Exercise hyperaemia: matching blood flow to metabolism
ATP turnover stimulates oxygen (O2) consumption in contracting skeletal muscles. For contractions to be sustained, as is the case during endurance exercise, there must be continuous supply of O2 to the contracting muscles. At the systemic level, ventilation and gas exchange in the lung, along with increases in cardiac output, are critical to meet the demands of the contracting skeletal muscle for oxygen, and within skeletal muscle there is typically an increase in blood flow (O2 delivery) that is proportional to what might generally be termed as ‘metabolic demand.’ Presented in Fig. 1 is an example of the tight matching between indices of O2 demand and increases in skeletal muscle blood flow during different modes of exercise (Mortensen et al. 2008).
Figure 1. Matching blood flow to metabolism during exercise.
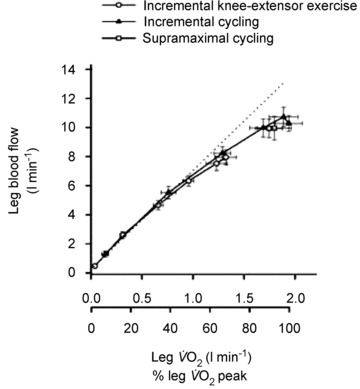
One-legged blood flow during incremental knee-extensor exercise and incremental and supramaximal cycling to exhaustion plotted against one-legged VO2. Adapted with permission from Mortensen et al. (2008).
It is important to note that at rest, skeletal muscle O2 consumption is quite low and blood flow is on the order of 3 ml (100 g)−1 min−1. This is consistent with the general concept that resting oxygen consumption in normal-sized humans is ∼250 ml min−1. However, during maximal exercise O2 consumption can increase 10- to 15-fold in untrained young subjects, and 20- to 25-fold in elite highly trained endurance athletes. These increases in O2 consumption are facilitated by vast increases in cardiac output and, more importantly, increases in skeletal muscle blood flow of 50- to 100-fold (Andersen & Saltin, 1985; Armstrong & Laughlin, 1985; Musch, 1988). So, exercising skeletal muscles can increase their O2 consumption markedly, and this increase in O2 consumption drives vast increases in blood flow.
The mechanisms responsible for increasing blood flow at the onset of exercise as well as maintaining it over time involve a complex interaction between mechanical and neural factors and various local metabolic and endothelial derived substances that influence vascular tone (Clifford, 2007). These potential mechanisms are listed in Table 1 and have previously been reviewed in detail (Shepherd, 1983; Clifford, 2007; Joyner & Wilkins, 2007).
Table 1.
Potential mechanisms responsible for increasing blood flow in contracting muscle
| 1. Mechanical |
| a. Muscle pump (↑ in the arterio-venous pressure gradient for flow) |
| b. Mechanical deformation of the vessel wall |
| 2. Neural |
| a. Blunting of sympathetic α-adrenergic vasoconstriction (functional sympatholysis) |
| b. Sympathetic cholinergic vasodilatation |
| c. Acetylcholine spillover from motor-end plates |
| 3. Metabolic |
| Substances produced and/or released from skeletal muscle, endothelial, and red blood cells (NO, adenosine, prostanoids, ATP, hydrogen ion, potassium, EDHF) |
| 4. Flow- or shear stress-induced vasodilatation |
| 5. Conducted vasodilatation |
What happens when O2 availability is limited?
The general principles outlined above reflect the synthesis of ideas and data generated in healthy humans with normal haematocrit levels, exercising at low altitude. This means that under most circumstances arterial O2 saturation remains high (95% or greater). What happens when arterial O2 content is lowered by hypoxia or anaemia, or when the ability of red cells to carry and release O2 is limited by carbon monoxide (CO)? Under these circumstances, the relationship between blood flow and O2 demand is shifted upward so that blood flow increases but O2 delivery to the exercising muscle remains constant. For example, if arterial O2 saturation falls from 100% to 80% as a result of hypoxia, there is a ∼20% increase in blood flow so that total O2 delivery to the skeletal muscles remains constant. The compensatory vasodilatation exceeds that predicted by a simple sum of the individual dilator responses to hypoxia alone and normoxic exercise. This general principle is also seen when O2 delivery is limited by CO or anaemia and the general concept of 1:1 compensation is seen under all three conditions (Roach et al. 1999; Gonzalez-Alonso et al. 2001).
Compensatory vasodilatation in the face of sympathetic vasoconstrictor activity
One especially interesting feature associated with compensatory vasodilatation is that during hypoxia there is increased sympathetic vasoconstrictor activity directed towards skeletal muscle (Hanada et al. 2003) (Fig. 2). This means that the signals associated with the compensatory vasodilatation are opposed by the vasoconstrictor activity. To better understand this interaction, we evaluated the effects of α-adrenergic blockade on the compensatory vasodilatation during hypoxic forearm exercise (Wilkins et al. 2008). Similar to findings under resting conditions (Weisbrod et al. 2001), α-adrenergic receptor blockade revealed a substantially greater vasodilatation during hypoxic exercise compared to control hypoxic exercise conditions (i.e. during saline infusion). However, despite elevated sympathetic vasoconstrictor activity during hypoxic exercise (compared to normoxic exercise or hypoxia alone), substantial compensatory vasodilatation persists.
Figure 2. Hypoxia induced increases in muscle sympathetic nerve activity (MSNA).
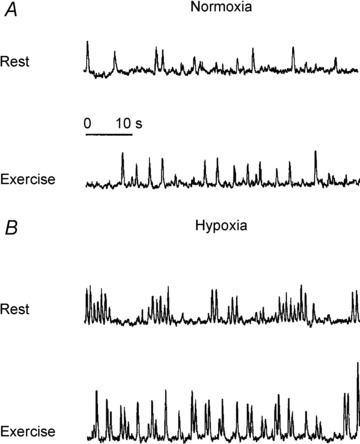
Representative recordings of leg MSNA at rest and during exercise under normoxia (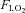 21%; A) and hypoxia (
21%; A) and hypoxia (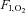 ) ∼10%; B). Adapted with permission from Hanada et al. (2003).
) ∼10%; B). Adapted with permission from Hanada et al. (2003).
Under normoxic conditions sympathetic vasoconstrictor responses are blunted in the vascular beds of contracting limbs (Remensnyder et al. 1962; Dinenno & Joyner, 2003), a phenomenon referred to as ‘functional sympatholysis’. Moreover vasoconstrictor responses to sympathetic stimulation and exogenous noradrenaline are attenuated during hypoxia (Heistad & Wheeler, 1970; Heistad et al. 1975). These findings raise the possibility that blunted vasoconstrictor responsiveness (augmented functional sympatholysis) contributes to the compensatory vasodilatation during hypoxic exercise. However, we found that the compensatory vasodilator response to hypoxic forearm exercise was not due to an augmented functional sympatholysis (Wilkins et al. 2006). These observations suggest that enhanced vasodilator signals and not blunted vasoconstriction are primarily responsible for the compensatory vasodilatation in skeletal muscle during hypoxic exercise.
Role of local vasodilator pathways
Our lab has conducted a series of studies to evaluate at least some of the putative metabolic vasodilating substances (adenosine, nitric oxide (NO), prostaglandins, etc.) as potential mediators of compensatory vasodilatation during hypoxia. During mild rhythmic hand gripping we found evidence that at least some of the compensatory vasodilatation was mediated by vasodilating β-adrenergic receptors in the active limb, stimulating the release of NO (Wilkins et al. 2008; Casey et al. 2010). As exercise intensity increased, there still appeared to be a role for NO but the β-adrenergic-NO pathway was absent and there must have been some other mechanism evoking NO release as a mediator of compensatory vasodilatation (Wilkins et al. 2008; Casey et al. 2011). These mechanisms may include direct release of NO from the endothelium as a result of luminal hypoxia (Pohl & Busse, 1989), shear stress mediated NO release (Kooijman et al. 2008), NO from erythrocytes in the form S-nitrosohaemoglobin (Stamler et al. 1989), and/or increased NO release via ATP and prostaglandins (Mortensen et al. 2007; Mortensen et al. 2009). Nonetheless, evidence that hypoxia increases plasma but not skeletal muscle interstitial NO in humans is likely to suggest an endovascular or endothelial source (Leuenberger et al. 2008).
Adenosine has also been suggested to play a role in compensatory vasodilator responses (Bryan & Marshall, 1999a,b). However, in several human studies we (Casey et al. 2009) and others (Heinonen et al. 2010) were unable to find clear evidence for a primary role for adenosine. Moreover, adenosine does not contribute to the compensatory vasodilatation after NO synthase inhibition, thus indicating that adenosine does not act through a NO-independent pathway in the compensatory vasodilator response during hypoxic exercise (Casey et al. 2010). This is important because in the coronary circulation adenosine appears to play an important role in regulating blood flow to ischaemic tissue in various models of coronary artery disease (Laxson et al. 1993). Likewise, in exercising human forearm muscles, inflation of a balloon in the brachial artery upstream from the forearm causes an immediate decrease in forearm blood flow followed by recovery to control values and we have evidence that adenosine does in fact contribute to the recovery of flow under these circumstances (Casey & Joyner, 2011).
Over the past several years it has been proposed that erythrocytes are not only responsible for sensing and carrying O2, but also participate in the regulation of blood flow and its distribution by releasing ATP (Ellsworth et al. 2009). In this context, acute exposure to hypoxia at rest as well as exercise under normoxic conditions leads to increases in venous plasma levels of ATP (Gonzalez-Alonso et al. 2002; Mortensen et al. 2011). Whether erythrocyte derived ATP contributes to the compensatory vasodilatation observed during hypoxic exercise is still unclear. Unfortunately, specific pharmacological antagonists for P2 receptors to address the role of ATP in the hypoxia-induced compensatory vasodilatation are currently unavailable for human use. Therefore, we are left to rely on plasma measures to gain insight into the contribution of ATP in the compensatory vasodilator response to hypoxic exercise. Along these lines, arterial and venous plasma levels of ATP measured via intravascular microdialysis are not greater during hypoxic compared to normoxic exercise (Mortensen et al. 2011). Thus the question whether ATP contributes to compensatory vasodilatation during hypoxic exercise remains somewhat elusive. Figure 3 illustrates well established as well as potential dilator signals that contribute to compensatory vasodilatation during submaximal hypoxic exercise.
Figure 3. Proposed mechanisms for hypoxia-induced vasodilatation at rest and during exercise.
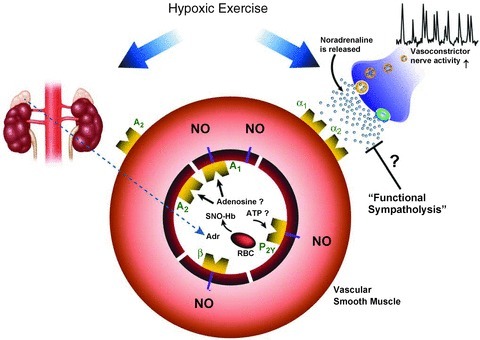
During hypoxic exercise NO is the final common pathway for the compensatory dilator response. Systemic adrenaline release, acting via β-adrenergic receptors, contributes to the NO-mediated vasodilatation at lower exercise intensities, but this β-adrenergic contribution decreases with increasing exercise intensity. ATP released from the red blood cell remains an attractive candidate for stimulating NO during higher intensity hypoxic exercise. Adenosine receptor activation does not appear to play a major role (either dependent or independent of NO) in the compensatory vasodilator response during hypoxic exercise in humans. Compensatory vasodilatation persists despite an increased sympathetic vasoconstrictor activity directed towards skeletal muscle during hypoxic exercise. Blunted vasoconstrictor responsiveness (augmented functional sympatholysis) does not appear to contribute to the compensatory vasodilatation during hypoxic exercise. α1 and α2 indicate α1 and α2 adrenergic receptors, respectively; A1 and A2, adenosine receptors; β, β2 adrenergic receptors; Adr, adrenaline.
What happens during whole body maximal exercise?
Under the assumption that increases in cardiac output during exercise are directed at the active skeletal muscle, there is evidence for compensatory vasodilatation during submaximal whole body exercise (Stenberg et al. 1966; Hughes et al. 1968; Vogel & Gleser, 1972). For example, when the arterial O2 content and saturation are reduced via acute hypoxia, there is an increase in cardiac output that keeps O2 delivery to the skeletal muscles constant (Hughes et al. 1968). However, during maximal exercise involving a large muscle mass (i.e. cycling or running), blood flow and vasodilatation in the exercising limbs are reduced under hypoxic compared to normoxic conditions (Calbet, 2000). The lower blood flow in exercising skeletal muscle is likely to be related to the significant reductions in maximal cardiac output observed during hypoxic exercise (Calbet et al. 2003, 2009a; Lundby et al. 2006). The potential mechanisms for reductions in maximal cardiac output during acute hypoxia have been recently reviewed by others (Calbet et al. 2009b). Conversely, when small muscle mass is activated and cardiac output is not limited, skeletal muscle blood flow is maintained at peak exercise under hypoxic conditions (Calbet et al. 2009a).
It is apparent that exercise intensity (submaximal vs. maximal), the amount of muscle mass, and the severity and duration of hypoxia are all important factors in determining the blood flow and compensatory vasodilator responses during hypoxic exercise. The studies related to the mechanisms responsible for compensatory vasodilatation during hypoxic exercise highlighted in this review have mainly been derived using a forearm exercise model and local infusions of various study drugs. This approach has allowed us, as well as others, to investigate local vascular control without additional confounding variables (i.e. cardiovascular reflexes) that occur with exercise involving larger muscle mass or systemic drug infusions. However, it is currently unclear whether the regulatory mechanisms involved in compensatory vasodilatation during forearm exercise continue to contribute to muscle blood flow as exercise intensity and/or the amount of active muscle mass increases. In this context, future studies will be needed to examine vascular responses to hypoxic exercise during ‘real world’ settings (such as whole body exercise at altitude).
Summary
It is clear that acute reductions in available O2 via systemic hypoxia promote compensatory vasodilatation and an augmented blood flow in contracting skeletal muscle during submaximal workloads. The compensatory response is essential to preserving muscle O2 delivery and ensuring it is matched to demand. Interestingly, the compensatory vasodilatation and augmented flow persist despite large increases in sympathetic vasoconstrictor activity directed towards skeletal muscle. Thus, the degree of vasodilatation prevails over the vasoconstrictor response in determining vasomotor tone during hypoxic exercise.
We have demonstrated that a key vasodilator signal in the compensatory response is NO. However, it appears that the NO-mediated component of the compensatory vasodilatation during hypoxic exercise is regulated through different pathways with increasing exercise intensity. That is a β-adrenergic receptor-stimulated NO component exists during low-intensity hypoxic exercise, whereas the source of NO contributing to compensatory dilatation is less dependent on β-adrenergic mechanisms as exercise intensity increases. It is currently unclear what the stimulus of NO release is with increasing intensity of muscle contraction but does not appear to be adenosine. ATP released from erythrocytes and/or endothelial derived prostaglandins remain attractive candidates for stimulating NO release during higher intensity hypoxic exercise.
References
- Andersen P, Saltin B. Maximal perfusion of skeletal muscle in man. J Physiol. 1985;366:233–249. doi: 10.1113/jphysiol.1985.sp015794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong RB, Laughlin MH. Rat muscle blood flows during high-speed locomotion. J Appl Physiol. 1985;59:1322–1328. doi: 10.1152/jappl.1985.59.4.1322. [DOI] [PubMed] [Google Scholar]
- Bryan PT, Marshall JM. Adenosine receptor subtypes and vasodilatation in rat skeletal muscle during systemic hypoxia: a role for A1 receptors. J Physiol. 1999a;514:151–162. doi: 10.1111/j.1469-7793.1999.151af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan PT, Marshall JM. Cellular mechanisms by which adenosine induces vasodilatation in rat skeletal muscle: significance for systemic hypoxia. J Physiol. 1999b;514:163–175. doi: 10.1111/j.1469-7793.1999.163af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calbet JA. Oxygen tension and content in the regulation of limb blood flow. Acta physiol Scand. 2000;168:465–472. doi: 10.1046/j.1365-201x.2000.00698.x. [DOI] [PubMed] [Google Scholar]
- Calbet JA, Boushel R, Radegran G, Sondergaard H, Wagner PD, Saltin B. Determinants of maximal oxygen uptake in severe acute hypoxia. Am J Physiol Regul Integr Comp Physiol. 2003;284:R291–303. doi: 10.1152/ajpregu.00155.2002. [DOI] [PubMed] [Google Scholar]
- Calbet JA, Radegran G, Boushel R, Saltin B. On the mechanisms that limit oxygen uptake during exercise in acute and chronic hypoxia: role of muscle mass. J Physiol. 2009a;587:477–490. doi: 10.1113/jphysiol.2008.162271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calbet JA, Robach P, Lundby C. The exercising heart at altitude. Cell Mol Life Sci. 2009b;66:3601–3613. doi: 10.1007/s00018-009-0148-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey DP, Curry TB, Wilkins BW, Joyner MJ. Nitric oxide-mediated vasodilation becomes independent of β-adrenergic receptor activation with increased intensity of hypoxic exercise. J Appl Physiol. 2011;110:687–694. doi: 10.1152/japplphysiol.00787.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey DP, Joyner MJ. Contribution of adenosine to compensatory dilation in hypoperfused contracting human muscles is independent of nitric oxide. J Appl Physiol. 2011;110:1181–1189. doi: 10.1152/japplphysiol.00836.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey DP, Madery BD, Curry TB, Eisenach JH, Wilkins BW, Joyner MJ. Nitric oxide contributes to the augmented vasodilatation during hypoxic exercise. J Physiol. 2010;588:373–385. doi: 10.1113/jphysiol.2009.180489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey DP, Madery BD, Pike TL, Eisenach JH, Dietz NM, Joyner MJ, Wilkins BW. Adenosine receptor antagonist and augmented vasodilation during hypoxic exercise. J Appl Physiol. 2009;107:1128–1137. doi: 10.1152/japplphysiol.00609.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifford PS. Skeletal muscle vasodilatation at the onset of exercise. J Physiol. 2007;583:825–833. doi: 10.1113/jphysiol.2007.135673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinenno FA, Joyner MJ. Blunted sympathetic vasoconstriction in contracting skeletal muscle of healthy humans: is nitric oxide obligatory. J Physiol. 2003;553:281–292. doi: 10.1113/jphysiol.2003.049940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellsworth ML, Ellis CG, Goldman D, Stephenson AH, Dietrich HH, Sprague RS. Erythrocytes: oxygen sensors and modulators of vascular tone. Physiology. 2009;24:107–116. doi: 10.1152/physiol.00038.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Alonso J, Olsen DB, Saltin B. Erythrocyte and the regulation of human skeletal muscle blood flow and oxygen delivery: role of circulating ATP. Circ Res. 2002;91:1046–1055. doi: 10.1161/01.res.0000044939.73286.e2. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Alonso J, Richardson RS, Saltin B. Exercising skeletal muscle blood flow in humans responds to reduction in arterial oxyhaemoglobin, but not to altered free oxygen. J Physiol. 2001;530:331–341. doi: 10.1111/j.1469-7793.2001.0331l.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada A, Sander M, Gonzalez-Alonso J. Human skeletal muscle sympathetic nerve activity, heart rate and limb haemodynamics with reduced blood oxygenation and exercise. J Physiol. 2003;551:635–647. doi: 10.1113/jphysiol.2003.044024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinonen IH, Kemppainen J, Kaskinoro K, Peltonen JE, Borra R, Lindroos M, Oikonen V, Nuutila P, Knuuti J, Boushel R, Kalliokoski KK. Regulation of human skeletal muscle perfusion and its heterogeneity during exercise in moderate hypoxia. Am J Physiol Regul Interg Comp Physiol. 2010;299:R72–79. doi: 10.1152/ajpregu.00056.2010. [DOI] [PubMed] [Google Scholar]
- Heistad DD, Abboud FM, Mark AL, Schmid PG. Effect of hypoxemia on responses to norepinephrine and angiotensin in coronary and muscular vessels. J Pharmacol Exp Ther. 1975;193:941–950. [PubMed] [Google Scholar]
- Heistad DD, Wheeler RC. Effect of acute hypoxia on vascular responsiveness in man. I. Responsiveness to lower body negative pressure and ice on the forehead. II. Responses to norepinephrine and angiotensin. 3. Effect of hypoxia and hypocapnia. J Clin Invest. 1970;49:1252–1265. doi: 10.1172/JCI106338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes RL, Clode M, Edwards RH, Goodwin TJ, Jones NL. Effect of inspired O2 on cardiopulmonary and metabolic responses to exercise in man. J Appl Physiol. 1968;24:336–347. doi: 10.1152/jappl.1968.24.3.336. [DOI] [PubMed] [Google Scholar]
- Joyner MJ, Wilkins BW. Exercise hyperaemia: is anything obligatory but the hyperaemia. J Physiol. 2007;583:855–860. doi: 10.1113/jphysiol.2007.135889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooijman M, Thijssen DH, de Groot PC, Bleeker MW, van Kuppevelt HJ, Green DJ, Rongen GA, Smits P, Hopman MT. Flow-mediated dilatation in the superficial femoral artery is nitric oxide mediated in humans. J Physiol. 2008;586:1137–1145. doi: 10.1113/jphysiol.2007.145722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laxson DD, Homans DC, Bache RJ. Inhibition of adenosine-mediated coronary vasodilation exacerbates myocardial ischemia during exercise. Am J Physiol Heart Circ Physiol. 1993;265:H1471–1477. doi: 10.1152/ajpheart.1993.265.5.H1471. [DOI] [PubMed] [Google Scholar]
- Leuenberger UA, Johnson D, Loomis J, Gray KS, MacLean DA. Venous but not skeletal muscle interstitial nitric oxide is increased during hypobaric hypoxia. Eur J Appl Physiol. 2008;102:457–461. doi: 10.1007/s00421-007-0601-x. [DOI] [PubMed] [Google Scholar]
- Lundby C, Sander M, van Hall G, Saltin B, Calbet JA. Maximal exercise and muscle oxygen extraction in acclimatizing lowlanders and high altitude natives. J Physiol. 2006;573:535–547. doi: 10.1113/jphysiol.2006.106765. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
Mortensen SP, Damsgaard R, Dawson EA, Secher NH, Gonzalez-Alonso J. Restrictions in systemic and locomotor skeletal muscle perfusion, oxygen supply and
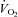 during high-intensity whole-body exercise in humans. J Physiol. 2008;586:2621–2635. doi: 10.1113/jphysiol.2007.149401. [DOI] [PMC free article] [PubMed] [Google Scholar]
during high-intensity whole-body exercise in humans. J Physiol. 2008;586:2621–2635. doi: 10.1113/jphysiol.2007.149401. [DOI] [PMC free article] [PubMed] [Google Scholar] - Mortensen SP, Gonzalez-Alonso J, Damsgaard R, Saltin B, Hellsten Y. Inhibition of nitric oxide and prostaglandins, but not endothelial-derived hyperpolarizing factors, reduces blood flow and aerobic energy turnover in the exercising human leg. J Physiol. 2007;581:853–861. doi: 10.1113/jphysiol.2006.127423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen SP, Nyberg M, Thaning P, Saltin B, Hellsten Y. Adenosine contributes to blood flow regulation in the exercising human leg by increasing prostaglandin and nitric oxide formation. Hypertension. 2009;53:993–999. doi: 10.1161/HYPERTENSIONAHA.109.130880. [DOI] [PubMed] [Google Scholar]
- Mortensen SP, Thaning P, Nyberg M, Saltin B, Hellsten Y. Local release of ATP into the arterial inflow and venous drainage of human skeletal muscle: insight from ATP determination with the intravascular microdialysis technique. J Physiol. 2011;589:1847–1857. doi: 10.1113/jphysiol.2010.203034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musch TI. Skeletal muscle blood flow in exercising dogs. Med Sci Sports Exerc. 1988;20:S104–108. doi: 10.1249/00005768-198810001-00002. [DOI] [PubMed] [Google Scholar]
- Pohl U, Busse R. Hypoxia stimulates release of endothelium-derived relaxant factor. Am J Physiol Heart Circ Physiol. 1989;256:H1595–1600. doi: 10.1152/ajpheart.1989.256.6.H1595. [DOI] [PubMed] [Google Scholar]
- Remensnyder JP, Mitchell JH, Sarnoff SJ. Functional sympatholysis during muscular activity. Observations on influence of carotid sinus on oxygen uptake. Circ Res. 1962;11:370–380. doi: 10.1161/01.res.11.3.370. [DOI] [PubMed] [Google Scholar]
- Roach RC, Koskolou MD, Calbet JA, Saltin B. Arterial O2 content and tension in regulation of cardiac output and leg blood flow during exercise in humans. Am J Physiol Heart Circ Physiol. 1999;276:H438–445. doi: 10.1152/ajpheart.1999.276.2.H438. [DOI] [PubMed] [Google Scholar]
- Shepherd JT. Circulation to skeletal muscle. In: Shepherd JT, Abboud FM, editors. Handbook of Physiology, section 2, The Cardiovascular System, vol. 3, Peripheral Circulation and Organ Blood Flow. Bethesda: American Physiological Society; 1983. pp. 319–370. [Google Scholar]
- Stamler J, Mendelsohn ME, Amarante P, Smick D, Andon N, Davies PF, Cooke JP, Loscalzo J. N-Acetylcysteine potentiates platelet inhibition by endothelium-derived relaxing factor. Circ Res. 1989;65:789–795. doi: 10.1161/01.res.65.3.789. [DOI] [PubMed] [Google Scholar]
- Stenberg J, Ekblom B, Messin R. Hemodynamic response to work at simulated altitude, 4,000 m. J Appl Physiol. 1966;21:1589–1594. doi: 10.1152/jappl.1966.21.5.1589. [DOI] [PubMed] [Google Scholar]
- Vogel JA, Gleser MA. Effect of carbon monoxide on oxygen transport during exercise. J Appl Physiol. 1972;32:234–239. doi: 10.1152/jappl.1972.32.2.234. [DOI] [PubMed] [Google Scholar]
- Weisbrod CJ, Minson CT, Joyner MJ, Halliwill JR. Effects of regional phentolamine on hypoxic vasodilatation in healthy humans. J Physiol. 2001;537:613–621. doi: 10.1111/j.1469-7793.2001.00613.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins BW, Pike TL, Martin EA, Curry TB, Ceridon ML, Joyner MJ. Exercise intensity-dependent contribution of beta-adrenergic receptor-mediated vasodilatation in hypoxic humans. J Physiol. 2008;586:1195–1205. doi: 10.1113/jphysiol.2007.144113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins BW, Schrage WG, Liu Z, Hancock KC, Joyner MJ. Systemic hypoxia and vasoconstrictor responsiveness in exercising human muscle. J Appl Physiol. 2006;101:1343–1350. doi: 10.1152/japplphysiol.00487.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]