

Abstract
Postprandially, the liver experiences an extensive metabolic reprogramming that is required for the switch from glucose production to glucose assimilation. Upon refeeding, the unfolded protein response (UPR) is rapidly, though only transiently, activated. Activation of the UPR results in a cessation of protein translation, increased chaperone expression, and increased ER-mediated protein degradation, but it is not clear how the UPR is involved in the postprandial switch to alternate fuel sources. Activation of the inositol-requiring enzyme 1 (IRE1) branch of the UPR signaling pathway triggers expression of the transcription factor Xbp1s. Using a mouse model with liver-specific inducible Xbp1s expression, we demonstrate that Xbp1s is sufficient to provoke a metabolic switch characteristic of the postprandial state, even in the absence of caloric influx. Mechanistically, we identified UDP-galactose-4-epimerase (GalE) as a direct transcriptional target of Xbp1s and as the key mediator of this effect. Our results provide evidence that the Xbp1s/GalE pathway functions as a novel regulatory nexus connecting the UPR to the characteristic postprandial metabolic changes in hepatocytes.
Introduction
Postprandially, the hepatic metabolic program undergoes extensive changes to switch from glucose production to glucose assimilation. A key feature of this metabolic reprogramming is an increase in protein synthesis, which in turn triggers the unfolded protein response (UPR) (1). In cultured cells, the activation of the UPR temporarily attenuates protein translation, increases chaperone expression, and stimulates ER-associated protein degradation (2, 3). However, little is known about the role of the UPR in the adjustment of the liver to the postprandial state in vivo. Among the three branches of UPR signaling pathways, the inositol-requiring enzyme 1 (IRE1) pathway is the most conserved. Activated IRE1 displays endoribonuclease activity, which cleaves its primary target, the mRNA encoding X-box-binding protein 1 (Xbp1). The specific excision of 26 nucleotides from the Xbp1 mRNA causes a frameshift, and the spliced Xbp1 form (referred to as Xbp1s) is translated into a potent transcription factor that stimulates expression of chaperones and components of the ER-associated protein degradation pathway.
Whereas insulin is the major regulator of the postprandial hepatic remodeling, Xbp1s has the potential to be a critical mediator as well. Xbp1s is induced by refeeding. Moreover, Xbp1s effectively enhances insulin sensitivity in the liver of ob/ob mice (4–7). One mechanism underlying the Xbp1s-mediated enhancement of insulin sensitivity relies on the nuclear translocation of Xbp1s (4, 5). In contrast, liver-specific deficiency of Xbp1 leads to reduced lipogenic gene expression and diminished hepatic lipid synthesis (8). The crosstalk between Xbp1s and metabolic responses is thus apparent in a number of models and cell types (7, 8). However, we lack any insight into how Xbp1s regulates hepatic adjustment to the postprandial state.
Using a liver-inducible Xbp1s–overexpressing mouse model (hereafter referred to as the the LIXs mouse), we found that persistent Xbp1s expression enhanced biosynthetic activity and reduced hepatic glucose release. We identified UDP-galactose-4-epimerase (GalE) as a direct target of Xbp1s and analyzed its contributions to the phenotypes apparent in LIXs mice, as well as its role in postprandial glucose assimilation in wild-type mice. GalE, which encodes an UDP-galactose-4-epimerase in the Leloir pathway (9), is the only major enzyme that catalyzes the interconversion of UDP-Glc and UDP-Gal as well as UDP-GlcNAc and UDP-GalNAc in mammals. Moreover, GalE is the rate-limiting enzyme under fed conditions to supply UDP-Gal for protein glycosylation in liver, suggesting that it is a potent regulator for protein glycosylation in vivo (10). We therefore identify the Xbp1s/GalE axis as a key regulatory nexus to connect the UPR, classically associated with events in the secretory pathway, to insulin-regulated glucose assimilation and anabolism, a hallmark of the postprandial metabolic adjustment in hepatocytes in vivo.
Results
Fasting-refeeding acutely activates hepatic Xbp1s and the UPR.
To define the role of Xbp1s in the postprandial adaptation, we performed a time course study of fasting-refeeding. Xbp1s mRNA increased more than 10-fold within the first hour of refeeding (Figure 1, A and B), overlapping with its pattern of protein expression (Figure 1C). This upregulation was transient. By 3 hours of refeeding, Xbp1s levels dropped to near basal values, only to increase again after 8 hours, when the animals started to eat again (Supplemental Figure 1A; supplemental material available online with this article; doi: 10.1172/JCI62819DS1). In addition to Xbp1s, other components of the UPR were also activated during the postprandial state. These include the induction of phosphorylation of eIF2α and the transcriptional activation of Bip, Atf6, CHOP, ERdj4, and Edem1 (Supplemental Figure 1, B–D). The ratio of Xbp1s to Xbp1 does not reflect the absolute increase in Xbp1 splicing, since unspliced Xbp1 is continually transcriptionally replenished. Therefore, even though the Xbp1s/Xbp1 ratio is not changed, there is strong reason to believe that IRE1α activation occurs in the postprandial state as well.
Figure 1. Postprandial activation of hepatic Xbp1s in WT mice and Xbp1s induction in LIXs mice.

(A and B) WT FVB mice (n = 3 per group) were fasted for 24 hours and refed up to 15 hours. The experiments in A and B were repeated twice. (A) A representative image of Xbp1 RT-PCR products from liver with GAPDH as a loading control. U, unspliced Xbp1; S, spliced Xbp1. (B) Xbp1s levels as determined by qPCR. (C) Xbp1s protein levels during fasting-refeeding in WT livers and by induction in LIXs mice with lamin as loading control for nuclear fraction. (D) RT-PCR analysis of liver Xbp1 from mice fed ad libitum with Dox chow diet. (E) The comparison of Xbp1s protein expression in WT by refeeding and LIXs mice by induction. *P < 0.05.
Inducible overexpression of Xbp1s in LIXs mice mimics postprandial features in WT mice.
The complex nature of the postprandial hepatic response and transient expression of Xbp1s make it difficult to dissect the role of Xbp1s. We therefore developed a strategy to induce Xbp1s in hepatocytes without the need to expose mice to refeeding (Supplemental Figure 1E). We put the Xbp1s transgene under the control of a tetracycline-responsive element (TRE) to allow inducible expression by doxycycline (Dox) in the presence of a tetracycline reverse transcriptional activator (rtTA). The rtTA transgene is driven by the Rosa26 promoter with a transcriptional stop cassette flanked by 2 loxP sites upstream of rtTA (11). Combined with an albumin promoter–driven Cre transgene, we obtained a mouse model with inducible hepato-specific expression of Xbp1s. When LIXs mice were fed a Dox-containing chow diet, Xbp1s mRNA was readily detected within 24 hours (Figure 1D). The endogenous full-length Xbp1 mRNA level dropped at 24 and 48 hours after induction, consistent with the observation that Xbp1s enhances the splicing of its own full-length Xbp1 precursor (12). In light of the very short half-life of Xbp1s (13), the induction of Xbp1s is readily reversible. After an overnight fast and with no alternative source of Dox provided, the Xbp1s mRNA completely disappeared, whereas the full-length endogenous Xbp1 mRNA was restored (Figure 1D). Upon 24 and 48 hours of induction with Dox, the protein levels of Xbp1s in LIXs mice reached levels about 5-fold greater than those in WT mice after 2 hours’ refeeding (Figure 1, C and E). The functionality of the transgenic Xbp1s was validated as judged by increased expression of Xbp1s target genes in LIXs mice (Supplemental Figure 1F). The LIXs model therefore allows us to acutely activate and maintain the hepatic expression of Xbp1s without the need to expose mice to a fasting-refeeding routine.
We employed metabolic cage studies to test whether Xbp1s expression effectively mimics the systemic effects of refeeding. In WT mice, the respiratory exchange ratio (RER) increased upon refeeding (Figure 2, A and B). RER reflects the ratio of CO2 release to O2 consumption, indicating the relative contributions of glycolysis and glucose oxidation versus β-oxidation toward whole-body energy consumption. A higher RER indicates increased glycolysis and glucose oxidation. As expected, refeeding of WT animals led to a rapid rise in RER, suggesting that glucose is the preferred fuel source for CO2 production, consistent with the established transition of fuel usage from fatty acids to glucose upon refeeding. Upon induction, LIXs mice show a significantly higher RER than WT mice during the dark phase (Figure 2C). Moreover, induction was associated with a trend toward increased food intake in LIXs mice (Figure 2D) and improved body weight recovery at the end of the metabolic cage study (Supplemental Figure 1G). These results suggest that Xbp1s expression in the liver can effectively mimic the global metabolic characteristics associated with refeeding in WT animals.
Figure 2. Hepatic Xbp1s overexpression mimics the metabolic effects of refeeding.

RER (A) and food intake (B) of WT mice (n = 5 per group) during refeeding after a 24-hour fast. Time 0 represents 12 p.m. RER (C) and food intake (D) of WT and LIXs mice after Dox induction (n = 4 per group). Mice were switched to a Dox chow diet at 2 p.m. on day 3 in the metabolic cage. Time 0 represents 12 p.m. of day 4 (22 hours after induction). *P < 0.05.
Xbp1s induction leads to rapid biomass accumulation and glycogen depletion.
Along with the systemic effects on whole-body RER, Xbp1s induction also leads to profound changes in the liver. Wet liver mass increased 1.7-fold in LIXs mice within 48 hours of induction (Table 1). Hepatic triglyceride (TG) content increased more dramatically relative to cholesterol and protein content. As a result of the increased TG content, LIXs mice show reduced dry mass content per gram wet liver after lipid extraction. This difference (216.98 – 160.06 = 56.92 mg/g wet liver) was comparable to the increase in TG content (55.83 – 5.8 = 50.03 mg/g wet liver). The weight increase was therefore due to TG and not due to edema formation. Importantly, per total liver, LIXs mice had greater dry mass (226.3 mg in WT vs. 275.4 mg in LIXs), with protein content comparable to controls (mg protein/mg dry liver). Taking the enhanced liver mass into account, this highlights an increase in total liver protein in LIXs mice.
Table 1.
Hepatic parameters after switch to Dox-containing diet for 48 hours
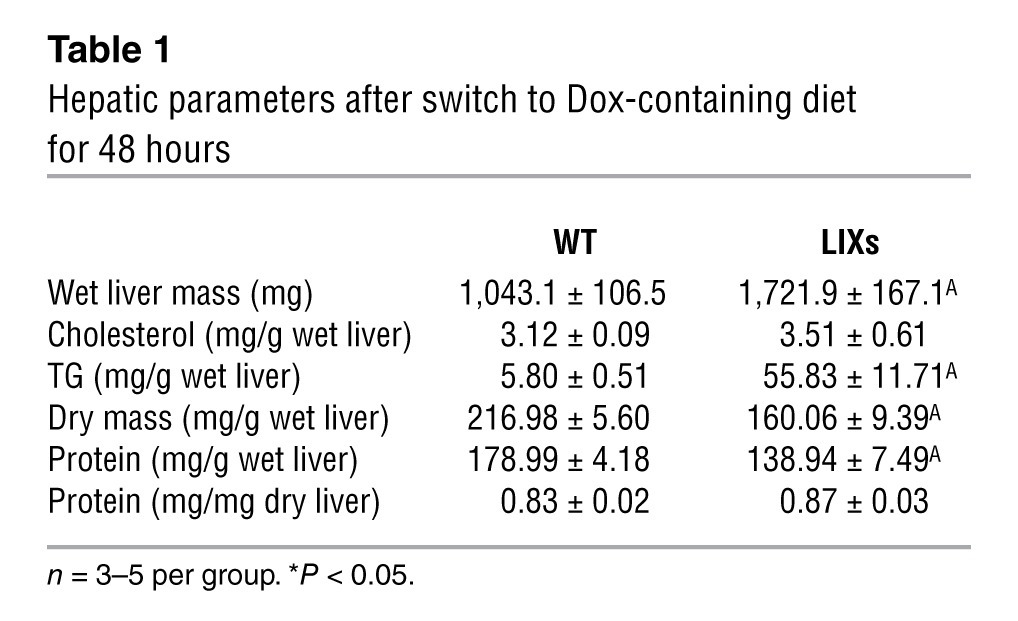
In contrast to the increased hepatic lipid and protein content, hepatic glycogen was severely depleted in LIXs mice after 48 hours of induction (Figure 3A). This suggests that the rapid accumulation of biomass (1.7-fold in 2 days) results from increased synthesis of macromolecules at the expense of hepatic glycogen/glucose. To test whether the reduced glycogen levels in LIXs mice were due to decreased glucose uptake, we performed immunofluorescence staining of GLUT2, the major glucose transporter in hepatocytes. There was no change upon 24 hours of Dox exposure, whereas LIXs hepatocytes showed an enlarged volume with increased surface staining for GLUT2 signal after 48 hours of induction (Figure 3B). The enhanced levels of GLUT2 argue against a defect in glucose uptake. In fact, the metabolic cage studies indicate higher glucose utilization in LIXs animals (Figure 2C). From a more global perspective, this suggests that one of the roles for endogenous Xbp1s in postprandial hepatic remodeling is to coordinate a broad anabolic program in the hepatocyte. Upon Xbp1s overexpression, however, cellular glucose is excessively consumed for these anabolic reactions, and as a result, hepatic glycogen storage is depleted.
Figure 3. Hepatic Xbp1s overexpression triggers hypoglycemia.

(A) Hepatic glycogen staining from mice on Dox chow diet for 48 hours. Scale bars: 50 μm. (B) GLUT2 immunofluorescence staining. Scale bars: 50 μm. (C–E) Mice (n = 3 per group) were fed Dox chow diet for 24–96 hours. Serum glucose levels (C), liver TG content (D), and serum free fatty acid levels (E) under fed or 6-hour fast conditions. *P < 0.05. (F) Serum glucose levels during a pyruvate tolerance test (n = 3 per group). *P < 0.05.
Xbp1s induction diminishes hepatic glucose release, which leads to hypoglycemia and triggers lipolysis from adipocytes for hepatic TG accumulation.
Along with the depletion of liver glycogen, the serum glucose levels started to decrease by 72 hours after induction in LIXs mice under fed conditions (Figure 3C). Moreover, a 6-hour fast caused severe hypoglycemia in LIXs mice within 48 hours after induction. The fed insulin levels started to decrease upon induction and were significantly lower in LIXs mice by 72 hours of induction (Supplemental Figure 2A). A 6-hour fast at that stage still reduced insulin levels in LIXs mice and WT mice to comparable levels. However, 96 hours after induction, fasted insulin levels were even lower in LIXs mice (Supplemental Figure 2B). These results suggest that LIXs mice have impaired adaptation to fasting, presumably due to reduced hepatic glucose release. In WT mice, fasting-induced fatty liver associated with increased TG content, but only a marginal increase in cholesterol (60 mg/g for TG vs. 0.3 mg/g for cholesterol, Supplemental Figure 2C). The fatty liver phenotype observed in LIXs mice (Table 1) is thus reminiscent of the changes in hepatic lipid content during fasting. Lipolysis in adipocytes increases as a response to reduced hepatic glucose release and associated low insulin levels. The fasting effects of adipocyte lipolysis on hepatic TG content are further exacerbated in LIXs mice. Indeed, a 6-hour fast significantly increased liver TG content in LIXs mice acutely, within 24 hours of Dox exposure (Figure 3D). Moreover, we detected a significant increase in serum free fatty acids in the fed state after 72 hours of induction (Figure 3E). Consistent with the increase in lipolysis, LIXs mice displayed a gradual reduction in the volume of adipose tissue, whereas an increase in hepatic lipid by CT scanning was apparent (Supplemental Figure 2D). As a control, when the mice were switched back to regular chow diet without Dox, the liver steatosis decreased rapidly, and the fat tissue volume increased correspondingly in LIXs mice. No significant changes in liver lipid or fat tissue volume were detected in WT control mice under the same conditions, indicating that the effects are specific for Xbp1s induction rather than indirect through Dox treatment.
Mammals maintain serum glucose levels within a very narrow range. This is mainly achieved through regulation of hepatic glucose release. We therefore examined hepatic gluconeogenesis using pyruvate tolerance tests. Dox exposure for 48 hours caused a reduction in glucose release in LIXs mice (Figure 3F). Impaired β-oxidation could be a possible mechanism, as it provides essential precursors for gluconeogenesis. Moreover, β-oxidation defects have been considered to be a feature associated with the UPR (14). The transcriptional levels of β-oxidation and gluconeogenic genes were indeed decreased in LIXs mice (Supplemental Figure 2, E and F). To address whether reduced β-oxidation is a causal factor, we measured the actual β-oxidation potential biochemically in LIXs animals. Surprisingly, we found a similar partitioning of radioactivity in LIXs and control mice for up to 48 hours of Dox induction (Supplemental Figure 2G). This suggests that the β-oxidation capacity in LIXs mice does not substantially differ from that in WT mice. Furthermore, administration of a PPARα agonist (15) exacerbated rather than rescued the hypoglycemic phenotype (Supplemental Figure 2H), strongly arguing against β-oxidation as a causal factor for the defective glucose release. In fact, G6pc, catalyzing the final step of glucose release, is downregulated in ob/ob mice by Xbp1s directly and indirectly through FoxO1 (6). However, the hypoglycemia observed in LIXs mice is unlikely to be solely a consequence of the suppression of G6pc, since hepatic G6pc deficiency is commonly associated with increased glycogen storage in hepatocytes, not with the reduced levels that we observe under the conditions examined here (Figure 3A and ref. 16).
Insulin action as the master regulator of hepatic glucose release in ob/ob mice is enhanced by Xbp1s (7). This was also observed in the LIXs mice, as LIXs livers showed a faster response to insulin exposure (Supplemental Figure 2I). Furthermore, the mice displayed an improvement in insulin sensitivity as indicated by insulin tolerance tests (Supplemental Figure 2J). In isolated hepatocytes, Xbp1s induction led to an enhanced insulin response, indicating a cell-autonomous effect of Xbp1s (Supplemental Figure 2K). LIXs mice therefore developed severe hypoglycemia along with a significant increase in hepatic biomass and hepatic insulin sensitivity. This enhanced local insulin sensitivity may in fact be a key mediator of the increase in the hepatic anabolic programs. Xbp1s activation therefore mimics a “permanently fed state.”
GalE, regulated by Xbp1s, links the UPR and postprandial remodeling.
Based on the analysis above, we performed a microarray screen for novel target genes of Xbp1s. These genes are expected to show a significant increase in mRNA levels in LIXs mice after induction, and upregulation in WT mice upon refeeding. GalE was one of the most highly upregulated among the genes that fulfilled both of these criteria (Supplemental Table 1).
GalE encodes the enzyme uridine diphosphate galactose-4-epimerase. It is the final and key enzyme of the Leloir pathway and acts in concert with GalK and GalT for utilization of dietary galactose (Supplemental Figure 3A and ref. 9). GalE catalyzes the interconversion of UDP-galactose (UDP-Gal) and UDP-glucose (UDP-Glc), as well as a pair of larger substrates, UDP-GalNAc and UDP-GlcNAc. GalE is the rate-limiting enzyme for cellular production of UDP-Gal and UPD-GalNAc (10). The microarray data suggest GalE as a key mediator of Xbp1s action, linking the UPR with cellular glucose metabolism, as UDP-galactosyl and UDP-glucosyl groups are critical substrates for both protein and lipid glycosylation in biosynthetic pathways and regulation of protein folding in the ER (17).
Consistent with the microarray analysis, GalE expression was drastically increased in LIXs mice regardless of caloric influx, as assessed by both qPCR and immunoblotting (Figure 4, A and B). Importantly, this is not a reflection of a general upregulation of the Leloir pathway. GalK, the first enzyme of the pathway, catalyzing the phosphorylation of galactose, remained unchanged in the 24-hour-fed LIXs mice and was significantly downregulated at later time points or under fasting conditions (Figure 4C), reflecting a compensatory adaptation to the increased GalE expression.
Figure 4. Hepatic Xbp1s overexpression induces GalE.

Gene transcription of GalE (A) and GalK (C) and immunoblotting of GalE (B) from mice (n = 3 per group) fed Dox food for 24–72 hours. GAPDH served as a loading control. *P < 0.05.
To assess the relationship between GalE and Xbp1s under normal physiological conditions, we first analyzed the effect of refeeding on GalE levels. We subjected WT animals to fasting of different durations (0, 6, 12, and 24 hours), followed by 2 hours of refeeding. Refeeding induced significant increases in Xbp1s expression in mice fasted for 12 and 24 hours (Figure 5, A and B). Under these conditions, GalE transcription mirrored the pattern of Xbp1s expression, a phenomenon not observed for galK. We then measured galE expression in LIXs mice after induction by Dox gavage in combination with fasting-refeeding. Xbp1s was robustly increased with a Dox gavage in LIXs mice (Figure 5, C and D). The regimen of 6-hour fasting followed by 2 hours of refeeding with regular chow increased galE expression significantly in LIXs mice, whereas no difference was observed in WT mice. In line with the greater increase in Xbp1s upon refeeding in 24-hour fasted mice, GalE expression reflected the same pattern (Figure 5D). Again, this behavior was not observed for GalK. Interestingly, the expression levels of Bip and ERdj4 were comparable in WT and LIXs mice in fasted groups under all conditions, and refeeding led to comparable changes regardless of genotype (Supplemental Figure 3B). These results indicate that the rapid increase in GalE transcripts is unlikely to be a secondary event. No changes in liver TG content, serum glucose levels, or liver acylcarnitine composition were detected between WT and LIXs mice under these conditions (Supplemental Figure 3, C and D), suggesting that the observed GalE upregulation occurs prior to the development of the LIXs phenotypes.
Figure 5. Hepatic GalE expression associates with Xbp1s induction in vivo.

(A and B) WT mice were fasted for different durations and refed for 2 hours. Expression of Xbp1s, GalE, and GalK (B) was examined by qPCR (n = 3 per group). *P < 0.05. (C and D) WT and LIXs mice were fasted for 6 or 24 hours before a 2-hour refeeding. Induction was performed once by Dox gavage 3 hours prior to refeeding. Sac, sacrifice. Expression of Xbp1s, GalE, and GalK (D) is shown (n = 3 per group). *P < 0.05. (E) Xbp1s expression in genetically (ob/ob) and diet-induced (HFD) obese mice (n = 3 per group) with actin as loading control. U, unspliced Xbp1; S, spliced Xbp1. Expression of GalE was determined by qPCR (F). *P < 0.05.
Since ER stress has been implicated in the development of obesity-related metabolic dysfunction (7), we assessed GalE expression in a high-fat diet–induced (HFD-induced) obese mouse model as well as in leptin-deficient ob/ob animals. Both conditions were associated with elevated Xbp1s (Figure 5E). GalE was upregulated 4-fold in the 10 week HFD-fed mice and doubled in ob/ob mice (Figure 5F). Thus, GalE is upregulated under pathological conditions associated with obesity tied to constitutive Xbp1s activation.
To dissect whether the activation of GalE by Xbp1s was cell autonomous, we infected primary mouse hepatocytes with a lentivirus overexpressing Xbp1s. GalE was significantly upregulated in Xbp1s-infected cells, whereas the lipogenic genes Fasn and Scd1 were not altered (Figure 6A). In another tissue culture model, tunicamycin treatment of Huh7 cells (a human hepatoma cell line) was sufficient to stimulate Xbp1s but failed to induce GalE (Figure 6B). However, when insulin was supplemented to mimic a postprandial setting, GalE expression was significantly increased. This is consistent with the observation that Xbp1s functions in the postprandial state when insulin is elevated. In fact, WT mice refed after a 24-hour fast showed a greater increase in GalE expression than LIXs mice refed after a 6-hour fast (Figure 5D). This is in contrast to the Xbp1s mRNA expression, as WT mice refed after a 24-hour fast showed a smaller increase in Xbp1s expression than LIXs mice refed after a 6-hour fast (Figure 5D). These results reflect an additive effect of insulin on GalE activation by Xbp1s, presumably through enhanced nuclear translocation (4, 5). Indeed, the insulin levels in WT mice refed after a 24-hour fast were much higher than in LIXs mice refed after a 6-hour fast (Figure 6C).
Figure 6. GalE expression is upregulated by Xbp1s in vitro.

(A) Gene expression in mouse primary hepatocytes (n = 5–6 per group) infected with lentivirus overexpressing LacZ or Xbp1s. *P < 0.05. (B) Gene expression in Huh7 cells after treatment with tunicamycin (TM) and insulin (n = 3 per group). *P < 0.05. (C) Serum insulin levels in mice from Figure 5C (n = 3 per group). *P < 0.05.
To test whether Xbp1s is not only sufficient but required for GalE activation, we performed siRNA-based knockdown of Xbp1s in isolated primary hepatocytes. GalE expression was significantly reduced under these conditions, indicating that Xbp1s is necessary for galE expression (Supplemental Figure 3E). Although tunicamycin induced Xbp1s expression, Xbp1s-mediated GalE activation was specifically blunted. This is intriguing, as, in fact, tunicamycin is an analog of UDP-GlcNAc. It inhibits the enzyme GlcNAc-1-phosphotransferase, which transfers GlcNAc-1-P from UDP-GlcNAc to membrane-bound Dol-P, forming GlcNAc-P-Dol, the N-glycan precursor synthesized on the cytoplasmic face of the ER. Endogenous UDP-GlcNAc therefore potentially serves as a potent inhibitor of Xbp1s-mediated activation of GalE. In contrast, when thapsigargin, another ER stress inducer, was used, both Xbp1s and GalE increased (Supplemental Figure 3F).
To test whether Xbp1s directly regulates GalE expression, we analyzed the promoter of GalE and found a highly conserved binding site for Xbp1s across species: TCCACGTC (Figure 7A and ref. 18). A luciferase assay with the GalE promoter containing the potential binding site showed dose-dependent upregulation by Xbp1s (Figure 7B). When the potential Xbp1s-binding site was mutated or deleted, the luciferase activity was reduced by 50%, indicating this site is critical for activation by Xbp1s, although other binding sites may also exist (Figure 7C). By ChIP assays, we found that hepatic Xbp1s is associated with the GalE promoter in WT mice 2 and 6 hours after refeeding. In contrast, Xbp1s protein was not detected on the GalE promoter of fasted mice (Figure 7D). Together, our results indicated a strong temporal correlation between Xbp1s induction and GalE expression both in vivo and in vitro. Importantly, the increase in GalE expression occurs prior to most of the other phenotypic and gene expression changes observed in LIXs mice. This suggests that GalE could be a major mediator for Xbp1s-induced early- and long-term metabolic consequences in LIXs mice. Considering the functional involvement of GalE in the interconversion of galactosyl and glucosyl groups, we propose that GalE plays a critical role in mediating Xbp1s action on both glucose metabolism and the UPR.
Figure 7. GalE is a target gene of Xbp1s.

(A) A consensus binding site of Xbp1s is indicated on alignment of GalE promoters from mouse, rat, and human. (B) Luciferase assays for GalE promoter regulated by Xbp1s in HEK293T cells (n = 3–6 per group). (C) Deletion or mutation of the Xbp1s binding site reduces luciferase activity (n = 3 per group). *P < 0.05. (D) Xbp1s protein association with Gale promoter examined by ChIP assay using WT livers under condition of fasting, 2 hours refeeding, and 6 hours refeeding. Ctrl, control.
In contrast to GalE, the lipogenic genes did not show a significant increase in LIXs mice under fed conditions, and in fact displayed a global decrease with fasting (Supplemental Figure 4A). Acute induction via Dox gavage, illustrated in Figure 5C, also did not cause a significant increase in the lipogenic program in LIXs mice with refeeding (Supplemental Figure 4B). Importantly, a biochemical assay for de novo lipid synthesis showed that LIXs mice maintained the same rate of fatty acid and cholesterol synthesis as controls (Supplemental Figure 4C). Our data therefore do not support Xbp1s as a direct transcriptional regulator of the lipogenic program under these conditions.
GalE, regulated by fasting-refeeding, provides a novel target for therapeutic intervention for metabolic dysfunction.
Although GalE is highly conserved from bacteria to mammals, little is known about its transcriptional regulation. We found that GalE expression was regulated by fasting-refeeding in WT mice: downregulated during a fast and upregulated upon refeeding (Figure 8A). Thus, we propose a model to explain the contributions of the Xbp1s/GalE axis in the context of refeeding of WT animals in comparison to the “pseudo-refeeding” status in LIXs mice (Figure 8B). In WT mice, refeeding activates Xbp1s, which in turn upregulates GalE transcription. This moderate increase in GalE during the first 1–3 hours of refeeding enhances the conversion of UDP-Gal to UDP-Glc to accelerate the restoration of hepatic glycogen by assimilation of diet-derived galactose. Consistent with this model, within 2 hours of refeeding, hepatic glycogen levels were fully restored in overnight-fasted mice (data not shown). After glycogen is replenished, the continuous increase in GalE then interconverts UDP-Glc and UDP-Gal to facilitate protein and lipid biosynthesis, which also competes for intracellular glucose and reduces its release from hepatocytes. However, in the LIXs mice, GalE transcription is maintained at high levels due to sustained Xbp1s activation, which, together with the increased anabolic rate in liver, constantly consumes cellular glucose 6-phosphate (G-6-P). As a result, hepatic glucose release is drastically suppressed, and remaining glycogen is broken down. Responding to the low systemic glucose levels in LIXs mice, adipocyte-mediated lipolysis is enhanced, which leads to the acute accumulation of hepatic TGs at high levels.
Figure 8. Regulation of GalE by fasting-refeeding.

(A) Expression of GalE in WT mice (n = 3 per group) during fasting-refeeding. Experiments were repeated twice. (B) Model for the role of GalE in glucose assimilation in WT mice by refeeding (left) and LIXs mice by induction (right). See text for details.
According to this model, GalE is one of the critical regulatory factors for the partitioning of glucose toward biosynthetic activity and away from hepatic glucose release. To test this, we injected an adenoviral preparation to achieve GalE overexpression in ob/ob mice, a model with excessive hepatic glucose production. In an oral glucose tolerance test, GalE adenovirus–infected ob/ob mice showed a significant improvement in glucose tolerance compared with the controls (LacZ adenovirus or no-virus group) (Figure 9A). The improvement was due to reduced hepatic glucose release, since the GalE adenovirus–infected mice also showed significantly reduced glucose release during a pyruvate tolerance test (Figure 9B). Similarly, in mice fed a HFD for 10 weeks, GalE adenoviral infection significantly lowered fasting glucose levels (Figure 9C).
Figure 9. Hepatic GalE overexpression improves insulin sensitivity.

(A and B) Serum glucose levels during an oral glucose tolerance test (A) and a pyruvate tolerance test (B) in ob/ob mice (n = 4–5 per group) assayed 7 days after adenoviral infection. *P < 0.05. (C) Fasting (6 hours) serum glucose levels from WT mice (n = 3–4 per group) assayed 7 days after adenoviral infection. The mice were fed a HFD for 10 weeks before infection. *P < 0.05. (D and E) Serum glucose and insulin levels during an oral glucose tolerance test (D) and serum glucose levels during an insulin tolerance test (E) in WT mice (n = 5–6 per group) assayed 7 days after infection. *P < 0.05.
We wanted to examine whether GalE activation causes phenotypic changes in lean WT animals kept on a chow diet (conditions comparable to experiments done in LIXs mice). We therefore injected the adenoviral preparation into WT mice. While an oral glucose tolerance test showed no difference in glucose clearance (Figure 9D), insulin sensitivity was significantly enhanced in GalE-infected mice (Figure 9E). This suggests that GalE overexpression alone can potently reduce hepatic glucose release even in WT animals. GalE overexpression, however, is insufficient to cause hypoglycemia in lean mice maintained on a chow diet. This is possibly due to the systemic ability to adjust insulin release, i.e., the GalE-infected group reduced insulin release during the glucose challenge to offset the effect of reduced hepatic glucose release. Indeed, we found that the insulin levels in GalE-infected mice were not only lower at baseline, but also remained so over the entire course of the oral glucose tolerance test (Figure 9D). Furthermore, we were examining a loss-of-function phenotype in these experiments (loss of hepatic glucose output). In contrast to our genetic Dox-inducible model, which potentially affects every hepatocyte, adenoviral infection is never 100% efficient, even at a high MOI, so that there is always a substantial number of hepatocytes remaining unaffected and responsive of compensation for euglycemia in WT mice.
GalE overexpression increases the rate of interconversion between UDP-glucosyl and UDP-galactosyl groups. If products are constantly removed from the equilibrium, the flux from G-6-P to UDP sugars can be maintained at high rates in hepatocytes. This will ultimately lead to hypoglycemia in LIXs mice. One major pathway for the consumption of UDP sugars is protein glycosylation. We therefore examined the levels of the N-linked glycans on hepatic proteins. LIXs mice showed an increase in N-glycosylation per gram protein in the fed state (Figure 10A) but no change in the fasted state (data not shown). Moreover, when cellular nucleotide sugars were analyzed, LIXs mice showed increases in cellular levels of UDP-Glc, UDP-Gal, UDP-GlcNAc, and UDP-GalNAc (Figure 10, B and C). In the fasting state, the LIXs mice retained higher levels of UDP-GlcNAc and UDP-GalNAc than controls, whereas the UDP-Glc and UDP-Gal levels were the same (Supplemental Figure 5A). These data suggest that GalE induction shunts more glucose into the pool of UDP sugars for anabolic reactions. Additionally, these observations also provide a mechanism for the synergistic effects of the activation of β-oxidation by fenofibrate in LIXs mice: the more acetyl-coA generated through β-oxidation, the more rapid the depletion of glucose due to the conversion to acetylated hexosamines.
Figure 10. GalE overexpression enhances biosynthesis of nucleotide sugars and protein glycosylation.

(A–C) Representative FACE images of total hepatic N-glycan with G4-G7 glucose oligomer standards (A) and nucleotide-sugar pools that have been hydrolyzed to allow detection of the corresponding free monosaccharides (B), and quantification of cellular UDP sugars (C) from mice (n = 4–5 per group) after 48 hours of induction. *P < 0.05. Man., mannose. (D) Immunofluorescence staining of hepatic LDL receptor 24 hours after induction. Scale bars: 50 μm.
Beyond the increased N-glycans in fed LIXs mice, we also found an increase in three single-sugar moieties from protein O-glycosylation, including GlcNAc, Glc, and Man (Supplemental Figure 5B). The LDL receptor is critically dependent on O-glycosylation for proper folding and function, with both Gal and GalNAc involved (19). Indeed, we found that the LIXs mice displayed strong hepatic staining for the LDL receptor after a 24-hour Dox exposure (Figure 10D). Furthermore, siRNA-mediated knockdown of galE in isolated primary hepatocytes led to a reduction in cellular levels of LDL receptor (data not shown). Consistent with the role of Xbp1s as an anabolic factor, induction of Xbp1s in primary hepatocytes increased protein synthesis (Figure 11A). siRNA-mediated knockdown of GalE caused a lower rate of protein synthesis in both WT and LIXs hepatocytes (Figure 11B). A possible mechanism for this reduced global protein synthesis is an impaired response to insulin. Indeed, a GalE siRNA-mediated knockdown reduced the insulin response in hepatocytes (Figure 11C). In contrast, overexpression of GalE improved insulin signaling (Figure 11D). Moreover, to test whether GalE is sufficient to enhance protein synthesis, we examined the effects of GalE overexpression on adiponectin production in a HEK293T cell system. When a GalE expression construct was co-transfected with an adiponectin expression construct, adiponectin protein levels significantly increased in cells (Figure 11E), with no significant change in mRNA level and adiponectin secretion rate (data not shown). These results suggest that GalE may be a rate-limiting factor for protein synthesis, presumably through enhanced supply of UDP sugars for glycosylation and increased ER capacity for protein folding.
Figure 11. GalE regulates protein synthesis and cellular response to insulin.

(A) Measurement of protein synthesis stimulated by Xbp1s in primary hepatocytes using 3H-leucine incorporation. *P < 0.05. (B) Knockdown of GalE reduces protein synthesis rate in both WT and LIXs hepatocytes. *P < 0.05. (C) Knockdown of GalE impairs insulin signaling in WT hepatocytes. (D) Overexpression of GalE enhances the insulin response in WT rat hepatocytes. WB, Western blot. (E) Adiponectin levels were examined by immunoblotting in HEK293T cells when co-transfected with a GalE expression plasmid (1 μg) or the vehicle plasmid (1 μg). Actin served as loading control. APN, adiponectin. GDI, guanosine nucleotide dissociation inhibitor. NA, no virus infection.
Discussion
Hepatic Xbp1s induction mimics a potent refeeding signal.
Refeeding causes a comprehensive metabolic response in the liver. Here, we characterized the LIXs mouse model to assess the specific role of Xbp1s in this process. This model offers a unique opportunity to study the effects of Xbp1s at very high temporal resolution in vivo. Upon induction, LIXs mice mimic the fed state. The LIXs mice display an acute increase in liver mass within 48 hours of induction. Per gram tissue, the enlarged livers still maintain a normal rate of de novo lipid biosynthesis, supporting a significant increase in total lipid production per liver. The increase in liver mass correlates with the depletion of hepatic glycogen and development of hypoglycemia, both of which suggest that Xbp1s promotes glucose assimilation for anabolic reactions in hepatocytes. As a result, glucose release from hepatocytes is reduced, causing systemic hypoglycemia. The actions of Xbp1s in LIXs mice are consistent with its insulin sensitization role (4–7). The Xbp1s-mediated enhancement of anabolic reactions in LIXs mice is further supported by the observation that mice with hepatic Xbp1 deficiency show resistance to body weight gain on an HFD (8). In addition, the Xbp1s-mediated anabolic action has also been reported for phospholipids (20) and secretory proteins (21). All of these observations support that postprandial induction of Xbp1s in liver is a critical component for efficient anabolic recovery of biomass loss experienced during fasting. In contrast to WT mice, which display the phenomenon only during refeeding, LIXs mice have greater anabolic activity, associated with an increase in UPR activity. This highlights that some components of the UPR can serve as integral components of the machinery that boosts biosynthesis during the postprandial response.
GalE plays an essential role in hepatic glucose assimilation.
GalE is an enzyme of the Leloir pathway catalyzing the interconversion of UDP-Gal and UDP-Glc (9, 22). The importance of the Leloir pathway is highlighted by genetic deficiencies in human patients. Whereas loss-of-function mutations in GalT or GalK, the other two genes of the Leloir pathway, have been described (23, 24), no cases of functional GalE-null mutants have been reported (25). Consistent with the essential nature of this enzyme, the GalE-deficient fruit fly is developmentally lethal (26). Our data show that GalE knockdown leads to impaired insulin signaling and protein biosynthesis in hepatocytes, implying a potential mechanism for the essential role of GalE in cell survival. Since mammals, unlike unicellular organisms, are not critically dependent on an exogenous galactose supply, GalE transcription is not responsive to the presence of dietary galactose (27). Here, we demonstrate that GalE is transcriptionally regulated by Xbp1s. Due to the unique role of GalE in maintaining global protein glycosylation, the knockdown of GalE leads to a general reduction of signal transduction due to a reduced biosynthetic rate of receptors. Conversely, we found that overexpression of GalE enhances insulin sensitivity in vitro and in vivo, providing an additional mechanism for enhanced insulin action beyond Xbp1s nuclear translocation (4, 5). Our data thus suggest that GalE plays a dual role in allowing Xbp1s to unfold its full anabolic potential. GalE accelerates the isomerization of nucleotide sugars, thereby increasing consumption of cellular glucose and directly enhancing glucose uptake. GalE also exerts indirect effects by increasing protein synthesis by facilitating glycosylation. Key products of this process include growth factor receptors and glucose transporters. The increases in these proteins are functionally instrumental for the cell to enhance glucose assimilation and allow these carbohydrates to flow into anabolic pathways. The discovery of the complex regulatory patterns for GalE and the profound role of GalE in glucose homeostasis indicate that GalE specifically, and galactose metabolism in general, may play a critical role in whole-body energy homeostasis, beyond the context of the classical Leloir pathway.
The Xbp1s/GalE axis contributes to the drastic phenotype observed in LIXs mice.
In LIXs mice, expression of Xbp1s mimics refeeding, even in the absence of caloric influx. This leads to reduced hepatic glucose release and eventually causes systemic hypoglycemia. Adipose tissue responds by increasing lipolysis, leading to hepatic steatosis. Rather than being a direct result of impaired β-oxidation or gluconeogenesis, we found that the lack of hepatic glucose release is caused by persistent diversion of intracellular glucose to biosynthetic pathways. Consistent with this mechanism, LIXs mice did not show evidence of decreased glucose uptake or changes in insulin signaling in the liver. In contrast to its physiological regulation by fasting and refeeding in WT mice, GalE is upregulated constitutively under both fed and fasted conditions in LIXs mice. On one hand, increased GalE causes a rapid interconversion of UDP-glusosyl and UDP-galactosyl groups, which are building blocks for glycosylation of proteins and lipids. On the other hand, increased insulin sensitivity and ensuing anabolic reactions in LIXs mice demand more UDP sugars. Therefore, the upregulation of GalE is a prerequisite for the rapid increase in liver mass, one of the most profound phenotypic changes associated with LIXs mice. Moreover, GalE overexpression enhances insulin signaling in hepatocytes, which is further exemplified in ob/ob animals. Notably, the effects of GalE overexpression recapitulate what has been previously reported for adenoviral Xbp1s in improving the glucose tolerance in ob/ob mice (6).
Since LIXs is an overexpression mouse model, there is a potential concern that overexpression of Xbp1s and GalE will cause cytotoxicity and lead to hepatomegaly. We therefore focused on the acute response in LIXs mice within 48 hours of induction, when the food intake and body weight of mice remain unchanged. Our studies showed that the acute response is unlikely to be caused by cytotoxicity for the following reasons: First, the liver mass rapidly increased without edema within 48 hours of induction. In addition, no cell death was observed in cultured LIXs hepatocytes after induction up to 72 hours. Instead, insulin signaling (a fundamental “pro-survival signal”) was enhanced in LIXs hepatocytes within 72 hours after induction both in vivo and in vitro. Furthermore, the LIXs hepatocytes showed widespread increases in anabolic reactions (except for glycogen synthesis), suggesting a controlled conversion from glycogen-glucose to other macromolecules. Last, the LIXs mice responded to fenofibrate to an even higher extent than WT mice, indicating the LIXs hepatocytes remain fully responsive in vivo. Moreover, consistent with our observations here, Xbp1s overexpression has recently been show to enhance chondrocyte hypertrophy (28).
GalE links the role of Xbp1s in the UPR with metabolism.
The fact that GalE is activated by Xbp1s provides a direct link between the UPR and cellular reprogramming in the postprandial state. UDP-Glc and UDP-Gal as well as UDP-GlcNAc and UDP-GalNAc are critical metabolites for glycosylation and protein folding. It is therefore critical to coordinate their production with the pool of glucose devoted to oxidation for energy or other intermediate metabolite production. Up to 35% of all polypeptides produced by a cell are either secretory or transmembrane proteins. This imparts a high demand for UDP sugars for post-translational modifications. Simultaneously, the other steps in the ER, such as disulfide bond formation and vesicular transport, consume glucose for the generation of ATP and oxidative power. It is therefore not surprising that the UPR exerts extensive influences on carbohydrate metabolism. Indeed, numerous metabolic changes have been found to be associated with ER stress (7, 8, 29, 30). GalE is the only enzyme known to catalyze the interconversion between glucosyl and galactosyl pools. Our results thus provide a mechanistic basis for these findings. Through activation of GalE, Xbp1s regulates the interconversion of UDP sugars. As an enzyme, GalE only accelerates this reaction, but does not change the equilibrium, which allows a bidirectional reaction based on the concentrations of UDP-Gal and UDP-Glc (22). Under nutrient-rich conditions, such as the postprandial state, upregulation of Xbp1s/GalE leads to a rapid equilibrium between UDP-glucosyl and UDP-galactosyl groups. This is coupled to glycogen deposition, protein synthesis, and lipogenesis. In this aspect, the increased UPR in LIXs mice is consistent with the higher rate of protein synthesis. During conditions when nutrients are limited, such as glucose deprivation, the major fate of UDP sugars is to be converted to glucose 1-phosphate and then to G-6-P, a starting point for glucose catabolism and ATP generation. Accordingly, the protein synthesis is reduced, which leads to reduced UPR activity.
The UPR is activated under various physiological and pathological conditions. The identification of the Xbp1s/GalE pathway prompted us to propose a common role of the UPR. Our results highlight the Xbp1s/GalE pathway as a mechanism enabling cells to assimilate glucose, which helps cells to adjust glucose flux in the context of either nutrient sufficiency or insufficiency. GalE thus plays essential role in cell growth and survival. Our data suggest that the postprandial induction of the Xbp1s/GalE axis is critical for recovering body weight loss from fasting. In this regard, we propose that the Xbp1s/GalE axis might be generally involved in conditions of acute body weight loss or gain, such as infectious disease, obesity, or cancer. Tumor cell growth and proliferation are associated with an increased demand for glucose partitioning toward biosynthetic pathways. Indeed, Xbp1s expression is induced in multiple cancers (31, 32), and GalE has been found to be upregulated in 100% of all tumor tissues examined in the Human Protein Atlas (33).
Our studies uncovered a novel function of Xbp1s in the postprandial state in vivo. Xbp1s orchestrates changes for the regulation of hepatic insulin sensitivity, with GalE as an integral component of this adaptive response. While it is unlikely that GalE is the sole mediator of all of the postprandial changes that Xbp1s exerts on the hepatocyte, our observations firmly establish GalE as a critical player in the process of postprandial adaptation. Furthermore, the Xbp1s/GalE axis provides a direct link between the UPR and carbohydrate metabolism. Future studies are warranted for a better understanding of the Xbp1s/GalE nexus under various pathological conditions, such as insulin resistance, obesity, and cancer.
Methods
Transgenic mice.
Mouse Xbp1s was cloned into the pTRE vector (Clontech) with a rabbit β-globin 3′ UTR. The TRE-Xbp1s transgenic mice were generated by the transgenic core facility at UTSW. Liver-specific albumin-Cre transgenic and Rosa26-loxP-STOP-loxP-rtTA transgenic (Jackson Laboratory) mice were backcrossed into the FVB background for at least 5 generations (11). Age-matched transgenic animals with albumin-Cre and Rosa26-loxP-STOP-loxP-rtTA but lacking the TRE-Xbp1s transgene were used as controls. Mice were maintained on a 12-hour dark/12-hour light cycle from 6 a.m. to 6 p.m. and housed in groups of no more than 5 with unlimited access to water and chow (2916, Teklad) or Dox-containing (200 mg/kg) chow diets (Bio-Serv).
Fatty acid and cholesterol synthesis in vivo.
Rates of fatty acid synthesis were measured using 3H-labeled H2O as previously described (34). Prior to the experiment, LIXs mice were kept on a control or Dox-containing diet for 3 days. The rates of fatty acid and cholesterol synthesis were calculated as nanomoles of 3H-radioactivity incorporated into fatty acid and cholesterol per hour per gram of wet tissue.
3H-triolein uptake and β-oxidation.
Tissue-specific lipid uptake and β-oxidation analysis were performed as previously described (35). Briefly, 3H-triolein was injected via tail vein (2 μCi/mouse in 100 μl of 5% intralipid) following a 6-hour fast. After 15 minutes, tissues were harvested, and lipids were extracted. Samples were then counted for radioactivity.
Tolerance tests and fenofibrate treatment.
For pyruvate tolerance tests, mice were fed Dox diets for 24 or 48 hours and fasted for 6 hours. Before fasting, a bolus of 1 mg Dox was gavaged, and the animals were maintained on water containing 0.1 mg/ml Dox. Animals received 2 mg/kg body weight of sodium pyruvate through intraperitoneal injection, and blood was withdrawn through tail vein. For glucose tolerance tests, after 3 hours of fasting, glucose (2.5 mg/kg body weight) was gavaged orally. For insulin tolerance tests, insulin (Novo Nordisk) of 1 IU/kg body weight was administrated intraperitoneally. To test insulin signaling in vivo, mice were starved overnight, and insulin of 1 IU/kg body weight was administrated intraperitoneally. Livers were harvested at 5 or 30 minutes after insulin injections. Fenofibrate (Sigma-Aldrich) was prepared in Tween 80 and diluted in 1% methylcellulose and administered to animals daily at 150 mg/kg body weight by oral gavage.
Metabolite measurements.
Frozen tissues of 100 mg were used for lipid extraction and measurement by the UTSW Metabolic Core Facility as described previously (36). Carnitine measurements were performed as before (37). Serum insulin was measured using mouse insulin ELISA kits (Millipore). Serum glucose levels were determined by an oxidase-peroxidase assay (Sigma-Aldrich). Metabolic cage studies were performed in the UTSW Metabolic Core facility. CT scanning was performed as described (36).
Liver nucleotide sugar and glycan analysis.
The nucleotide sugars were analyzed as previously described (38). Briefly, the water fraction was analyzed for nucleotide sugars with AMAC labeling and fluorophore-assisted carbohydrate electrophoresis (FACE) gel. The remaining pellet was analyzed for N-glycans and O-glycans. For N-glycans, 1 U of peptide N-glycanase F (Prozyme) was added to 1 ml of pellet solution. The released N-glycans was labeled with ANDS for FACE gel analysis. The O-linked glycan was released from glycoprotein by β-elimination as described previously (39). For total O-glycan analysis, the O-glycans released above were labeled with ANDS and AMAC and resolved on FACE gel. For O-GlcNAc, O-mannose, and O-glucose analysis, the released O-glycans were further cleaned with ion exchange resin AG1-X8 to remove all the negatively charged O-glycans, then labeled with AMAC and resolved on FACE gel.
Histology.
Livers fixed in 10% neutralized formalin were processed for paraffin sections. After de-paraffinization, liver sections were incubated in antigen retrieval buffer (Dako), followed by blocking in PBS with 1% BSA and non-immune donkey antibodies. Primary antibodies used were LDL receptor (40) and GLUT2 (Chemicon). Cy3-labeled or Cy5-labeled secondary antibodies (Jackson ImmunoResearch Laboratories Inc.) were used, and slides were imaged using a TCS SP5 confocal microscope (Leica) or AxioObserver epifluorescence microscope (Zeiss). The H&E and glycogen PAS staining were performed by the Histology Core at UTSW. Both were imaged using a Coolscope (Nikon).
ChIP assay.
WT mice were fasted for 24 hours and then refed for 2 or 6 hours. Livers were processed to isolate nuclei using an NE-PER kit (Pierce). Immunoprecipitation reactions were performed using an anti-Xbp1 and control TNP sera (41). After reverse crosslinking and protease K digestion, DNA was purified with a QIAquick kit (Qiagen). Promoter occupancy of Xbp1 was analyzed by amplification of a fragment (290 bp) corresponding to Xbp1-binding element. Genomic DNA isolated before immunoprecipitation was used as input.
Primary hepatocyte and Huh7 cell experiments.
Primary rat hepatocytes were isolated as previously described (42). On day 0, isolated hepatocytes were plated and infected with adenovirus at an MOI of 10 in DMEM. On day 1, cells were treated with insulin (final concentration, 16 nM) and incubated for 10 minutes or 6 hours. Primary mouse hepatocytes were isolated from WT or LIXs animals. On day 1, tunicamycin (0.5 μg/ml) or thapsigargin (0, 100, 250 nM) was added to cells for 5 hours. Dox (0.5 μg/ml) was used to induce Xbp1s in culture. GalE siRNA (Sigma-Aldrich) was applied to primary hepatocytes using RNAiMAX reagent (Invitrogen) on day 0. On day 1, cells were serum starved for 4 hours and then stimulated by insulin and harvested for immunoblotting. Hepatocytes were supplemented with 3H-leucine (PerkinElmer) on day 0 at 0.4 μCi/ml and cultured for 24–48 hours. The proteins were precipitated and counted with a scintillation counter. The Huh7 cells were treated with tunicamycin (500 ng/ml), insulin (160 nM), or both during a 4-hour serum starvation period.
galE promoter cloning, luciferase assay, and adiponectin synthesis assay.
The mouse galE promoter (1 kb upstream of start codon) was cloned into a pGL3 vector. After 36 hours of transfection using HEK293T cells, luciferase assays were performed (Promega). β-Galactosidase expression construct was co-transfected for normalization of luciferase activity (ABI). HEK293T cells were cultured in a 6-well plate until they reached 70%–80% confluency before transfection using Trans-IT 293 reagent (Mirus) to examine the intracellular levels of adiponectin by immunoblotting.
Lentivirus generation and infection.
Xbp1s was engineered into a lentiviral expression plasmid (pCCL.PPT.hPGK.dNGFR.IRES.EGFP) and packaged into lentiviral particles. The lentivirus was concentrated using Concentrator and titrated by qRT-PCR (Clontech). Hepatocytes were infected with an MOI of 5–10 supplemented with 9 μg/ml Polybrene (Sigma-Aldrich).
GalE adenovirus generation and infection.
Mouse GalE was cloned from a fetal mouse brain cDNA library and inserted into a DUAL-CCM vector for adenovirus generation (Vector BioLabs). Virus was diluted in PBS and administrated through tail vein injection at 109 virus particles per mouse. Measurements were performed 7 days after infection.
RNA isolation, RT-PCR, qPCR, and microarray studies.
Total RNA was isolated from frozen tissues using NucleoSpin RNA II mini columns (Macherey-Nagel). cDNA was generated with iScript (Bio-Rad). Primers used are listed in Supplemental Table 2. Xbp1 PCR was done as described previously (43). 18s rRNA was used as loading control, and the expression of each gene was calculated relative to the lowest level within that data set. Microarray studies were performed with pooled samples on an Illumina mouse-6 V2 BeadChip by the Microarray Core of UTSW. Analysis was done with Partek Genomics Suite and Ingenuity Pathways Analysis, and the original microarray data have been deposited in the GEO database (GSE39301).
Immunoblot analysis.
Frozen samples were homogenized in T-PER lysis buffer (Pierce) supplemented with protease inhibitor (Roche) and phosphatase inhibitor (Sigma-Aldrich). The nuclear extract was prepared using an NE-PER kit (Pierce). An equal amount of total protein from each sample was resolved on 4%–20% Tris-glycine gels (Bio-Rad) and transferred to nitrocellulose membranes (Bio-Rad). The antibodies used were GAPDH, tubulin, actin, total AKT, Xbp1 (Santa Cruz Biotechnology Inc.), GalE (Abcam), p-AKT, lamin (Cell Signaling Technology), p-eIF2α, and total eIF2α (Invitrogen). IRDye 800– or IRDye 700–conjugated secondary antibodies (Rockland) were used, and membranes were scanned and processed using the LI-COR Odyssey infrared imaging system.
Statistics.
Data are expressed as mean ± SEM. Student’s t test (2-tailed) was used for comparisons between 2 groups. For tolerance tests, 2-way ANOVA and subsequent Tukey tests were performed. A P value less than 0.05 was considered significant.
Study approval.
The Institutional Animal Care and the Use Committee of UTSW approved all animal experiments.
Supplementary Material
Acknowledgments
We thank Bob Hammer and the Transgenic Core Facility for generation of the mouse models in this study; Quan Li and the Microarray Core Facility for the microarray studies; Lilja Kjalarsdottir for cell reporter assays; Xiaorong Fu and Shawn Burgess for the carnitine measurements; Victor Pashkov and Thomas Wilkie for help with mouse hepatocyte preparation; John Shelton and the Histology Core for assistance with histology; and the Metabolic Phenotyping Core for metabolic studies. We thank Steven Connell, Yukiko Mayauchi, Amy Song, Xinyu Wu, Steven Spurgin, and Virginia Liu for technical assistance. This study was supported by NIH grants R01-DK55758 and P01DK088761 (to P.E. Scherer), 5PLDK081182 (to J.D. Horton), and R01-GM38545 (to M.A. Lehrman) and a grant from the Welch Foundation I-1168 (to M.A. Lehrman). Y. Deng is supported by a postdoctoral fellowship from the American Diabetes Association (7-08-MN-53). Z.V. Wang is supported by a postdoctoral fellowship from the American Heart Association (10POST4320009).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(1):455–468. doi:10.1172/JCI62819.
References
- 1.Pfaffenbach KT, et al. Rapamycin inhibits postprandial-mediated X-box-binding protein-1 splicing in rat liver. J Nutr. 2010;140(5):879–884. doi: 10.3945/jn.109.119883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 3.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334(6059):1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 4.Winnay JN, Boucher J, Mori MA, Ueki K, Kahn CR. A regulatory subunit of phosphoinositide 3-kinase increases the nuclear accumulation of X-box-binding protein-1 to modulate the unfolded protein response. Nat Med. 2010;16(4):438–445. doi: 10.1038/nm.2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Park SW, et al. The regulatory subunits of PI3K, p85alpha and p85beta, interact with XBP-1 and increase its nuclear translocation. Nat Med. 2010;16(4):429–437. doi: 10.1038/nm.2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou Y, et al. Regulation of glucose homeostasis through a XBP-1-FoxO1 interaction. Nat Med. 2011;17(3):356–365. doi: 10.1038/nm.2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ozcan U, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306(5695):457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 8.Lee AH, Scapa EF, Cohen DE, Glimcher LH. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science. 2008;320(5882):1492–1496. doi: 10.1126/science.1158042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leloir LF. The enzymatic transformation of uridine diphosphate glucose into a galactose derivative. Arch Biochem Biophys. 1951;33(2):186–190. doi: 10.1016/0003-9861(51)90096-3. [DOI] [PubMed] [Google Scholar]
- 10.Hellerstein MK, Munro HN. Glycoconjugates as noninvasive probes of intrahepatic metabolism: III. Application to galactose assimilation by the intact rat. Metabolism. 1988;37(4):312–317. doi: 10.1016/0026-0495(88)90129-1. [DOI] [PubMed] [Google Scholar]
- 11.Belteki G, et al. Conditional and inducible transgene expression in mice through the combinatorial use of Cre-mediated recombination and tetracycline induction. Nucleic Acids Res. 2005;33(5):e51. doi: 10.1093/nar/gni051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23(21):7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107(7):881–891. doi: 10.1016/S0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 14.Rutkowski DT, et al. UPR pathways combine to prevent hepatic steatosis caused by ER stress-mediated suppression of transcriptional master regulators. Dev Cell. 2008;15(6):829–840. doi: 10.1016/j.devcel.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knopp RH. Drug treatment of lipid disorders. N Engl J Med. 1999;341(7):498–511. doi: 10.1056/NEJM199908123410707. [DOI] [PubMed] [Google Scholar]
- 16.Chou JY, Jun HS, Mansfield BC. Glycogen storage disease type I and G6Pase-beta deficiency: etiology and therapy. Nat Rev Endocrinol. 2010;6(12):676–688. doi: 10.1038/nrendo.2010.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fang M, et al. The ER UDPase ENTPD5 promotes protein N-glycosylation, the Warburg effect, and proliferation in the PTEN pathway. Cell. 2010;143(5):711–724. doi: 10.1016/j.cell.2010.10.010. [DOI] [PubMed] [Google Scholar]
- 18.Yamamoto K, Yoshida H, Kokame K, Kaufman RJ, Mori K. Differential contributions of ATF6 and XBP1 to the activation of endoplasmic reticulum stress-responsive cis-acting elements ERSE, UPRE and ERSE-II. J Biochem. 2004;136(3):343–350. doi: 10.1093/jb/mvh122. [DOI] [PubMed] [Google Scholar]
- 19.Kingsley DM, Krieger M, Holton JB. Structure and function of low-density-lipoprotein receptors in epimerase-deficient galactosemia. N Engl J Med. 1986;314(19):1257–1258. [PubMed] [Google Scholar]
- 20.Sriburi R, et al. Coordinate regulation of phospholipid biosynthesis and secretory pathway gene expression in XBP-1(S)-induced endoplasmic reticulum biogenesis. J Biol Chem. 2007;282(10):7024–7034. doi: 10.1074/jbc.M609490200. [DOI] [PubMed] [Google Scholar]
- 21.Jin F, Kretschmer PJ, Harkins RN, Hermiston TW. Enhanced protein production using HBV X protein (HBx), and synergy when used in combination with XBP1s in BHK21 cells. Biotechnol Bioeng. 2009;105(2):341–349. doi: 10.1002/bit.22536. [DOI] [PubMed] [Google Scholar]
- 22.Frey PA. The Leloir pathway: a mechanistic imperative for three enzymes to change the stereochemical configuration of a single carbon in galactose. FASEB J. 1996;10(4):461–470. [PubMed] [Google Scholar]
- 23.Novelli G, Reichardt JK. Molecular basis of disorders of human galactose metabolism: past, present, and future. Mol Genet Metab. 2000;71(1–2):62–65. doi: 10.1006/mgme.2000.3073. [DOI] [PubMed] [Google Scholar]
- 24.Bosch AM. Classical galactosaemia revisited. J Inherit Metab Dis. 2006;29(4):516–525. doi: 10.1007/s10545-006-0382-0. [DOI] [PubMed] [Google Scholar]
- 25.Walter JH, et al. Generalised uridine diphosphate galactose-4-epimerase deficiency. Arch Dis Child. 1999;80(4):374–376. doi: 10.1136/adc.80.4.374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanders RD, Sefton JM, Moberg KH, Fridovich-Keil JL. UDP-galactose 4’ epimerase (GALE) is essential for development of Drosophila melanogaster. Dis Model Mech. 2010;3(9–10):628–638. doi: 10.1242/dmm.005058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Timson DJ. The structural and molecular biology of type III galactosemia. IUBMB Life. 2006;58(2):83–89. doi: 10.1080/15216540600644846. [DOI] [PubMed] [Google Scholar]
- 28.Liu Y, et al. XBP1S associates with RUNX2 and regulates chondrocyte hypertrophy. J Biol Chem. 2012;287(41):34500–34513. doi: 10.1074/jbc.M112.385922. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29.Lee AS. The glucose-regulated proteins: stress induction and clinical applications. Trends Biochem Sci. 2001;26(8):504–510. doi: 10.1016/S0968-0004(01)01908-9. [DOI] [PubMed] [Google Scholar]
- 30.Csala M, Margittai E, Banhegyi G. Redox control of endoplasmic reticulum function. Antioxid Redox Signal. 2010;13(1):77–108. doi: 10.1089/ars.2009.2529. [DOI] [PubMed] [Google Scholar]
- 31.Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer. 2008;8(11):851–864. doi: 10.1038/nrc2501. [DOI] [PubMed] [Google Scholar]
- 32.Romero-Ramirez L, et al. XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res. 2004;64(17):5943–5947. doi: 10.1158/0008-5472.CAN-04-1606. [DOI] [PubMed] [Google Scholar]
- 33.Uhlen M, et al. Towards a knowledge-based Human Protein Atlas. Nat Biotechnol. 2010;28(12):1248–1250. doi: 10.1038/nbt1210-1248. [DOI] [PubMed] [Google Scholar]
- 34.Shimano H, Horton JD, Hammer RE, Shimomura I, Brown MS, Goldstein JL. Overproduction of cholesterol and fatty acids causes massive liver enlargement in transgenic mice expressing truncated SREBP-1a. J Clin Invest. 1996;98(7):1575–1584. doi: 10.1172/JCI118951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laplante M, et al. Tissue-specific postprandial clearance is the major determinant of PPARgamma-induced triglyceride lowering in the rat. Am J Physiol Regul Integr Comp Physiol. 2009;296(1):R57–R66. doi: 10.1152/ajpregu.90552.2008. [DOI] [PubMed] [Google Scholar]
- 36.Asterholm IW, Scherer PE. Enhanced metabolic flexibility associated with elevated adiponectin levels. Am J Pathol. 2010;176(3):1364–1376. doi: 10.2353/ajpath.2010.090647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sunny NE, et al. Progressive adaptation of hepatic ketogenesis in mice fed a high-fat diet. Am J Physiol Endocrinol Metab. 2010;298(6):E1226–E1235. doi: 10.1152/ajpendo.00033.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gao N, Lehrman MA. Non-radioactive analysis of lipid-linked oligosaccharide compositions by fluorophore-assisted carbohydrate electrophoresis. Methods Enzymol. 2006;415:3–20. doi: 10.1016/S0076-6879(06)15001-6. [DOI] [PubMed] [Google Scholar]
- 39.Huang Y, Mechref Y, Novotny MV. Microscale nonreductive release of O-linked glycans for subsequent analysis through MALDI mass spectrometry and capillary electrophoresis. Anal Chem. 2001;73(24):6063–6069. doi: 10.1021/ac015534c. [DOI] [PubMed] [Google Scholar]
- 40.Lagace TA, et al. Secreted PCSK9 decreases the number of LDL receptors in hepatocytes and in livers of parabiotic mice. J Clin Invest. 2006;116(11):2995–3005. doi: 10.1172/JCI29383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ferdous A, Sikder D, Gillette T, Nalley K, Kodadek T, Johnston SA. The role of the proteasomal ATPases and activator monoubiquitylation in regulating Gal4 binding to promoters. Genes Dev. 2007;21(1):112–123. doi: 10.1101/gad.1493207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li S, Brown MS, Goldstein JL. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc Natl Acad Sci U S A. 2010;107(8):3441–3446. doi: 10.1073/pnas.0914798107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iwakoshi NN, Lee AH, Vallabhajosyula P, Otipoby KL, Rajewsky K, Glimcher LH. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat Immunol. 2003;4(4):321–329. doi: 10.1038/ni907. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.