

Abstract
We previously reported that statin myopathy is associated with impaired carbohydrate (CHO) oxidation in fast-twitch rodent skeletal muscle, which we hypothesised occurred as a result of forkhead box protein O1 (FOXO1) mediated upregulation of pyruvate dehydrogenase kinase-4 (PDK4) gene transcription. Upregulation of FOXO gene targets known to regulate proteasomal and lysosomal muscle protein breakdown was also evident. We hypothesised that increasing CHO oxidation in vivo, using the pyruvate dehydrogenase complex (PDC) activator, dichloroacetate (DCA), would blunt activation of FOXO gene targets and reduce statin myopathy. Female Wistar Hanover rats were dosed daily for 12 days (oral gavage) with either vehicle (control, 0.5% w/v hydroxypropyl-methylcellulose 0.1% w/v polysorbate-80; n = 9), 88 mg kg−1 day−1 simvastatin (n = 8), 88 mg kg−1 day−1 simvastatin + 30 mg kg−1 day−1 DCA (n = 9) or 88 mg kg−1 day−1 simvastatin + 40 mg kg−1 day−1 DCA (n = 9). Compared with control, simvastatin reduced body mass gain and food intake, increased muscle fibre necrosis, plasma creatine kinase levels, muscle PDK4, muscle atrophy F-box (MAFbx) and cathepsin-L mRNA expression, increased PDK4 protein expression, and proteasome and cathepsin-L activity, and reduced muscle PDC activity. Simvastatin with DCA maintained body mass gain and food intake, abrogated the myopathy, decreased muscle PDK4 mRNA and protein, MAFbx and cathepsin-L mRNA, increased activity of PDC and reduced proteasome activity compared with simvastatin. PDC activation abolished statin myopathy in rodent skeletal muscle, which occurred at least in part via inhibition of FOXO-mediated transcription of genes regulating muscle CHO utilisation and protein breakdown.
Key points
Statin myopathy impairs phosphatidylinositol 3-kinase/Akt signalling and activates forkhead box protein O (FOXO) transcription factors in vivo in rodent skeletal muscle. This is associated with upregulation of downstream gene targets known to increase proteasomal and lysosomal-mediated protein breakdown, oxidative stress and inflammation, and inhibit muscle carbohydrate (CHO) oxidation.
We hypothesised that forcibly increasing muscle CHO oxidation in vivo, using the pyruvate dehydrogenase complex activator, dichloroacetate (DCA), would blunt statin-mediated increases in mRNA expression of these FOXO gene targets, thereby reducing statin myopathy.
Chronic administration of DCA with simvastatin dampened statin-mediated increases in muscle atrophy F-box (MAFbx), cathepsin-L and pyruvate dehydrogenase kinase-4 mRNA in a dose-dependent manner, which was corroborated by protein activity and expression measurements, and blunted statin myopathy.
These results provide convincing evidence that pharmacologically increasing muscle CHO oxidation reduces simvastatin-induced myopathy by dampening the upregulation of genes known to increase proteasomal and lysosomal protein breakdown and inhibit CHO oxidation.
Introduction
Statins inhibit synthesis of mevalonate by inhibiting 3-hydroxy-3-methyl-glutaryl coenzyme A reductase, the rate-limiting step in hepatic cholesterol and isoprenoid biosynthesis. Statins are used clinically for cholesterol reduction in hypercholesterolaemia, and their therapy has been associated with a 30% reduction in cardiovascular events in patients with vascular disease (Vaughan & Gotto, 2004). Statins have also been found to be protective against heart failure in patients where cholesterol levels are not elevated (Ridker et al. 2008). Furthermore, because of reported pleiotropic effects, statin use has expanded to the treatment of other conditions, including ventricular arrhythmias, idiopathic dilated cardiomyopathy, cancer, osteoporosis and diabetes (Dirks & Jones, 2006).
Statins are generally well tolerated, but can have severe myopathic effects, albeit relatively infrequently (Thompson et al. 2003). Statin-related myopathy has varying degrees of severity, ranging from muscle myositis and myalgia (muscle aches or weaknesses with and without increased serum creatine kinase (CK) concentration, respectively) and, in the severest case, rhabdomyolysis (>10 times the upper limit of normal serum CK). Randomised trials suggest rhabdomyolysis is a rare event, and even myalgia and myositis, although more common, are not seen as being highly prevalent (Thompson et al. 2003). Nevertheless, in general practices the incidence of adverse muscular events associated with the most commonly prescribed statin, simvastatin, is high, particularly at higher doses (>40 mg; 18%; Bruckert et al. 2005). Furthermore, recent meta-data analyses have associated intensive high-dose statin therapy with increased risk of new-onset diabetes compared with moderate-dose therapy (Sattar et al. 2010; Preiss et al. 2011). Given the recommendation by the National Institute of Clinical Excellence (NICE) to prescribe simvastatin over other statins (NICE, 2006), and to prescribe high-dose statin therapy (80 mg day−1) to older patients (Deedwania et al. 2007) when in fact older age per se has been reported to be a major myopathy risk factor (Nichols & Koro, 2007), adverse muscular events could potentially further increase with increased use. Current treatment for statin myopathy is the discontinuation of statin use, in which symptoms are most often reversible (Dirks & Jones, 2006), and therefore it is important to identify the mechanism of statin-induced myopathy.
Using an animal model of statin-induced myopathy (Westwood et al. 2005), we have previously shown simvastatin administration impairs phosphatidylinositol 3-kinase (PI3k)/Akt signalling in muscle, resulting in the dephosphorylation (and activation of) the forkhead box protein O (FOXO) (1 and 3) transcription factors, and the induction of ubiquitin and lysosomal proteolysis through upregulation of the FOXO downstream target genes muscle atrophy F-box (MAFbx), muscle RING finger-1 (MuRF-1) and cathepsin-L mRNA. Furthermore, these changes preceded a statin-induced decline in muscle mass reported as the protein:DNA ratio (Mallinson et al. 2009). Carbohydrate (CHO) oxidation was also impaired in simvastatin-treated animals, reflected by a decline in muscle glycogen utilisation and a marked increase in the FOXO downstream target pyruvate dehydrogenase kinase-4 (PDK4) mRNA expression above control (Mallinson et al. 2009). Combined muscle protein loss and impairment of muscle glycogen oxidation, in parallel with blunted Akt signalling and activated FOXO and its target genes, have also been reported with fibrate therapy (Motojima & Seto, 2003; Constantin et al. 2007), in an in vivo model of endotoxaemia (Crossland et al. 2008) and in critical care patients (Constantin et al. 2011). Additionally, exposure of human rhabdomyosarcoma cells to statins has been shown to increase protein degradation by induction of autophagy (Araki & Motojima, 2008). A hallmark of muscle autophagy during in vivo catabolic conditions, which include starvation (Lecker et al. 2004; Mammucari et al. 2007), denervation (Mammucari et al. 2007), cancer cachexia and diabetes (Lecker et al. 2004), is upregulation of the microtubule-associated protein 1, light chain 3 (LC3) gene. However, to date, no study has examined the role of autophagy in an in vivo model of statin-induced myopathy.
Upregulation of PDK2 and PDK4, the main isoforms identified in skeletal muscle (Bowker-Kinley et al. 1998), phosphorylate and inactivate muscle pyruvate dehydrogenase complex (PDC; Wieland, 1983), thereby limiting the rate of CHO oxidation. PDC is a multi-enzyme complex located on the inner mitochondrial membrane that catalyses the irreversible conversion of pyruvate to acetyl-CoA (Denton et al. 1972). Dichloroacetate (DCA) is a halogenated carboxylic acid that has been shown to increase the activity of the PDC (Whitehouse & Randle, 1973; Whitehouse et al. 1974) by competitively inhibiting PDK2 and PDK4 (Bowker-Kinley et al. 1998). Several studies have demonstrated DCA increases CHO oxidation and reduces muscle lactate accumulation, through increased PDC activation, in rat (Clark et al. 1987), canine (Timmons et al. 1996, 1998c; Grassi et al. 2002) and human skeletal muscle (Timmons et al. 1998a; Constantin-Teodosiu et al. 1999, 2012). In keeping with this, DCA has also been shown to decrease blood glucose concentration in healthy humans (Wells et al. 1980; Jahoor et al. 1989) and patients with diabetes (Stacpoole et al. 1978), probably due to increased muscle and liver oxidative glucose disposal (Shangraw et al. 1989; Constantin-Teodosiu et al. 1999).
Interestingly, DCA administration has also been linked to the regulation of muscle protein turnover. Goodman et al. reported DCA attenuated muscle branched-chain amino acid (BCAA) accumulation in the perfused rat hind limb in the fed state, which may reflect a diminished net muscle proteolysis. Furthermore, DCA administration has been used to increase muscle glutamine concentrations in an attempt to increase muscle protein synthesis in burn injury (Ferrando et al. 1998), sepsis (Vary et al. 1988) and diabetes (Goodman et al. 1978), although the acute elevation did not affect muscle protein metabolism.
In the present study we aimed to test the hypothesis that forcibly increasing muscle CHO oxidation by increasing PDC activity and flux, using DCA, would suppress statin-mediated upregulation of FOXO target genes, thereby blunting muscle necrosis and the rise in plasma CK levels in an in vivo rodent model of statin myopathy. To do this we measured muscle PDC activity and flux through the PDC reaction, mRNA and protein expression levels of muscle PDK4, mRNA expression levels of the other FOXO gene targets MAFbx and cathepsin-L, and also muscle proteasome and cathepsin-L activity, and related these events to functional end-points, namely body mass and food intake, and the incidence of muscle necrosis and plasma CK concentration. Using this approach we hoped to dampen the myopathic effects associated with simvastatin, thereby providing novel and important insight into a pharmacological approach directed towards the treatment of statin-induced myopathy.
Methods
Ethical approval
This study was planned in accordance with the standards of animal care and ethics described in ‘Guidance on the Operations of the Animal (Scientific Procedures) Act 1986’ issued by the UK Home Office, and was conducted so that any clinical expression of toxicity remained within a moderate severity limit as described in guidelines agreed with the UK Home Office Inspector.
Animals and treatments
Simvastatin lactone (purity 98.4%) was obtained from Wuhan S&M Biochemie (Wuhan Hubei, China). The statin was formulated for dosing as a suspension in water containing 0.5% w/v hydroxypropyl methylcellulose and 0.1% w/v polysorbate 80. Sodium DCA was obtained from Sigma-Aldrich (St Louis, MO, USA). Female Wistar Hanover rats, substrain Crl:WI (Glx/BRL/Han) BR (Charles River, UK), aged 8 weeks, were multiple-housed appropriate to each study and were acclimatised for 6 days. The animal rooms were illuminated in a 12 h light/dark cycle, and temperature and humidity were controlled (limits 21 ± 2°C and 55 ± 15% relative humidity). Pelleted RM1(E) SQC rodent diet and drinking water were freely available. Animals were dosed with 88 mg kg−1 body weight per day (mg kg−1 day−1) of simvastatin for 12 days (simvastatin, n = 8), 88 mg kg−1 day−1 simvastatin and 30 mg kg−1 day−1 DCA for 12 days (simvastatin + DCA 30 mg, n = 9), 88 mg kg−1 day−1 simvastatin and 40 mg kg−1 day−1 DCA for 12 days (simvastatin + DCA 40 mg, n = 9) or vehicle (0.5% w/v hydroxypropyl methylcellulose and 0.1% w/v polysorbate 80) for 12 days (control, n = 9) by oral gavage. The animals in the simvastatin + DCA groups were primed with 50 mg kg−1 day−1 DCA for 5 days prior to the administration of simvastatin, and DCA was given for a further 24 h after the simvastatin administration ceased at 12 days to ensure PDC was fully activated (Fig. 1).
Figure 1. Protocol of simvastatin and dichloroacetate (DCA) dosing during the study.
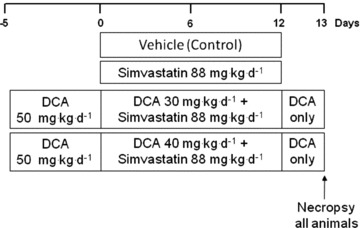
Vehicle (0.5% w/v hydroxypropyl methylcellulose and 0.1% w/v polysorbate 80, control) and simvastatin (88 mg kg−1 day−1) were given daily for 12 days. In the DCA groups, animals were primed with DCA (50 mg kg−1 day−1) for 5 days before dosing with either DCA 30 mg kg−1 day−1 + simvastatin 88 mg kg−1 day−1 or DCA 40 mg kg−1 day−1 + simvastatin 88 mg kg−1 day−1 for 12 days. DCA dosing continued for a further 24 h after the simvastatin administration ceased at 12 days. All animals were necropsied on day 13.
Statin administration is known to predominantly affect type IIB fibres (Westwood et al. 2005, 2008), and therefore biceps femoris muscle, composed of approximately 70% type IIB fibres (Armstrong & Phelps, 1984), was investigated in this study. Muscles were harvested under terminal anaesthesia with halothane, immediately snap-frozen and stored in liquid nitrogen.
Quantification of muscle necrosis
Muscle necrosis was quantified in biceps femoris sections using the method previously described (Westwood et al. 2005). The magnitude of necrosis was graded as follows: minimal, up to 10 muscle fibres affected in whole section; mild, up to 20% of fibres in section affected; moderate, up to 50% of fibres in section affected; and severe, more than 50% of fibres in section affected (Westwood et al. 2005). Detailed histological images depicting muscle necrosis in the model used in the present study were presented in Westwood et al. (2005). Plasma CK concentration (IU l−1) was also determined using a spectrophotometric assay (Szasz et al. 1976).
Muscle cathepsin-L, MAFbx, LC3 and PDK4 mRNA expression
RNA was extracted from approximately 30 mg snap-frozen tissue using RNA Plus (Qbiogene, Uxbridge, UK). First strand cDNA was synthesised from 1 μg total RNA, using Powerscript reverse transcriptase (BD Biosciences, Oxford, UK) and random primers (Promega, Southampton, UK). A reaction was also carried out with the reverse transcriptase omitted to assess genomic DNA contamination.
All reactions were performed in the ABI Prism 7000 Sequence Detection System (Applied Biosystems, Foster City, CA, USA). Each well contained 2 μl cDNA, 12.5 μl Universal Taqman 2× PCR master mix (Eurogentec, Liege, Belgium), 18 μm primer, 5 μm probe and 9.25 μl RNase-free water to make a volume of 25 μl. Each sample was measured in duplicate. The housekeeping gene used was hydroxymethylbilane synthase (HMBS), which was selected because it was not affected by the statin treatment (data not shown). The thermal cycling conditions used were 2 min at 50°C, 10 min at 95°C, followed by 40 cycles at 95°C for 15 s and 60°C for 1 min. Genes analysed were cathepsin-L (Rn01442127), MAFbx (Rn00591730), LC3 (Rn00563181) and PDK4 (Rn00585577; Applied Biosystems). Relative quantification of the genes was determined from linear interpolation of standard curves constructed using pooled cDNA synthesised from extracted RNA from control and treated tissue. The relative values for the gene of interest were normalised with the relative values of HMBS (Rn00565886). The control group was used as calibrator with a value of 1.
Muscle PDK4 protein expression
Protein was extracted from approximately 30 mg snap-frozen tissue using a method modified from Blough et al. (1999). Samples were homogenised in 50 mm Tris buffer pH 7.5 with the addition of protease and phosphatase inhibitors (Sigma-Aldrich, UK), centrifuged and the supernatant containing the cytosolic protein fraction was collected. Protein content was measured using the Bradford assay. Protein samples were run on a 4–12% Bis-tris acrylamide gel (Invitrogen, Paisley, UK) at 200 V for 1 h and transferred onto a polyvinylidene difluoride membrane for 1 h 45 min. Protein transfer was checked with Ponceau S staining before being blocked in 5% BSA-TBS-Tween. Membranes were probed with PDK4 primary antibody (1:500; Abcam, Cambridge, UK) overnight at 4°C. Membranes were washed with TBS-Tween and incubated with the secondary antibody HRP-linked anti-rabbit IgG (GE Healthcare, Amersham, UK; 1:2000) in 1% BSA-TBS-Tween. Membranes were incubated with ECL chemiluminescence detection reagent (Pierce, Cramlington, UK) and exposed to X-ray film. Blots were scanned using a Duoscan T1200, Agfa, Mortsel, Belgium. Bands were identified with Genetools from Syngene (Cambridge, UK), and the density volume was adjusted by subtracting the local background and then normalised with actin (1:1000; Sigma-Aldrich, UK).
Muscle PDC activity
A small portion of frozen ‘wet’ muscle was used to determine PDC activity as previously described (Constantin-Teodosiu et al. 1991). Briefly, the activity of PDC in its dephosphorylated active form (PDCa) was assayed in a buffer containing NaF and DCA, and was expressed as a rate of acetyl-CoA formation (mmol min−1 kg−1 wet muscle) at 37°C.
Muscle acetylcarnitine content
After removal of visible blood and connective tissue, freeze-dried muscle samples were powdered and extracted with 0.5 mol l−1 perchloric acid (containing 1 mmol l−1 EDTA), and neutralised with 2.2 mmol l−1 KHCO3. Acetylcarnitine was measured as an index of PDC flux by a radioisotope enzymatic assay as previously described (Cederblad et al. 1990), as an indicator of the magnitude of flux through the PDC reaction (Constantin-Teodosiu et al. 1993).
Muscle 20S proteasome and cathepsin-L activity
Frozen muscle (15–25 mg) was homogenised, and the soluble muscle extract used to measure the chymotrypsin-like activity of the 20S proteasome using the fluorogenic substrate N-Suc-Leu-Leu-Val-Tyr-7-amido-4-methyl-coumarin (Sigma-Aldrich), according to Dawson et al. (1995). The same homogenate was used to determine cathepsin-L activity using the substrate Z-Phe-Arg-7-amido-4-methyl-coumarin (Sigma-Aldrich), according to Bergmeyer (1983).
Statistics
Data are expressed as mean ± SEM. Treatment effects were investigated using one-way ANOVA. When a significant F-ratio was found, a least significance difference post hoc test was performed to locate specific differences. A significance level of P < 0.05 was used.
Results
DCA prevents the reduced food intake and body mass loss associated with high-dose simvastatin administration
Average daily food intake declined from day 5 in the simvastatin and simvastatin + DCA 30 mg groups (P < 0.05 compared with control; Fig. 2A). By the end of the study, food consumption was similar in the control and simvastatin + DCA 30 mg groups, but approximately 25% lower in the simvastatin group (P < 0.001, compared with control). There was a decline in the average body mass of both the simvastatin and simvastatin + DCA 30 mg groups from day 7 (P < 0.05 compared with control; Fig. 2B). In both control and simvastatin + DCA 40 mg groups body mass had increased by 10% at the end of the study (P < 0.001, P < 0.05, respectively, compared with simvastatin).
Figure 2. Daily food intake (A, g) and body mass (B, g) of control, simvastatin- and simvastatin plus dichloroacetate (DCA)-treated animals.
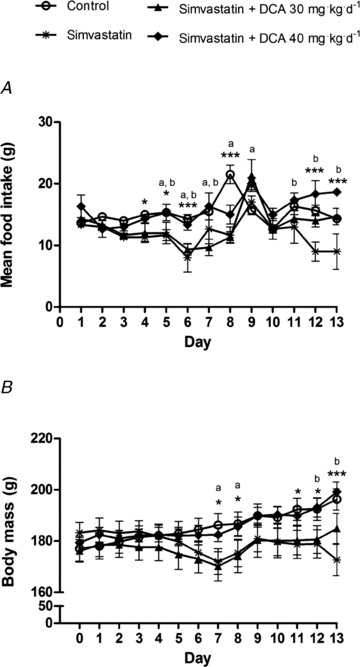
Values expressed as mean ± SEM. *P < 0.05, ***P < 0.001 when control compared with simvastatin, aP < 0.05 when control compared with simvastatin + DCA 30 mg, bP < 0.05 when simvastatin compared with simvastatin + DCA 40 mg.
DCA administration protects against simvastatin-induced myopathy
Simvastatin increased mean plasma CK levels 95-fold above control (P < 0.05; Fig. 3). There was a trend (P = 0.10) for plasma CK to be reduced in simvastatin + DCA 30 mg, whilst the administration of simvastatin + DCA 40 mg completely abolished this increase in plasma CK with simvastatin (P < 0.05 vs. simvastatin). In line with these observations, histological analysis revealed evidence of muscle damage (necrosis) in simvastatin compared with control (Table 1). The severity grade and frequency of muscle necrosis was unchanged from simvastatin to simvastatin + DCA 30 mg. However, the severity of muscle necrosis was similar to control in simvastatin + DCA 40 mg.
Figure 3. Plasma creatine kinase (CK) concentration in control, simvastatin- and simvastatin plus dichloroacetate (DCA)- treated animals.
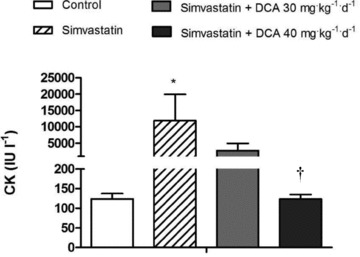
Values expressed as mean ± SEM. *P < 0.05 when compared with control, and †P < 0.05 when compared with simvastatin.
Table 1.
Incidence and severity of muscle fibre necrosis for control, simvastatin- and simvastatin plus DCA-treated animals
| Animal number | CT | Sol | BF | Gas | |
|---|---|---|---|---|---|
| Group 1 | 1 | ||||
| Vehicle (control) | 2 | * | |||
| 3 | * | ||||
| 4 | |||||
| 5 | |||||
| 6 | * | * | |||
| 7 | |||||
| 8 | |||||
| 9 | * | ||||
| Group 2 | 10 | *** | ** | *** | |
| Simvastatin, 88 mg kg−1 day−1 | 11 | ** | ** | *** | |
| 12 | ** | ** | ** | ||
| 13 | * | * | ** | ||
| 14 | |||||
| 15 | * | ||||
| 16 | |||||
| 17 | |||||
| 18 | |||||
| Group 3 | 19 | ** | — | ** | ** |
| Simvastatin + sodium DCA, | 20 | * | ** | ** | |
| 88/30 mg kg−1 day−1 | 21 | * | * | * | |
| 22 | ** | ||||
| 23 | * | * | |||
| 24 | * | ||||
| 25 | * | ||||
| 26 | |||||
| 27 | |||||
| Group 4 | 28 | ||||
| Simvastatin + sodium DCA, | 29 | * | |||
| 88/40 mg kg−1 day−1 | 30 | ||||
| 31 | * | ||||
| 32 | * | ||||
| 33 | * | ||||
| 34 | * | ||||
| 35 | * | ||||
| 36 | * |
BF, biceps femoris; CT, cranial tibial; DCA, dichloroacetate; Gas, gastrocnemius; Sol, soleus. —: muscle not sampled; no symbol: muscle necrosis not present. *Minimal necrosis: up to 10 muscle fibres affected in whole section; **mild necrosis: up to 20% of fibres in section affected; ***moderate necrosis: up to 50% of fibres in section affected.
DCA reverses the simvastatin-induced increase in muscle PDK4 mRNA and protein expression, and inhibition of muscle PDC activity and acetylcarnitine accumulation
Muscle PDK4 mRNA expression in the simvastatin-treated group was elevated threefold above control (P < 0.001; Fig. 4A). In keeping with this, PDK4 protein expression was also increased 0.5-fold above control (P < 0.05; Fig. 4B). Compared with simvastatin, mRNA and protein expression levels were dampened in simvastatin + DCA 30 mg (P < 0.001, P < 0.05, respectively) and simvastatin + DCA 40 mg (P < 0.001, P < 0.05, respectively), and were no different from expression levels seen in control. Muscle PDC activity was reduced in simvastatin compared with control (P < 0.05; Fig. 4C). However, muscle PDC activity was unchanged from control in simvastatin + DCA 30 mg and simvastatin + DCA 40 mg. There was a trend for muscle acetylcarnitine content to decrease in simvastatin compared with control (P = 0.10; Fig. 4D). Muscle acetylcarnitine content increased above control and simvastatin alone with simvastatin + DCA 30 mg (P < 0.05, P < 0.05, respectively) and simvastatin + DCA 40 mg (P < 0.01, P < 0.01, respectively; Fig. 4D), demonstrating PDC flux was increased.
Figure 4. Muscle pyruvate dehydrogenase kinase-4 (PDK4) mRNA and protein expression, pyruvate dehydrogenase complex (PDC) activity and acetylcarnitine content after simvastatin and dichloroacetate (DCA) administration.
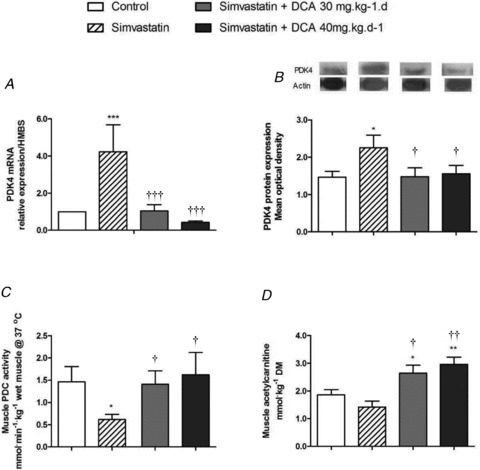
A, PDK4 mRNA expression, values expressed as mean fold changes relative to control. B, PDK4 protein expression, values normalised to actin and expressed as mean ± SEM. C, muscle PDC activity, values expressed as mean ± SEM. D, muscle acetylcarnitine content, values expressed as mean ± SEM. *P < 0.05 and **P < 0.01 when compared with control, †P < 0.05 and ††P < 0.01 when compared with simvastatin.
The upregulation of transcriptional markers of ubiquitin proteasome, lysosomal and autophagy-mediated protein breakdown with simvastatin is attenuated with DCA administration
MAFbx mRNA expression was sixfold greater in simvastatin compared with control (P < 0.001; Fig. 5A). Simvastatin + DCA 30 mg administration resulted in a fourfold decrease in MAFbx mRNA expression compared with simvastatin (P < 0.05), and simvastatin + DCA 40 mg returned MAFbx mRNA levels to that of the control (P < 0.001, compared with simvastatin). Muscle proteasome activity was increased above control with simvastatin administration (P < 0.05; Fig. 6A). There was a strong trend (P = 0.08) for simvastatin + DCA 40 mg to reduce proteasome activity levels to that of the control. Simvastatin alone increased cathepsin-L mRNA expression fivefold above control (P < 0.001; Fig. 5B), and simvastatin + DCA 40 mg reduced expression to that seen in control (P < 0.001). In keeping with this, cathepsin-L activity was increased in simvastatin (P < 0.05; Fig. 6B), and both simvastatin + 30 mg DCA and simvastatin + 40 mg DCA brought cathepsin-L activity levels to that of control. LC3 mRNA expression was upregulated sixfold above control in simvastatin (P < 0.001; Fig. 5C), and both simvastatin + DCA 30 mg (P < 0.001) and simvastatin + DCA 40 mg (P < 0.001) significantly decreased LC3 mRNA expression when compared with simvastatin alone.
Figure 5. mRNA expression of markers of proteasome-, lysosomal- and autophagy-mediated proteolysis (fold changes relative to control) in biceps femoris muscle of animals treated with simvastatin or simvastatin plus dichloroacetate (DCA).
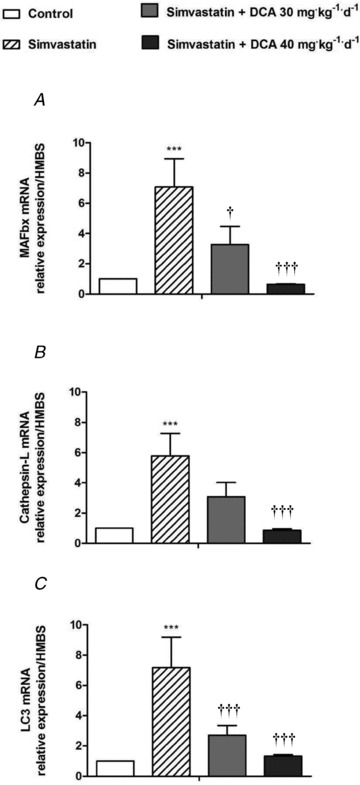
A, muscle atrophy F-box (MAFbx) expression. B, cathepsin-L expression. C, LC3 expression. Values expressed as mean fold changes relative to control ± SEM. ***P < 0.001 when compared with control (which is set at 1), †P < 0.05 and †††P < 0.001 when compared with simvastatin.
Figure 6. Proteasome (A) and cathepsin-L (B) activity in biceps femoris muscle of animals treated with simvastatin or simvastatin plus dichloroacetate (DCA).
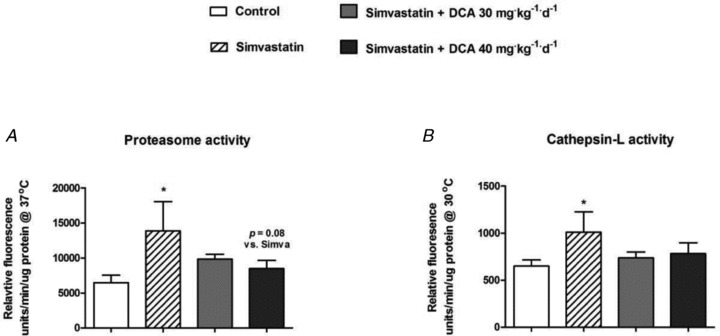
Values expressed as mean ± SEM. *P < 0.05 compared with control.
Discussion
The present study provides novel and impactful insight into the inhibition of statin-mediated myopathy by demonstrating that forcibly increasing muscle CHO oxidation with DCA can prevent the myopathic effects seen with simvastatin in an animal model of statin-induced myopathy. Specifically, DCA reversed the statin-mediated inhibition of muscle PDC activity and flux, abolished statin-mediated upregulation of FOXO target genes MAFbx and cathepsin-L, decreased PDK4 mRNA and protein expression, and normalised statin-mediated increases in muscle cathepsin-L and proteasome activity, which collectively was associated with a blunting of muscle necrosis and myopathy and the maintenance of body mass and food intake. To our knowledge, this is the first study to date to identify such a promising pharmacological approach in the prevention of statin-induced myopathy.
Our previous work (Mallinson et al. 2009) showed one of the main effects of simvastatin on muscle metabolism is the inhibition of CHO oxidation, specifically by upregulation of PDK4 mRNA, via blunted Akt signalling and activation of FOXO transcription factors. Indeed, a similar switch in muscle fuel metabolism is prominent in other animal models of muscle atrophy, namely sepsis (Crossland et al. 2008), peroxisome proliferator-activated receptor delta agonism (Constantin et al. 2007; Constantin-Teodosiu et al. 2009) and in critically ill patients (Constantin et al. 2011), where in all scenarios PDK4 mRNA and protein expression are upregulated and PDC activity decreased. In the present study, as hypothesized, simvastatin administration increased muscle PDK4 mRNA and protein expression (Fig. 4A and B), and inhibited muscle PDC activity (Fig. 4C) and flux (Fig. 4D). In keeping with this, work by Motojima and Seto showed increased PDK4 mRNA expression in muscle of statin-treated rodents (Motojima & Seto, 2003). However, the present study is the first to our knowledge to demonstrate that simvastatin-mediated upregulation of PDK4 protein inhibits PDC activity and flux, clearly identifying the metabolic impact of statin-mediated PDK4 upregulation. Importantly, we also showed simultaneous treatment with DCA and simvastatin completely reversed simvastatin-mediated upregulation of PDK4 mRNA and protein expression (Fig. 4A and B), and inhibition of muscle PDC activity (Fig. 4C) and flux (Fig. 4D) to that seen in control. We feel that the effect of DCA on PDK4 mRNA and protein expression was unlikely to be an immediate or direct effect, but rather occurred secondary to chronically increased muscle glucose uptake and CHO oxidation, Akt phosphorylation and downstream inhibition of FOXO transcription factors. Indeed, as outlined above, DCA is known to decrease blood glucose concentration (Stacpoole et al. 1997), probably by increasing muscle glucose uptake and CHO oxidation in vivo. Furthermore, PDK4 expression is known to be controlled through activation of FOXO transcription factors (Furuyama et al. 2003), which in turn are activated via decreased phosphorylation of Akt (Zhao et al. 2007). In keeping with the acute effect of DCA on PDC activation and flux (rather than PDK4 mRNA and protein expression levels), studies have also shown DCA can bind to and inhibit PDK2 and PDK4 activity in vitro (Li et al. 2009), and inhibit PDK activity in mitochondrial extracts of rat skeletal muscle (Vary & Hazen, 1999).
In addition to having myopathic effects, statins are known to cause muscle weakness and premature fatigue in patients (Phillips et al. 2002), and based on the findings of the present study, it is plausible that statin-mediated inhibition of PDC and CHO oxidation may contribute to these side-effects. Indeed, inhibition of muscle PDC has been shown to impair muscle CHO oxidation and contractile function (Constantin-Teodosiu et al. 2009). Furthermore, PDK inhibition in instances of statin-mediated muscle weakness and premature fatigue may prove to be a viable strategy in reducing these symptoms. It is also interesting to note there is increasing evidence of a positive association between high-dose statin administration and new-onset type II diabetes (Sattar et al. 2010; Preiss et al. 2011), which could be, at least partly, explained by statin-mediated upregulation of muscle PDK4 and PDC inhibition; both of which have been advocated as being causative factors in skeletal muscle insulin resistance (Wu et al. 1999). Pertinent to the latter contention, work from our laboratory has recently shown that pharmacological activation of PDC through DCA administration can abrogate the high-fat diet-mediated inhibition of CHO oxidation during exercise in humans (Constantin-Teodosiu et al. 2012).
Another key event possibly involved in the development of statin-induced myopathy is the upregulation of proteasomal and lysosomal proteolysis, specifically by decreased Akt/FOXO signaling, and increased transcription of MAFbx, MuRF-1 and cathepsin-L, which occurs before evidence of extensive muscle damage becomes apparent (Mallinson et al. 2009). We therefore aimed to determine whether forcibly increasing muscle CHO oxidation using DCA would suppress statin-mediated upregulation of FOXO downstream target genes, thereby blunting muscle proteolysis and simvastatin-induced muscle myopathy. In keeping with this, the highest dose of DCA administered (40 mg kg−1 day−1) blunted the development of muscle myopathy, and blocked the reduction in food intake and body mass loss observed with simvastatin (Table 1; Fig. 2). High-dose statin administration has been associated with body mass loss and decreased food intake in this model (Westwood et al. 2005). Furthermore, these events were paralleled by a marked 95-fold decline in plasma CK concentration, an accepted marker of muscle damage, such that levels returned to that seen in control (Fig. 3). This 40 mg kg−1 day−1 dose of DCA also abolished the simvastatin-induced increase in MAFbx (Fig. 5A) and cathepsin-L (Fig. 5B) mRNA, and attenuated proteasome and cathepsin-L activity (Fig. 6). Of note, the dampening of the mRNA expression levels with DCA occurred in a dose-dependent manner (Fig. 5). Collectively, these data provide convincing evidence that increasing CHO oxidation with DCA reduces simvastatin-induced necrosis by simultaneously inhibiting proteasomal- and lysosomal-mediated protein breakdown. In vitro studies have shown that upregulation of MAFbx and cathepsin-L mRNA expression appears to occur via Akt signaling-mediated changes in FOXO transcription factors (Sandri et al. 2004; Stitt et al. 2004). Indeed, we have previously shown that statin administration decreases muscle Akt phosphorylation and increases expression of FOXO target genes similar to the present in vivo model of statin myopathy (Mallinson et al. 2009). In line with the present reported effect of DCA on PDK4 mRNA and protein expression, it is suggested that a chronic DCA-mediated increase in CHO oxidation was associated with increased Akt phosphorylation, leading to inactivation of FOXO transcription factors, and thereby the decreased expression of MAFbx and cathepsin-L mRNA and associated protein activity observed. However, more work is needed to clarify the exact mechanism by which forcibly increasing CHO oxidation with DCA prevents statin-induced upregulation of proteasomal and lysosomal protein breakdown. Previous studies have suggested that DCA treatment can blunt muscle catabolism, as evidenced by reduced levels of BCAA and phenylalanine in rodent muscle during sepsis (Vary et al. 1988), and reduced necrosis in rabbit tibialis muscle during acute limb ischaemia (Platz et al. 2007). Furthermore, Goodman et al. reported attenuated BCAA release from the perfused hind limb of fed healthy animals following DCA administration, suggesting DCA may either decrease protein breakdown and/or increase protein synthesis (Goodman et al. 1978). Because intramuscular glutamine concentrations have been correlated to muscle protein synthesis, a possible mechanism by which DCA exerts its effect on muscle protein metabolism is through its ability to increase skeletal muscle-free glutamine levels (Ferrando et al. 1998). Alternatively, increased glutamine availability has been shown to increase signalling through the PI3k/Akt pathway in HepG2 cells (van Meijl et al. 2010) and in muscle of rats with diet-induced obesity (Prada et al. 2007). Furthermore, new findings indicate that glutamine availability also relates strongly to the induction of apoptosis, working both as a nutrient and as a signalling molecule, acting directly or indirectly against the pathways leading to programmed cell death (Mates et al. 2006). However, it is not possible to distinguish from the present findings the exact mechanisms involved in the blunting of simvastatin-induced muscle myopathy by DCA.
Statins have also been shown in human rhabdomyosarcoma cells to mediate protein degradation by the induction of autophagy (Araki & Motojima, 2008). However, to date, no study has examined the role of autophagy in an in vivo model of statin-induced myopathy. Autophagy is a cellular process where intracellular components are degraded within lysosomes (Meijer & Codogno, 2004). Consistent with many in vivo models of muscle mass loss, including starvation (Lecker et al. 2004; Mammucari et al. 2007), denervation (Mammucari et al. 2007), cancer cachexia and diabetes (Lecker et al. 2004), is the upregulation of the gene LC3. Our data show, for the first time, that simvastatin administration increases the expression of LC3 mRNA in biceps femoris muscle, which suggests autophagy is involved in the muscle myopathy seen with statins. Furthermore, co-administration of simvastatin with DCA inhibited this increase in LC3 and returned mRNA expression levels to that seen in control, and in a dose-dependent manner (Fig. 5C). Furthermore, this was paralleled by decreased muscle necrosis (Table 1) and plasma CK (Fig. 3). Interestingly, autophagy has been shown to be induced by activated FOXO3a (Mammucari et al. 2007), and it may therefore be suggested that DCA administration inhibited induction of autophagy in statin-induced myopathy in a similar manner to the FOXO1-mediated inhibition of MAFbx and cathepsin-L mRNA expression.
Given PDC is key to the regulation of mitochondrial CHO oxidation, it is interesting to note that in line with our observations reported here mitochondrial dysfunction is evident in models of statin myopathy (Bouitbir et al. 2011; Mullen et al. 2011; Galtier et al. 2012; Kwak et al. 2012). Specifically, skeletal muscle tissue from patients receiving statin therapy shows impaired mitochondrial respiration restricted to mainly complex I of the respiratory chain (Sirvent et al. 2012). Such mitochondrial dysfunction is proposed to increase mitochondrial NADH (Sharma et al. 2011), thereby increasing the mitochondrial NADH/NAD+ ratio, which in turn could activate PDK (Bowker-Kinley et al. 1998) or directly inhibit flux through the PDC reaction (Constantin-Teodosiu et al. 1993). Furthermore, statin-induced mitochondrial dysfunction could lead to increased oxidative stress (Bouitbir et al. 2011; Kwak et al. 2012) and apoptosis (Kwak et al. 2012), which could be at least partly responsible for the statin-related muscle necrosis seen in statin myopathy.
The results of this study suggest the use of DCA could offer potential to prevent statin myopathy in the clinic, particularly as the highest dose administered (40 mg kg−1 day−1) is less than that previously administered to humans. DCA has been used clinically for the treatment of several conditions, including lactic acidosis (100 mg kg−1 day−1; Stacpoole et al. 1988), diabetes mellitus (50 mg kg−1 day−1; Stacpoole et al. 1978), hyperlipoproteinaemia (50 mg kg−1 day−1; Stacpoole et al. 1978), mitochondrial encephalomyopathies (2.4 g day−1; Curto et al. 2006) and to offset muscle fatigue associated with anaerobic ATP production during ischaemic muscle contraction (50 mg kg−1; Timmons et al. 1996, 1998a, 1998b). Chronic use (months to a year) of DCA is, however, associated with adverse side-effects, which has detracted from its widespread use (Kurlemann et al. 1995; Stacpoole et al. 1998; Spruijt et al. 2001). Nonetheless, it has been administered to adults at a dose of 50 mg kg−1 day−1 for up to 5 years to treat mitochondrial encephalomyopathy, which resulted in only mild side-effects that did not warrant discontinuation (Mori et al. 2004). A lower dose of 25 mg kg−1 day−1 was administered to children for up to 6 months in a controlled trial for treatment of lactic acidosis with no signs of any side-effects (Stacpoole et al. 2006). Clearly dose–response studies would be needed to determine whether a tolerated dose of DCA (or any other potential PDK inhibitor) could prevent statin myopathy in humans, and any clinical evaluation of DCA as a preventative treatment for statin myopathy would need careful consideration of its side-effect profile.
In summary, we have presented novel insight into the inhibition of statin myopathy by pharmacological means by demonstrating that increasing muscle CHO oxidation with DCA can reverse the myopathic effects seen with simvastatin in an animal model of statin-induced myopathy. Specifically, DCA increased muscle PDC activity and flux, abolished statin-mediated upregulation of FOXO target genes MAFbx and cathepsin-L, decreased PDK4 mRNA and protein expression, and decreased cathepsin-L and proteasome activity, which occurred concomitantly with the maintenance of food intake and body mass, and the blunting of muscle necrosis and myopathy. The finding that simvastatin activates the transcription and translation of PDK4 and thereby inhibits PDC-mediated CHO oxidation may account for the recent association between statin use and development of diabetes. Further studies are warranted to identify the precise mechanisms by which increased oxidative use of CHO prevents the myopathic effects of simvastatin administration.
Translational perspective
Statins are used widely to reduce hypercholesterolaemia in people with a heightened cardiovascular risk. Nevertheless, although rhabdomyolysis is rare, myositis and myalgia commonly accompany statin use, with a major symptom being muscle weakness and fatigue. Furthermore, increasing evidence suggests long-term, high-dose statin use is associated with an increased incidence of new-onset diabetes. The molecular mechanisms by which statins bring about these adverse effects are not fully resolved, but we have previously shown upregulation of downstream gene targets of the transcription factor FOXO, and evidence of increased proteasomal- and lysosomal-mediated muscle protein breakdown and inhibition of muscle CHO oxidation in an animal model of statin myopathy. The present study demonstrates that increasing CHO oxidation in vivo, using the PDK inhibitor, DCA, blunted activation of FOXO gene targets regulating muscle CHO utilisation and protein breakdown, which inhibited statin myopathy. This is the first demonstration that pharmacological intervention can prevent statin myopathy. The impairment of muscle CHO oxidation is a feature of many forms of muscle atrophy, and therefore the use of a pharmacological intervention to forcibly drive muscle CHO use could be of therapeutic benefit. However, further studies are warranted to assess the impact of long-term DCA use, or any other PDK inhibitor, and to determine the efficacy and minimal dose required to elicit similar responses in patients experiencing statin myopathy symptoms.
Acknowledgments
This study was supported by a grant from AstraZeneca Pharmaceuticals (UK), and The Dunhill Medical Trust supported J.E.M. whilst writing the manuscript (R101/0209). The authors are grateful to the laboratory personnel of Safety Assessment (Alderley Park) for conducting the animal studies.
Glossary
- BCAA
branched-chain amino acid
- CHO
carbohydrate
- CK
creatine kinase
- DCA
dichloroacetate
- FOXO
forkhead box protein O
- HMBS
hydroxymethylbilane synthase
- LC3
microtubule-associated protein 1, light chain 3
- MAFbx
muscle atrophy F-box
- MuRF-1
muscle RING finger-1
- PDC
pyruvate dehydrogenase complex
- PDK
pyruvate dehydrogenase kinase
- PI3k
phosphatidylinositol 3-kinase
Author contributions
J.E.M.: data analysis and interpretations, drafting and revising the manuscript. D.C.-T.: conception and experimental design, data interpretation, and revising the manuscript. P.D.G.: experimental design and revising the manuscript. E.A.M.: performed in vivo experiments and revised the manuscript. W.J.D.: performed in vivo experiments and revising the manuscript. F.R.W.: performed histological analysis and revised the manuscript. J.E.S.: conception and experimental design, data interpretation, and revising the manuscript. P.L.G.: conception and experimental design, data interpretation and major revising. All authors approved the final version of the manuscript.
References
- Araki M, Motojima K. Hydrophobic statins induce autophagy in cultured human rhabdomyosarcoma cells. Biochem Biophys Res Commun. 2008;367:462–467. doi: 10.1016/j.bbrc.2007.12.166. [DOI] [PubMed] [Google Scholar]
- Armstrong RB, Phelps RO. Muscle fiber type composition of the rat hindlimb. Am J Anat. 1984;171:259–272. doi: 10.1002/aja.1001710303. [DOI] [PubMed] [Google Scholar]
- Bergmeyer H. Methods in Enzymatic Analysis. John Wiley; 1983. [Google Scholar]
- Blough E, Dineen B, Esser K. Extraction of nuclear proteins from striated skeletal muscle. Biotechniques. 1999;26:202–206. doi: 10.2144/99262bm05. [DOI] [PubMed] [Google Scholar]
- Bouitbir J, Charles AL, Rasseneur L, Dufour S, Piquard F, Geny B, Zoll J. Atorvastatin treatment reduces exercise capacities in rats: involvement of mitochondrial impairments and oxidative stress. J Appl Physiol. 2011;111:1477–1483. doi: 10.1152/japplphysiol.00107.2011. [DOI] [PubMed] [Google Scholar]
- Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J. 1998;329:191–196. doi: 10.1042/bj3290191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruckert E, Hayem G, Dejager S, Yau C, Begaud B. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients – the PRIMO study. Cardiovasc Drugs Ther. 2005;19:403–414. doi: 10.1007/s10557-005-5686-z. [DOI] [PubMed] [Google Scholar]
- Cederblad G, Carlin JI, Constantin-Teodosiu D, Harper P, Hultman E. Radioisotopic assays of CoASH and carnitine and their acetylated forms in human skeletal muscle. Anal Biochem. 1990;185:274–278. doi: 10.1016/0003-2697(90)90292-h. [DOI] [PubMed] [Google Scholar]
- Clark AS, Mitch WE, Goodman MN, Fagan JM, Goheer MA, Curnow RT. Dichloroacetate inhibits glycolysis and augments insulin-stimulated glycogen synthesis in rat muscle. J Clin Invest. 1987;79:588–594. doi: 10.1172/JCI112851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantin D, Constantin-Teodosiu D, Layfield R, Tsintzas K, Bennett AJ, Greenhaff PL. PPAR delta agonism induces a change in muscle fuel metabolism and activation of an atrophy programme, but does not impair mitochondrial function. J Physiol. 2007;583:381–390. doi: 10.1113/jphysiol.2007.135459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantin D, McCullough J, Mahajan RP, Greenhaff PL. Novel events in the molecular regulation of muscle mass in critically ill patients. J Physiol. 2011;589:3883–3895. doi: 10.1113/jphysiol.2011.206193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantin-Teodosiu D, Baker DJ, Constantin D, Greenhaff PL. PPARδ agonism inhibits skeletal muscle PDC activity, mitochondrial ATP production and force generation during prolonged contraction. J Physiol. 2009;587:231–239. doi: 10.1113/jphysiol.2008.164210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantin-Teodosiu D, Cederblad G, Hultman E. A sensitive radioisotopic assay of pyruvate dehydrogenase complex in human muscle tissue. Anal Biochem. 1991;198:347–351. doi: 10.1016/0003-2697(91)90437-x. [DOI] [PubMed] [Google Scholar]
- Constantin-Teodosiu D, Cederblad G, Hultman E. PDC activity and acetyl group accumulation in skeletal muscle during isometric contraction. J Appl Physiol. 1993;74:1712–1718. doi: 10.1152/jappl.1993.74.4.1712. [DOI] [PubMed] [Google Scholar]
- Constantin-Teodosiu D, Constantin D, Stephens F, Laithwaite D, Greenhaff PL. The role of FOXO and PPAR transcription factors in diet-mediated inhibition of PDC activation and carbohydrate oxidation during exercise in humans and the role of pharmacological activation of PDC in overriding these changes. Diabetes. 2012;61:1017–1024. doi: 10.2337/db11-0799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantin-Teodosiu D, Simpson EJ, Greenhaff PL. The importance of pyruvate availability to PDC activation and anaplerosis in human skeletal muscle. Am J Physiol Endocrinol Metab. 1999;276:E472–E478. doi: 10.1152/ajpendo.1999.276.3.E472. [DOI] [PubMed] [Google Scholar]
- Crossland H, Constantin-Teodosiu D, Gardiner SM, Constantin D, Greenhaff PL. A potential role for Akt/FOXO signalling in both protein loss and the impairment of muscle carbohydrate oxidation during sepsis in rodent skeletal muscle. J Physiol. 2008;586:5589–5600. doi: 10.1113/jphysiol.2008.160150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curto N, Tremolizzo L, Mattavelli L, Piatti ML, Marzorati L, Guerra L, Grassi MG, Ferrarese C. A case of Melas (A3243G) on chronic dichloroacetate treatment. Eur Neurol. 2006;55:37–38. doi: 10.1159/000091424. [DOI] [PubMed] [Google Scholar]
- Dawson SP, Arnold JE, Mayer NJ, Reynolds SE, Billett MA, Gordon C, Colleaux L, Kloetzel PM, Tanaka K, Mayer RJ. Developmental changes of the 26 S proteasome in abdominal intersegmental muscles of Manduca sexta during programmed cell death. J Biol Chem. 1995;270:1850–1858. doi: 10.1074/jbc.270.4.1850. [DOI] [PubMed] [Google Scholar]
- Deedwania P, Stone PH, Bairey Merz CN, Cosin-Aguilar J, Koylan N, Luo D, Ouyang P, Piotrowicz R, Schenck-Gustafsson K, Sellier P, Stein JH, Thompson PL, Tzivoni D. Effects of intensive versus moderate lipid-lowering therapy on myocardial ischemia in older patients with coronary heart disease. Circulation. 2007;115:700–707. doi: 10.1161/CIRCULATIONAHA.106.654756. [DOI] [PubMed] [Google Scholar]
- Denton RM, Randle PJ, Martin BR. Stimulation by calcium ions of pyruvate dehydrogenase phosphate phosphatase. Biochem J. 1972;128:161–163. doi: 10.1042/bj1280161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirks AJ, Jones KM. Statin-induced apoptosis and skeletal myopathy. Am J Physiol Cell Physiol. 2006;291:C1208–C1212. doi: 10.1152/ajpcell.00226.2006. [DOI] [PubMed] [Google Scholar]
- Ferrando AA, Tipton KD, Doyle D, Phillips SM, Cortiella J, Wolfe RR. Testosterone injection stimulates net protein synthesis but not tissue amino acid transport. Am J Physiol Endocrinol Metab. 1998;275:E864–E871. doi: 10.1152/ajpendo.1998.275.5.E864. [DOI] [PubMed] [Google Scholar]
- Furuyama T, Kitayama K, Yamashita H, Mori N. Forkhead transcription factor FOXO1 (FKHR)-dependent induction of PDK4 gene expression in skeletal muscle during energy deprivation. Biochem J. 2003;275:365–371. doi: 10.1042/BJ20030022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galtier F, Mura T, Raynaud de Mauverger E, Chevassus H, Farret A, Gagnol JP, Costa F, Dupuy A, Petit P, Cristol JP, Mercier J, Lacampagne A. Effect of a high dose of simvastatin on muscle mitochondrial metabolism and calcium signaling in healthy volunteers. Toxicol Appl Pharmacol. 2012;263:281–286. doi: 10.1016/j.taap.2012.06.020. [DOI] [PubMed] [Google Scholar]
- Goodman MN, Ruderman NB, Aoki TT. Glucose and amino acid metabolism in perfused skeletal muscle. Effect of dichloroacetate. Diabetes. 1978;27:1065–1074. doi: 10.2337/diab.27.11.1065. [DOI] [PubMed] [Google Scholar]
- Grassi B, Hogan MC, Greenhaff PL, Hamann JJ, Kelley KM, Aschenbach WG, Constantin-Teodosiu D, Gladden LB. Oxygen uptake on-kinetics in dog gastrocnemius in situ following activation of pyruvate dehydrogenase by dichloroacetate. J Physiol. 2002;538:195–207. doi: 10.1113/jphysiol.2001.012984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahoor F, Shangraw RE, Miyoshi H, Wallfish H, Herndon DN, Wolfe RR. Role of insulin and glucose oxidation in mediating the protein catabolism of burns and sepsis. Am J Physiol Endocrinol Metab. 1989;257:E323–E331. doi: 10.1152/ajpendo.1989.257.3.E323. [DOI] [PubMed] [Google Scholar]
- Kurlemann G, Paetzke I, Moller H, Masur H, Schuierer G, Weglage J, Koch HG. Therapy of complex I deficiency: peripheral neuropathy during dichloroacetate therapy. Eur J Pediatr. 1995;154:928–932. doi: 10.1007/BF01957508. [DOI] [PubMed] [Google Scholar]
- Kwak HB, Thalacker-Mercer A, Anderson EJ, Lin CT, Kane DA, Lee NS, Cortright RN, Bamman MM, Neufer PD. Simvastatin impairs ADP-stimulated respiration and increases mitochondrial oxidative stress in primary human skeletal myotubes. Free Radic Biol Med. 2012;52:198–207. doi: 10.1016/j.freeradbiomed.2011.10.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, Price SR, Mitch WE, Goldberg AL. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 2004;18:39–51. doi: 10.1096/fj.03-0610com. [DOI] [PubMed] [Google Scholar]
- Li J, Kato M, Chuang DT. Pivotal role of the C-terminal DW-motif in mediating inhibition of pyruvate dehydrogenase kinase 2 by dichloroacetate. J Biol Chem. 2009;284:34458–34467. doi: 10.1074/jbc.M109.065557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallinson J, Constantin-Teodosiu D, Sidaway J, Westwood FR, Greenhaff P. Blunted Akt/FOXO signalling and activation of genes controlling atrophy and fuel use in statin myopathy. J Physiol. 2009;587:219–230. doi: 10.1113/jphysiol.2008.164699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, Goldberg AL, Schiaffino S, Sandri M. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6:458–471. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- Mates JM, Segura JA, Alonso FJ, Marquez J. Pathways from glutamine to apoptosis. Front Biosci. 2006;11:3164–3180. doi: 10.2741/2040. [DOI] [PubMed] [Google Scholar]
- Meijer AJ, Codogno P. Regulation and role of autophagy in mammalian cells. Int J Biochem Cell Biol. 2004;36:2445–2462. doi: 10.1016/j.biocel.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Mori M, Yamagata T, Goto T, Saito S, Momoi MY. Dichloroacetate treatment for mitochondrial cytopathy: long-term effects in MELAS. Brain Dev. 2004;26:453–458. doi: 10.1016/j.braindev.2003.12.009. [DOI] [PubMed] [Google Scholar]
- Motojima K, Seto K. Fibrates and statins rapidly and synergistically induce pyruvate dehydrogenase kinase 4 mRNA in the liver and muscles of mice. Biol Pharm Bull. 2003;26:954–958. doi: 10.1248/bpb.26.954. [DOI] [PubMed] [Google Scholar]
- Mullen PJ, Zahno A, Lindinger P, Maseneni S, Felser A, Krahenbuhl S, Brecht K. Susceptibility to simvastatin-induced toxicity is partly determined by mitochondrial respiration and phosphorylation state of Akt. Biochim Biophys Acta. 2011;1813:2079–2087. doi: 10.1016/j.bbamcr.2011.07.019. [DOI] [PubMed] [Google Scholar]
- NICE. 2006. Cardiovascular Disease – Statins. Retrieved 25th April, 2008, from http://www.nice.org.uk.
- Nichols GA, Koro CE. Does statin therapy initiation increase the risk for myopathy? An observational study of 32,225 diabetic and nondiabetic patients. Clin Ther. 2007;29:1761–1770. doi: 10.1016/j.clinthera.2007.08.022. [DOI] [PubMed] [Google Scholar]
- Phillips PS, Haas RH, Bannykh S, Hathaway S, Gray NL, Kimura BJ, Vladutiu GD, England JD. Statin-associated myopathy with normal creatine kinase levels. Ann Intern Med. 2002;137:581–585. doi: 10.7326/0003-4819-137-7-200210010-00009. [DOI] [PubMed] [Google Scholar]
- Platz TA, Wilson JS, Kline JA, Rushing G, Parker JL, Moore EM, Southern FN. The beneficial effects of dichloroacetate in acute limb ischemia. Mil Med. 2007;172:628–633. doi: 10.7205/milmed.172.6.628. [DOI] [PubMed] [Google Scholar]
- Prada PO, Hirabara SM, de Souza CT, Schenka AA, Zecchin HG, Vassallo J, Velloso LA, Carneiro E, Carvalheira JB, Curi R, Saad MJ. L-glutamine supplementation induces insulin resistance in adipose tissue and improves insulin signalling in liver and muscle of rats with diet-induced obesity. Diabetologia. 2007;50:1949–1959. doi: 10.1007/s00125-007-0723-z. [DOI] [PubMed] [Google Scholar]
- Preiss D, Seshasai SRK, Welsh P, Murphy SA, Ho JE, Waters DD, DeMicco DA, Barter P, Cannon CP, Sabatine MS, Braunwald E, Kastelein JJP, de Lemos JA, Blazing MA, Pedersen TR, Tikkanen MJ, Sattar N, Ray KK. Risk of incident diabetes with intensive-dose compared with moderate-dose statin therapy. JAMA. 2011;305:2556–2564. doi: 10.1001/jama.2011.860. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM, Jr, Kastelein JJ, Koenig W, Libby P, Lorenzatti AJ, MacFadyen JG, Nordestgaard BG, Shepherd J, Willerson JT, Glynn RJ. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359:2195–2207. doi: 10.1056/NEJMoa0807646. [DOI] [PubMed] [Google Scholar]
- Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117:399–412. doi: 10.1016/s0092-8674(04)00400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattar N, Preiss D, Murray HM, Welsh P, Buckley BM, de Craen AJ, Seshasai SR, McMurray JJ, Freeman DJ, Jukema JW, Macfarlane PW, Packard CJ, Stott DJ, Westendorp RG, Shepherd J, Davis BR, Pressel SL, Marchioli R, Marfisi RM, Maggioni AP, Tavazzi L, Tognoni G, Kjekshus J, Pedersen TR, Cook TJ, Gotto AM, Clearfield MB, Downs JR, Nakamura H, Ohashi Y, Mizuno K, Ray KK, Ford I. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet. 2010;375:735–742. doi: 10.1016/S0140-6736(09)61965-6. [DOI] [PubMed] [Google Scholar]
- Shangraw RE, Jahoor F, Miyoshi H, Neff WA, Stuart CA, Herndon DN, Wolfe RR. Differentiation between septic and postburn insulin resistance. Metabolism. 1989;38:983–989. doi: 10.1016/0026-0495(89)90010-3. [DOI] [PubMed] [Google Scholar]
- Sharma LK, Fang H, Liu J, Vartak R, Deng J, Bai Y. Mitochondrial respiratory complex I dysfunction promotes tumorigenesis through ROS alteration and AKT activation. Hum Mol Genet. 2011;20:4605–4616. doi: 10.1093/hmg/ddr395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirvent P, Fabre O, Bordenave S, Hillaire-Buys D, Raynaud De Mauverger E, Lacampagne A, Mercier J. Muscle mitochondrial metabolism and calcium signaling impairment in patients treated with statins. Toxicol Appl Pharmacol. 2012;259:263–268. doi: 10.1016/j.taap.2012.01.008. [DOI] [PubMed] [Google Scholar]
- Spruijt L, Naviaux RK, McGowan KA, Nyhan WL, Sheean G, Haas RH, Barshop BA. Nerve conduction changes in patients with mitochondrial diseases treated with dichloroacetate. Muscle Nerve. 2001;24:916–924. doi: 10.1002/mus.1089. [DOI] [PubMed] [Google Scholar]
- Stacpoole PW, Barnes CL, Hurbanis MD, Cannon SL, Kerr DS. Treatment of congenital lactic acidosis with dichloroacetate. Arch Dis Child. 1997;77:535–541. doi: 10.1136/adc.77.6.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacpoole PW, Henderson GN, Yan Z, Cornett R, James MO. Pharmacokinetics, metabolism and toxicology of dichloroacetate. Drug Metab Rev. 1998;30:499–539. doi: 10.3109/03602539808996323. [DOI] [PubMed] [Google Scholar]
- Stacpoole PW, Kerr DS, Barnes C, Bunch ST, Carney PR, Fennell EM, Felitsyn NM, Gilmore RL, Greer M, Henderson GN, Hutson AD, Neiberger RE, O’Brien RG, Perkins LA, Quisling RG, Shroads AL, Shuster JJ, Silverstein JH, Theriaque DW, Valenstein E. Controlled clinical trial of dichloroacetate for treatment of congenital lactic acidosis in children. Pediatrics. 2006;117:1519–1531. doi: 10.1542/peds.2005-1226. [DOI] [PubMed] [Google Scholar]
- Stacpoole PW, Lorenz AC, Thomas RG, Harman EM. Dichloroacetate in the treatment of lactic acidosis. Ann Intern Med. 1988;108:58–63. doi: 10.7326/0003-4819-108-1-58. [DOI] [PubMed] [Google Scholar]
- Stacpoole PW, Moore GW, Kornhauser DM. Metabolic effects of dichloroacetate in patients with diabetes mellitus and hyperlipoproteinemia. N Engl J Med. 1978;298:526–530. doi: 10.1056/NEJM197803092981002. [DOI] [PubMed] [Google Scholar]
- Stitt TN, Drujan D, Clarke BA, Panaro F, Timofeyva Y, Kline WO, Gonzalez M, Yancopoulos GD, Glass DJ. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell. 2004;14:395–403. doi: 10.1016/s1097-2765(04)00211-4. [DOI] [PubMed] [Google Scholar]
- Szasz G, Gruber W, Bernt E. Creatine kinase in serum: 1. Determination of optimum reaction conditions. Clin Chem. 1976;22:650–656. [PubMed] [Google Scholar]
- Thompson PD, Clarkson P, Karas RH. Statin associated myopathy. JAMA. 2003;289:1681–1690. doi: 10.1001/jama.289.13.1681. [DOI] [PubMed] [Google Scholar]
- Timmons JA, Gustafsson T, Sundberg CJ, Jansson E, Greenhaff PL. Muscle acetyl group availability is a major determinant of oxygen deficit in humans during submaximal exercise. Am J Physiol Endocrinol Metab. 1998a;274:E377–E380. doi: 10.1152/ajpendo.1998.274.2.E377. [DOI] [PubMed] [Google Scholar]
- Timmons JA, Gustafsson T, Sundberg CJ, Jansson E, Hultman E, Kaijser L, Chwalbinska-Moneta J, Constantin-Teodosiu D, Macdonald IA, Greenhaff PL. Substrate availability limits human skeletal muscle oxidative ATP regeneration at the onset of ischemic exercise. J Clin Invest. 1998b;101:79–85. doi: 10.1172/JCI1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmons JA, Poucher SM, Constantin-Teodosiu D, Macdonald IA, Greenhaff PL. Regulation of skeletal muscle carbohydrate oxidation during steady-state contraction. Am J Physiol Regul Integr Comp Physiol. 1998c;274:R1384–R1389. doi: 10.1152/ajpregu.1998.274.5.R1384. [DOI] [PubMed] [Google Scholar]
- Timmons JA, Poucher SM, Constantin-Teodosiu D, Worrall V, Macdonald IA, Greenhaff PL. Increased acetyl group availability enhances contractile function of canine skeletal muscle during ischemia. J Clin Invest. 1996;97:879–883. doi: 10.1172/JCI118490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meijl LE, Popeijus HE, Mensink RP. Amino acids stimulate Akt phosphorylation, and reduce IL-8 production and NF-κB activity in HepG2 liver cells. Mol Nutr Food Res. 2010;54:1568–1573. doi: 10.1002/mnfr.200900438. [DOI] [PubMed] [Google Scholar]
- Vary TC, Hazen S. Sepsis alters pyruvate dehydrogenase kinase activity in skeletal muscle. Mol Cell Biochem. 1999;198:113–118. doi: 10.1023/a:1006993910781. [DOI] [PubMed] [Google Scholar]
- Vary TC, Siegel JH, Zechnich A, Tall BD, Morris JG, Placko R, Jawor D. Pharmacological reversal of abnormal glucose regulation, BCAA utilization, and muscle catabolism in sepsis by dichloroacetate. J Trauma. 1988;28:1301–1311. [PubMed] [Google Scholar]
- Vaughan CJ, Gotto AM. Update on statins 2003. Circulation. 2004;110:886–892. doi: 10.1161/01.CIR.0000139312.10076.BA. [DOI] [PubMed] [Google Scholar]
- Wells PG, Moore GW, Rabin D, Wilkinson GR, Oates JA, Stacpoole PW. Metabolic effects and pharmacokinetics of intravenously administered dichloroacetate in humans. Diabetologia. 1980;19:109–113. doi: 10.1007/BF00421855. [DOI] [PubMed] [Google Scholar]
- Westwood FR, Bigley A, Randall K, Marsden AM, Scott RC. Statin-induced muscle necrosis in the rat: distribution, development and fibre selectivity. Toxicol Pathol. 2005;33:246–257. doi: 10.1080/01926230590908213. [DOI] [PubMed] [Google Scholar]
- Westwood FR, Scott RC, Marsden AM, Bigley A, Randall K. Rosuvastatin: characterization of induced myopathy in the rat. Toxicol Pathol. 2008;36:345–352. doi: 10.1177/0192623307311412. [DOI] [PubMed] [Google Scholar]
- Whitehouse S, Cooper RH, Randle PJ. Mechanism of activation of pyruvate dehydrogenase by dichloroacetate and other halogenated carboxylic acids. Biochem J. 1974;141:761–774. doi: 10.1042/bj1410761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehouse S, Randle PJ. Activation of pyruvate dehydrogenase in perfused rat heart by dichloroacetate (Short Communication) Biochem J. 1973;134:651–653. doi: 10.1042/bj1340651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieland OH. The mammalian pyruvate dehydrogenase complex: structure and regulation. Rev Physiol Biochem Pharmacol. 1983;96:123–170. doi: 10.1007/BFb0031008. [DOI] [PubMed] [Google Scholar]
- Wu P, Inskeep K, Bower-Kinley M, Popov K, Harris R. Mechanism responsible for inactivation of skeletal muscle pyruvate dehydrogenase complex in starvation and diabetes. Diabetes. 1999;48:1593–1599. doi: 10.2337/diabetes.48.8.1593. [DOI] [PubMed] [Google Scholar]
- Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, Lecker SH, Goldberg AL. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscles. Cell Metab. 2007;6:472–483. doi: 10.1016/j.cmet.2007.11.004. [DOI] [PubMed] [Google Scholar]