

Abstract
Enhancement of contractile force (inotropy) occurs in skeletal muscle following neuroendocrine release of catecholamines and activation of muscle β-adrenergic receptors. Despite extensive study, the molecular mechanism underlying the inotropic response in skeletal muscle is not well understood. Here we show that phosphorylation of a single serine residue (S2844) in the sarcoplasmic reticulum (SR) Ca2+ release channel/ryanodine receptor type 1 (RyR1) by protein kinase A (PKA) is critical for skeletal muscle inotropy. Treating fast twitch skeletal muscle from wild-type mice with the β-receptor agonist isoproterenol (isoprenaline) increased RyR1 PKA phosphorylation, twitch Ca2+ and force generation. In contrast, the enhanced muscle Ca2+, force and in vivo muscle strength responses following isoproterenol stimulation were abrogated in RyR1-S2844A mice in which the serine in the PKA site in RyR1 was replaced with alanine. These data suggest that the molecular mechanism underlying skeletal muscle inotropy requires enhanced SR Ca2+ release due to PKA phosphorylation of S2844 in RyR1.
Key points
Under conditions of acute adrenergic stress (i.e. fight or flight response), the contractile force of muscle is enhanced, a phenomenon known as inotropy.
The molecular determinant of the inotropic mechanism is poorly understood but involves potentiated release of calcium within the muscle cell.
Here we report that adrenergic receptor-dependent phosphorylation of a single amino acid in the calcium release channel (ryanodine receptor 1) mediates the increased calcium and force that is seen in the muscle following acute stress.
These findings further our understanding of the molecular mechanisms of muscular force regulation, and the importance for exercise physiology and muscle weakness (dynopenia).
Introduction
Skeletal muscle displays increased force upon adrenergic stimulation (Brown et al. 1948; Cairns & Dulhunty, 1993b); however, despite the well-established role of this physiological response, the molecular mechanism is not known. Under conditions of stress, stimulation of Gs-protein coupled β-adrenergic receptors (β1- and β2-receptors) activates adenylyl cyclase and causes generation of the second messenger cAMP, which in turn activates protein kinase A (PKA) (Rockman et al. 2002; Lynch & Ryall, 2008). PKA-mediated phosphorylation of Ca2+ handling proteins is a key mechanism underlying increased contraction of cardiac myocytes (Bers, 2001; Shan et al. 2010b). In skeletal muscle, unlike in cardiac muscle, contraction does not depend on Ca2+ entry across the plasma membrane (e.g. via opening of L-type Ca2+ channels) and the Ca2+ needed to activate the myofilaments comes from the release of sarcoplasmic reticulum (SR) Ca2+ (Allen et al. 2008). Fast twitch skeletal muscle fibres (type II fibres) lack the SR Ca2+ ATPase (SERCA2a)-associated protein phospholamban, a target of PKA phosphorylation that regulates SR Ca2+ load in cardiomyocytes. β-receptor stimulation in the absence of phospholamban does not lead to changes in SERCA activity or SR Ca2+ load in fast twitch muscles (Cairns et al. 1993; Liu et al. 1997). Furthermore, neither myofilament Ca2+ sensitivity nor sarcolemmal Ca2+ fluxes are affected by β-adrenergic activation in fast twitch skeletal muscle (Cairns et al. 1993). None the less, fast twitch muscle fibres display a robust inotropic response to β-receptor agonists due to a potentiated increase in cytoplasmic [Ca2+] (Cairns & Dulhunty, 1993a).
Using phosphopeptide mapping or site-directed mutagenesis of recombinant ryanodine receptor type 1 (RyR1), we (Reiken et al. 2003), and others (Suko et al. 1993), have identified the PKA phosphorylation site of human RyR1 as RyR1-serine (S) 2843 (RyR1-S2844 in rodents). We have now used a novel genetically altered mouse (RyR1-S2844A), which expresses a mutant RyR1 that cannot be PKA phosphorylated, to show that β-receptor agonist-dependent increase in twitch [Ca2+] and force in mammalian fast twitch skeletal muscle are dependent on phosphorylation of a single amino acid, S2844 in RyR1.
Methods
Generation of ryanodine receptor type 1-serine 2844A mice
A PCR fragment carrying the point mutation, RyR1-2844 S > A, was used to replace the wild-type (WT) sequence by conventional cloning methods. The targeting vector was electroporated into embryonic stem cells derived from hybrid C57BL/6N × 129SvEv mice (Taconic, Germantown, NY, USA), which after selection were implanted into blastocysts in C57BL/6 mice. Chimeras were crossed with C57BL/6 to generate the F1. After confirmation of germline transmission and crossing with EIIa cre mice to excise the neomycin cassette, mice were backcrossed for more than six generations into the C57BL/6J strain. Embryonic stem cell preparation and implantation was performed at inGenious Targeting Laboratory, Ronkonkoma, New York, USA.
All animal experiments were approved by Columbia University's Institutional Animal Care and Use Committee. Killing of the experimental animals was performed using CO2 asphyxiation followed by cervical dislocation.
Muscle function
Extensor digitorum longus (EDL) muscles were dissected from hind limbs. Stainless steel hooks were tied to the muscle tendons using nylon sutures and the muscles were mounted between a force transducer (Harvard Apparatus, Holliston, MA, USA) and an adjustable hook. The muscles were immersed in a stimulation chamber containing O2/CO2 (95/5%) bubbled Tyrode solution (in mm: NaCl 121, KCl 5.0, CaCl2 1.8, MgCl2 0.5, NaH2PO4 0.4, NaHCO3 24, EDTA 0.1, glucose 5.5). The muscle was stimulated to contract using an electrical field between two platinum electrodes (Aurora Scientific, Aurora, Ontario, Canada).
At the start of each experiment the muscle length was adjusted to yield the maximum force. Isometric twitch force was produced by stimulating the muscle using a 0.5 ms pulse at supra-threshold voltage. Force was measured from each muscle before and after application of isoproterenol (ISO) (1 μm; 15 min). At the end of force measurement, the length (L0) and weight of the muscle was measured and the muscle was snap frozen in liquid N2.
Ca2+ imaging using fluorescent indicators in flexor digitorum brevis fibres
Single flexor digitorum brevis (FDB) fibres were obtained by enzymatic dissociation as previously described (Aydin et al. 2009). FDB muscles from both hind limbs were incubated for ∼2 h at 37°C in ∼4 ml Dulbecco's Modified Eagles medium containing 0.3% collagenase 1 (Sigma, St Louis, MO, USA) and 10% fetal bovine serum. The muscles were transferred to a culture dish containing fresh Dulbecco's modified Eagle's medium (∼4 ml) and gently triturated using a 1000 μl pipette until the muscles were dissociated. The cell suspension was stored in an incubator at 37°C/5% CO2 until the start of the experiment.
FDB fibres were loaded with the fluorescent Ca2+ indicator fluo-4 acetoxymethyl ester (AM; 5 μm, Invitrogen/Molecular Probes, Grand Island, NY, USA) for 15 min at room temperature. The cells were allowed to attach to a laminin-coated glass coverslip that formed the bottom of a perfusion chamber. The cells were then superfused with Tyrode solution [in mm: NaCl 121, KCl 5.0, CaCl2 1.8, MgCl2 0.5, NaH2PO4 0.4, NaHCO3 24, EDTA 0.1, glucose 5.5; bubbled with O2/CO2 (95/5%)]. The fibres were triggered to twitch contraction using electrical field stimulation (pulses of 0.5 ms at supra-threshold voltage) and fluo-4 fluorescence was monitored using a confocal microscope system (Zeiss LSM 5 Live, 40× oil immersion lens, excitation wavelength was 488 nm and the emitted fluorescence was recorded between 495 and 525 nm). The use of single excitation/emission dye Fluo-4 necessitates normalizing to pre-stimulation values to negate possible differences in dye loading and excitation strength. Only fibres attached to the bottom of the perfusion chamber throughout the twitch stimulation were measured from. All experiments were performed at room temperature (∼20°C).
In some experiments, FDB fibres were loaded with 6 μm of fura2-AM for 30 min and allowed to de-esterify in imaging buffer without fura for an additional 20 min. Cells were plated on glass coverslips mounted on a Nikon Diaphot microscope. Fura-2 was excited sequentially at 340 nm and 380 nm via a DeltaRamV high-speed random access monochromator (Photon Technology International, Birmingham, NJ, USA). Emitted fluorescence was recorded using an 814 photomultipler detection system connected to a PC running Felix32 (Photon Technology International).
Ryanodine receptor type 1 immunoprecipitation and immunoblotting
EDLs were isotonically lysed in 0.5 ml of buffer containing 50 mm Tris–HCl (pH 7.4), 150 mm NaCl, 20 mm NaF, 1.0 mm Na3VO4 and protease inhibitors. This muscle lysate was used for either immunoblotting with cAMP response element-binding protein (CREB/pCREB) antibodies or immunoprecipitation of the RyR1. In immunoprecipitation, an anti-RyR antibody (4 μg 5029 antibody) was used to immunoprecipitate RyR1 from 250 μg of tissue homogenate. The samples were incubated with the antibody in 0.5 ml of a modified RIPA buffer (50 mm Tris–HCl pH 7.4, 0.9% NaCl, 5.0 mm NaF, 1.0 mm Na3VO4, 1% Triton-X100 and protease inhibitors) for 1 h at 4°C. The immune complexes were incubated with protein A Sepharose beads (Sigma) at 4°C for 1 h and the beads were washed three times with buffer. Proteins were separated on SDS–PAGE gels (6% for RyR1, 15% for calstabin1) and transferred on to nitrocellulose membranes for 1 h at 200 mA (SemiDry transfer blot, Bio-Rad, Hercules, CA, USA). After incubation with blocking solution (LICOR Biosciences, Lincoln NE, USA) to prevent non-specific antibody binding, immunoblots were developed with anti-RyR (Affinity Bioreagents, Bolder, CO, USA; 1:2000), anti-phospho-RyR1-pSer2844 (1:5000). All immunoblots were developed and quantified using the Odyssey Infrared Imaging System (LICOR Biosystems) and infrared-labelled secondary antibodies.
Grip strength
Forelimb grip strength was measured in stressed WT and S2844A mice using an automated grip strength meter (BIOSEB In Vivo Research Instruments, Vitrolles, France). Mouse forelimbs were allowed to grab on to a metal grid (attached to a force transducer). Thereafter, mice were pulled by the tail until losing grip. Maximum force was recorded. The grip strength was calculated from the average of three repeated measurements in each mouse. This procedure activated stress signalling, as the mice resisted and displayed spontaneous defecation and/or urination. All measurements were performed blinded with respect to the mouse genotype.
Results
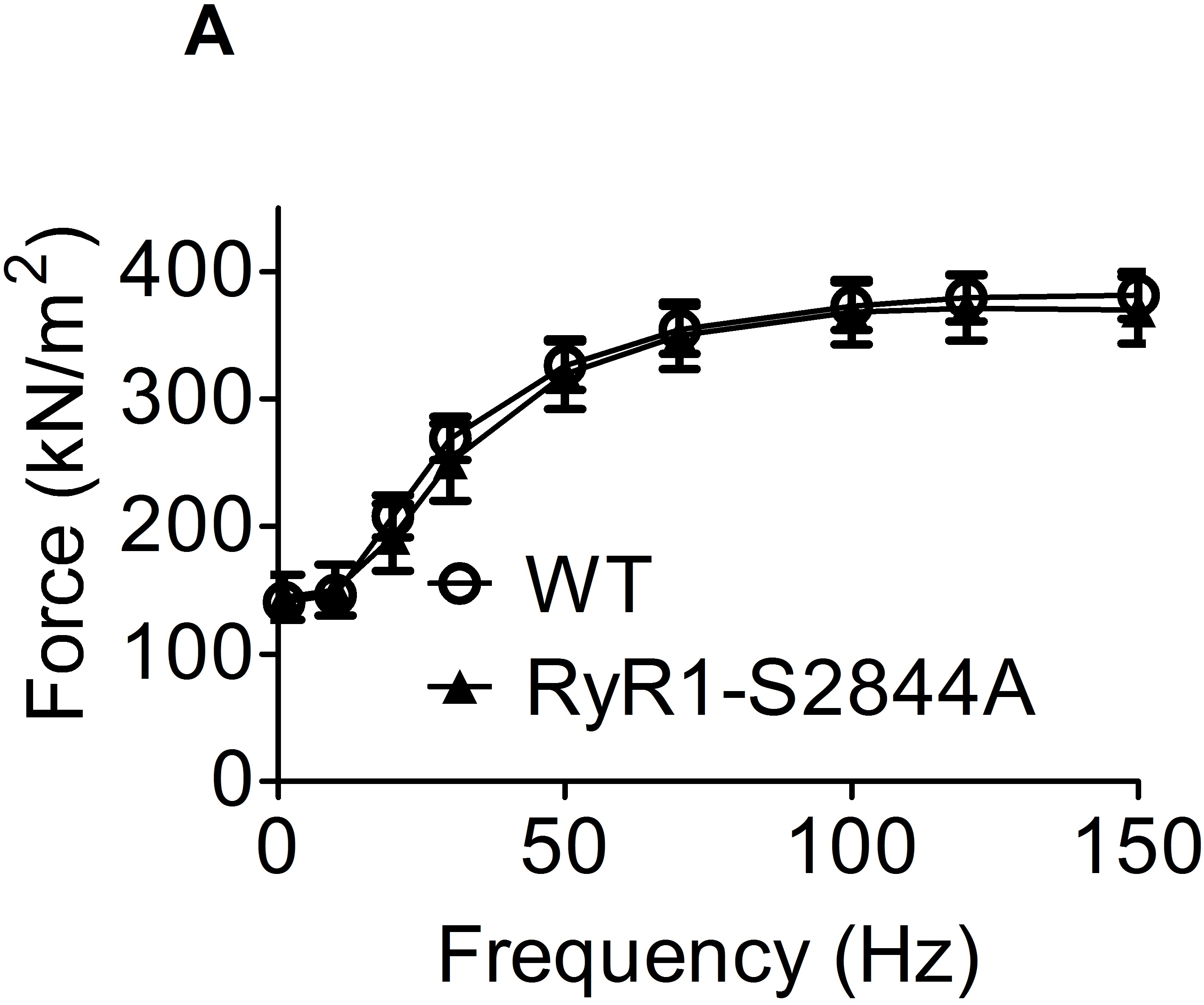
To examine the role of PKA-dependent RyR1 phosphorylation in skeletal muscle inotropy we generated a mutant mouse (RyR1-S2844A) in which the serine at the RyR1 PKA phosphorylation site was substituted with alanine, which is an amino acid that cannot be phosphorylated (Reiken et al. 2003). There was no difference in body weight between the WT and RyR1-S2844A mice [WT, 28 ± 1.1 g, n= 8; RyR1-S2844A, 26 ± 1.3 g, n= 9; mean ± SEM, P= NS (unpaired t test)] and no overt difference in skeletal muscle histology (Supplementary Fig. S1). Force generation in isolated fast twitch EDL muscles from the RyR1-S2844A mice was compared to WT mice. At baseline, force-generating capacity in the RyR1-S2844A and WT EDL was not different, as evidenced by the similar force–frequency curves (Supplementary Fig. S2). Moreover, isometric twitch force kinetics were similar in the two strains, as seen for both the force development rate (WT, 5.6 ± 0.5 N s−1; S2844A, 6.2 ± 0.7 N s−1; mean ± SEM, n= 8, P= NS) and half-relaxation time (WT, 42 ± 4 ms; S2844A, 37 ± 3 ms; mean ± SEM, n= 8, P= NS).
Brief application of the β-receptor agonist ISO (1 μm, 15 min) to WT EDL muscles caused a significant increase in twitch force (control, 140 ± 7.8 kPa; ISO, 159 ± 7.6 kPa; mean ± SEM, n= 8, P < 0.01, paired t test; Fig. 1A and B). In contrast, EDL muscles from the RyR1-S2844A mouse did not display any increase in twitch force in response to ISO (RyR1-S2844A control, 145 ± 18 kPa; RyR1-S2844A ISO, 142 ± 16 kPa; mean ± SEM, n= 8, P= NS, paired t test; Fig. 1C and D). Furthermore, in the presence of ISO the rate of twitch force development in WT muscle was faster (control, 5.6 ± 0.5 N s−1; ISO, 7.3 ± 0.3 N s−1; mean ± SEM, n= 8, P < 0.05; Fig. 1E and F). The RyR1-S2844A muscles did not display any significant change in the rate of force development when ISO was present (control, 6.2 ± 0.7 N s−1; ISO, 5.6 ± 0.8; mean ± SEM, n= 8, P= NS; Fig. 1E). However, the half-relaxation time was not significantly altered in any of the two strains following ISO exposure [WT control, 42 ± 4 ms, WT ISO, 39 ± 3 ms, P= NS; S2844A control, 37 ± 3 ms, 33 ± 2 ms, P= NS; mean ± SEM, n= 8 (in all groups)]. Taken together, these data indicate that RyR1-S2844 phosphorylation plays a key role in both the increase in peak force as well as the rate of force production in fast twitch skeletal muscle following adrenergic stimulation.
Figure 1. β-adrenergic inotropy in skeletal muscle is dependent on phosphorylation of serine 2844 in the RyR1.
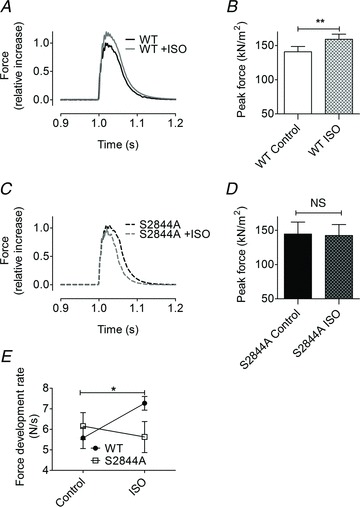
A and C, representative twitch force in extensor digitorum longus muscle from (A) wild-type (WT) and (C) S2844A mice. Force was normalized to the maximal twitch force before application of isoproterenol (ISO), which was set to 1. B and D, average specific force (twitch force normalized to cross-sectional area) before and after ISO in (B) WT and (D) S2844A EDL muscles. E, rate of force development before and after ISO in WT and S2844A extensor digitorum longus muscles. All pooled data are presented as mean ± SEM, n= 8 muscles for all groups. *P < 0.05, **P < 0.01 (paired t test for comparisons before and after ISO in the same muscle), ##P < 0.01 (unpaired t test, for comparison of WT and S2844A groups). NS, difference between control and ISO in the S2844A group was not significant (paired t test).
Muscle force is graded by SR Ca2+ release, which determines cytoplasmic [Ca2+], and β-adrenergic agonists have been shown to increase cytoplasmic [Ca2+] in fast twitch FDB muscle. However, the mechanism underlying this inotropic response remains obscure (Cairns et al. 1993). To examine ISO-induced enhancement of twitch Ca2+, we loaded isolated FDB muscle fibres with a cell permeable fluorescent indicator for Ca2+ (fluo-4) and triggered twitch contractions by electrically stimulating the fibres (Andersson et al. 2011). There were, however, no significant differences in peak twitch Ca2+ amplitudes in fibres from WT and mutant mice before ISO treatment (F/F0 of 3.4 ± 0.5 for WT fibres and 3.9 ± 0.2 for RyR1-S2844A fibres; mean ± SEM, n= 8–10, P= 0.3) nor were there any differences in the Ca2+ transient time to peak (TTP) (WT, 6.1 ± 0.7; S2844A, 5.1 ± 0.3; mean ± SEM, n= 8–10, P= 0.2). To test differences in resting Ca2+ between WT and S2844A muscles, we conducted a series of experiments using FDB fibres loaded with the ratiometric Ca2+ indicator fura-2 AM, which is a preferred indicator when comparing baseline fluorescence values from different muscles. We found no significant differences in resting Ca2+ levels when we compared fibres from WT and RyR1-S2844A mice [WT, 0.134 ± 0.003 a.u., n= 11 fibres (from two mice); S2844A, 0.128 ± 0.001 a.u., n= 9 fibres (from two mice); mean fura-2 AM ratios (340 nm/380 nm) ± SEM, P= NS].
Changes in fluo-4 fluorescence were measured in the same muscle fibres under control conditions and after 10 min in the presence of ISO (1 μm). In WT fibres, the peak fluo-4 signal increased significantly following ISO treatment (119 ± 4% of the pre-exposure control; n= 8 cells from three animals, P < 0.01; Fig. 2A and C). Basal fluo-4 fluorescence was not changed following ISO exposure when measured within the same fibre (WT control, 15.0 ± 1.9 a.u.; WT ISO, 14.8 ± 1.9 a.u.; mean ± SEM, n= 8 cells from three animals, P= NS), indicating that resting Ca2+ was unaltered. Moreover, the Ca2+ TTP was shorter with ISO (control, 6.1 ± 0.7 ms; vs. ISO, 4.5 ± 0.3; n= 8, P < 0.05; Fig. 2D). Thus, SR Ca2+ release amplitude is increased and occurs more rapidly when the muscle is exposed to acute adrenergic stimulation. This response could possibly explain the increased rate of force production under stress conditions.
Figure 2. Blocking phosphorylation of RyR1 at serine 2844 inhibits the β-adrenergic effect on Ca2+ transients in FDB muscle fibers.
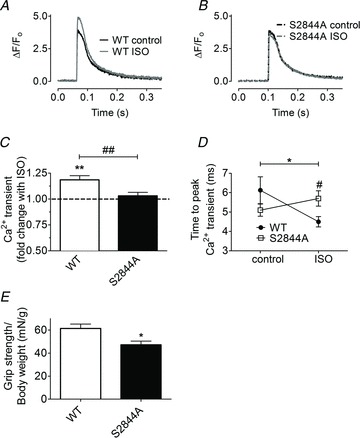
A and B, figures show fluorescence signals from the Ca2+ indicator fluo-4 during twitch stimulation before and after application of the β-receptor agonist isoproterenol (ISO) in wild-type (WT) (A) and S2844A (B) flexor digitorum brevis (FDB) muscle fibres. The fluo-4 signal was normalized to the minimum fluorescence signal before onset of the Ca2+ transient, which was set to zero. C, pooled data showing the fold change of the peak Ca2+ transient after application of ISO. The dashed line indicates no change in the presence of ISO. D, time to peak Ca2+ (the time from the beginning of a Ca2+ transient to its peak) before and after ISO in WT and S2844A FDB muscle. For pooled data (C and D): WT, n= 8 cells from three animals, S2844A, n= 10 cells from three animals. *P < 0.05, **P < 0.01, NS = not significant (paired t test, comparison before and after ISO), #P < 0.05, ##P < 0.01 (unpaired t test WT vs. S2844A). E, forelimb grip strength normalized to body weight in stressed WT and S2844A mice. Mouse forelimbs were allowed to grab on to a metal grid (attached to a force transducer). The maximum grip strength was measured as the mouse was pulled by its tail until it lost the grip. Maximum force was recorded and grip strength was calculated from the average of three repeated measurements in each mouse. WT, n= 8 mice; S2844A, n= 9 mice; *P < 0.05 (unpaired t test).
To test the hypothesis that PKA-dependent RyR1 phosphorylation underlies adrenergic stimulationmediated enhancement of cytoplasmic Ca2+ transients in skeletal muscle, we examined FDB muscle from RyR1-S2844A knock-in mice. There was no change in either fluo-4 peak fluorescence (ISO, 103 ± 4% of the pre-exposure control; n= 10 cells from three animals, P= NS; Fig. 2B and C) or Ca2+ TTP (control, 5.1 ± 0.3 ms; vs. ISO, 5.7 ± 0.4 ms; n= 10 cells from three animals, P= NS; Fig. 2D) following ISO stimulation of RyR1-S2844A skeletal muscle. Similar to WT fibres, resting Ca2+ was not changed by ISO exposure, as indicated by unaltered basal fluo-4 fluorescence (S2844A control, 17.1 ± 2.4 a.u.; S2844A ISO, 18.0 ± 2.4 a.u.; mean ± SEM, n= 10 cells from three animals, P= NS). Taken together, these data indicate that phosphorylation of a single amino acid, S2844, in RyR1 is required for the physiological changes in Ca2+ handling that occur in skeletal muscle following adrenergic stimulation.
To confirm the phosphorylation state of the Ca2+ release channel after ISO treatment, immunoprecipitated RyR1 from EDL muscle homogenates were immunoblotted using an RyR1 PKA phosphorylation site (RyR1-S2844) epitope-specific antibody (Andersson et al. 2011). ISO treatment of WT muscles led to an increase in phosphorylation of the RyR1, whereas no such increase was detected in the RyR1-S2844A skeletal muscles (Fig. 3A). In support of this finding using RyR1 isolated from RyR1-S2844A skeletal muscles we previously reported that heterologously expressed RyR1-S2844A channels cannot be PKA phosphorylated (Reiken et al. 2003). We have previously reported that under conditions of chronic adrenergic drive, such as in heart failure, excessive PKA phosphorylation of the skeletal muscle RyR1 is associated with depletion of the RyR1 stabilizing protein FK506 binding protein 12 (FKBP12 or calstabin1) (Reiken et al. 2003; Ward et al. 2003). We therefore measured binding of calstabin1 to the RyR1 in WT and S2844A muscle following acute ISO treatment. Under these experimental conditions, calstabin1 binding was not noticeably altered in either of the groups (Fig. 3A). To show that the absence of RyR1 PKA phosphorylation was not due to down-regulation of the β-receptor-cAMP-PKA signalling pathway, we measured the phosphorylation level of CREB/p-CREB (Lynch & Ryall, 2008). After ISO treatment, the WT and RyR1-S2844A muscles displayed the same degree of CREB phosphorylation (Fig. 3B) indicating that the β-receptor signalling cascade was intact in the RyR1-S2844A mutant mice.
Figure 3. Abolished phosphorylation of RyR1, but not of CREB, following β-adrenergic stimulation in S2844A muscles.
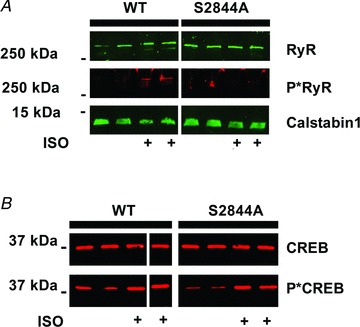
A, immunoblot of immunoprecipitated ryanodine receptor type 1 (RyR1) from wild-type (WT) and S2844A extensor digitorum longus muscles. The muscles treated with isoproterenol (ISO) are indicated with +. Treatment with ISO caused the WT, but not the S2844A, RyR1 to be phosphorylated (P*RyR1). The immunoblot also shows the amount of RyR1 stabilizing protein calstabin1 that was bound to the immunoprecipitated RyR1. Calstabin1 is also known as FKBP12. B, the transcription factor cAMP response element-binding protein (CREB) is a target of protein kinase A-mediated phosphorylation. ISO treatment caused CREB phosphorylation (P*CREB) in both WT and S2844A muscle, indicating that the β-receptor signalling system was intact in S2844A mice.
To determine the effect of adrenergic signalling on skeletal muscle strength in vivo, we measured forelimb grip strength in WT and RyR1-S2844A mice (Bellinger et al. 2009). Compared to WT, grip strength was lower in the RyR1-S2844A mice [WT, 1.7 ± 0.1 N, n= 8; vs. RyR1-S2844A, 1.2 ± 0.1 N, n= 9; mean ± SEM, P < 0.01 (unpaired t test)]. Moreover, the RyR1-S2844A muscle displayed reduced grip strength also when the strength measurement was normalized to body weight, [WT, 62 ± 4 mN g−1, n= 8; RyR1-S2844A, 47 ± 3 mN g−1, n= 9; mean ± SEM, P < 0.01 (unpaired t test); Fig. 2E].
Discussion
During exercise sympathetic nervous system activation results in catecholamine release that enhances skeletal muscle function. Indeed, enhanced twitch and tetanic cytoplasmic [Ca2+] in response to the β2-receptor agonist terbutaline have been described in both fast and slow twitch muscle fibres (Cairns et al. 1993; Ha et al. 1999). However, the molecular basis of the catecholamine-mediated stress-induced increase in skeletal muscle force production has remained unknown.
We have previously shown that chronic stress induces remodelling of the RyR1 macromolecular complex such that its components, including phosphodiesterase PDE4D3 (due to oxidation of the channel) and the Ca2+ release channel stabilizing protein calstabin1 are depleted from the RyR1 leading to PKA hyperphosphorylation of the channel and a diastolic SR Ca2+ leak (Reiken et al. 2003; Ward et al. 2003; Lehnart et al. 2005; Shan et al. 2010a,b). These changes, combined with other complex maladaptations that occur under prolonged pathological stress could contribute to defective muscle function. In this study, however, we investigated the mechanism whereby acute adrenergic stress causes enhanced skeletal muscle function and how this well established physiological process could be explained by PKA phosphorylation of RyR1 at S2844. Our data show that brief exposure of WT muscles to ISO leads to transient RyR1-S2844 PKA phosphorylation, providing a mechanism for physiological stress signalling downstream of the β-adrenergic receptor.
Though numerous components of the excitation–contraction coupling machinery have been excluded as underlying the adrenergic stimulation-mediated increase in skeletal muscle force production including the voltage sensor, L-type Ca2+ channel, SR Ca2+ load or myofilament Ca2+ sensitivity (Cairns & Dulhunty, 1993a,b; Cairns et al. 1993; Liu et al. 1997; Ha et al. 1999), the role of RyR1 in adrenergic stimulation-mediated increase of skeletal muscle force production has not been definitively addressed. Before the present study, the tools have not been available to determine if the mechanism that underlies stress-induced increase in SR Ca2+ release is dependent on RyR1 PKA phosphorylation. To resolve this gap in our understanding we generated a genetic model in which the PKA phosphorylation site in RyR1 was ablated by replacement of S2844 with alanine. Using this new tool, we now show that the stress-induced inotropic response in skeletal muscle is dependent on increased SR Ca2+ release via RyR1, and that PKA phosphorylation of a single amino acid of the RyR1, S2844, is the key signal. Thus, we have identified the β-receptor downstream target responsible for increased SR Ca2+ release and skeletal muscle force during sympathetic stimulation as occurs with exercise. The present findings elucidate a fundamental mechanism regulating muscle function and provide a basis for further exploration of the mechanisms underlying pathological conditions of muscle.
Acknowledgments
This study was financed by NIH grant 1R01AR060037 to A.R.M. D.C.A. was supported by grants from the Swedish Research Council (Vetenskapsrådet) and the Swedish Heart Lung Foundation (Hjärt-lungfonden). A.U. was supported by a fellowship (AHA 11PRE7810019) from the American Heart Association. A.R.M. is a consultant for a start-up company, ARMGO Pharma Inc., which is targeting RyR1 to improve exercise capacity in muscle diseases.
Glossary
- EC coupling
excitation–contraction coupling
- EDL
extensor digitorum longus (muscle)
- FDB
flexor digitorum brevis
- ISO
isoproterenol
- PKA
protein kinase A
- RyR1
ryanodine receptor type 1
- SR
sarcoplasmic reticulum
- WT
wild-type
Author contributions
D.C.A. and A.R.M. contributed to the conception and design of the experiments, interpretation of the data and writing of the manuscript. D.C.A., M.J.B., S.R., A.U. and T.S. performed experiments and analysed the data. All authors approved the final version of the manuscript.
Author's present address
D. C. Andersson: Department of Medicine, Karolinska Institutet, Stockholm, Sweden.
Supplementary material
Supplementry Figure S1
{kind=link}
Supplementry Figure S2
{kind=link}
References
- Allen DG, Lamb GD, Westerblad H. Skeletal muscle fatigue: cellular mechanisms. Physiol Rev. 2008;88:287–332. doi: 10.1152/physrev.00015.2007. [DOI] [PubMed] [Google Scholar]
- Andersson DC, Betzenhauser MJ, Reiken S, Meli AC, Umanskaya A, Xie W, Shiomi T, Zalk R, Lacampagne A, Marks AR. Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging. Cell Metab. 2011;14:196–207. doi: 10.1016/j.cmet.2011.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aydin J, Andersson DC, Hanninen SL, Wredenberg A, Tavi P, Park CB, Larsson NG, Bruton JD, Westerblad H. Increased mitochondrial Ca2+ and decreased sarcoplasmic reticulum Ca2+ in mitochondrial myopathy. Hum Mol Genet. 2009;18:278–288. doi: 10.1093/hmg/ddn355. [DOI] [PubMed] [Google Scholar]
- Bellinger AM, Reiken S, Carlson C, Mongillo M, Liu X, Rothman L, Matecki S, Lacampagne A, Marks AR. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat Med. 2009;15:325–330. doi: 10.1038/nm.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. Kluwer Academic Publishers; 2001. [Google Scholar]
- Brown GL, Bülbring E, Burns BD. The action of adrenaline on mammalian skeletal muscle. J Physiol. 1948;107:115–128. doi: 10.1113/jphysiol.1948.sp004255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns SP, Dulhunty AF. Beta-adrenergic potentiation of E-C coupling increases force in rat skeletal muscle. Muscle Nerve. 1993a;16:1317–1325. doi: 10.1002/mus.880161208. [DOI] [PubMed] [Google Scholar]
- Cairns SP, Dulhunty AF. The effects of beta-adrenoceptor activation on contraction in isolated fast- and slow-twitch skeletal muscle fibres of the rat. Br J Pharmacol. 1993b;110:1133–1141. doi: 10.1111/j.1476-5381.1993.tb13932.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns SP, Westerblad H, Allen DG. Changes of tension and [Ca2+]i during beta-adrenoceptor activation of single, intact fibres from mouse skeletal muscle. Pflugers Arch. 1993;425:150–155. doi: 10.1007/BF00374515. [DOI] [PubMed] [Google Scholar]
- Ha TN, Posterino GS, Fryer MW. Effects of terbutaline on force and intracellular calcium in slow-twitch skeletal muscle fibres of the rat. Br J Pharmacol. 1999;126:1717–1724. doi: 10.1038/sj.bjp.0702482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnart SE, Wehrens XH, Reiken S, Warrier S, Belevych AE, Harvey RD, Richter W, Jin SL, Conti M, Marks AR. Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell. 2005;123:25–35. doi: 10.1016/j.cell.2005.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Kranias EG, Schneider MF. Regulation of Ca2+ handling by phosphorylation status in mouse fast- and slow-twitch skeletal muscle fibers. Am J Physiol Cell Physiol. 1997;273:C1915–C1924. doi: 10.1152/ajpcell.1997.273.6.C1915. [DOI] [PubMed] [Google Scholar]
- Lynch GS, Ryall JG. Role of beta-adrenoceptor signaling in skeletal muscle: implications for muscle wasting and disease. Physiol Rev. 2008;88:729–767. doi: 10.1152/physrev.00028.2007. [DOI] [PubMed] [Google Scholar]
- Reiken S, Lacampagne A, Zhou H, Kherani A, Lehnart SE, Ward C, Huang F, Gaburjakova M, Gaburjakova J, Rosemblit N, Warren MS, He KL, Yi GH, Wang J, Burkhoff D, Vassort G, Marks AR. PKA phosphorylation activates the calcium release channel (ryanodine receptor) in skeletal muscle: defective regulation in heart failure. J Cell Biol. 2003;160:919–928. doi: 10.1083/jcb.200211012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockman HA, Koch WJ, Lefkowitz RJ. Seven-transmembrane-spanning receptors and heart function. Nature. 2002;415:206–212. doi: 10.1038/415206a. [DOI] [PubMed] [Google Scholar]
- Shan J, Betzenhauser MJ, Kushnir A, Reiken S, Meli AC, Wronska A, Dura M, Chen BX, Marks AR. Role of chronic ryanodine receptor phosphorylation in heart failure and beta-adrenergic receptor blockade in mice. J Clin Invest. 2010a doi: 10.1172/JCI37649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan J, Kushnir A, Betzenhauser MJ, Reiken S, Li J, Lehnart SE, Lindegger N, Mongillo M, Mohler PJ, Marks AR. Phosphorylation of the ryanodine receptor mediates the cardiac fight or flight response in mice. J Clin Invest. 2010b;120:4388–4398. doi: 10.1172/JCI32726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suko J, Maurer-Fogy I, Plank B, Bertel O, Wyskovsky W, Hohenegger M, Hellmann G. Phosphorylation of serine 2843 in ryanodine receptor-calcium release channel of skeletal muscle by cAMP-, cGMP- and CaM-dependent protein kinase. Biochim Biophys Acta. 1993;1175:193–206. doi: 10.1016/0167-4889(93)90023-i. [DOI] [PubMed] [Google Scholar]
- Ward CW, Reiken S, Marks AR, Marty I, Vassort G, Lacampagne A. Defects in ryanodine receptor calcium release in skeletal muscle from post-myocardial infarct rats. FASEB J. 2003;17:1517–1519. doi: 10.1096/fj.02-1083fje. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.