

Abstract
Ca2+-dependent signaling is highly regulated in cardiomyocytes and determines the force of cardiac muscle contraction. Ca2+ cycling refers to the release and reuptake of intracellular Ca2+ that drives muscle contraction and relaxation. In failing hearts, Ca2+ cycling is profoundly altered, resulting in impaired contractility and fatal cardiac arrhythmias. The key defects in Ca2+ cycling occur at the level of the sarcoplasmic reticulum (SR), a Ca2+ storage organelle in muscle. Defects in the regulation of Ca2+ cycling proteins including the ryanodine receptor 2, cardiac (RyR2)/Ca2+ release channel macromolecular complexes and the sarcoplasmic/endoplasmic reticulum Ca2+ ATPase 2a (SERCA2a)/phospholamban complex contribute to heart failure. RyR2s are oxidized, nitrosylated, and PKA hyperphosphorylated, resulting in “leaky” channels in failing hearts. These leaky RyR2s contribute to depletion of Ca2+ from the SR, and the leaking Ca2+ depolarizes cardiomyocytes and triggers fatal arrhythmias. SERCA2a is downregulated and phospholamban is hypophosphorylated in failing hearts, resulting in impaired SR Ca2+ reuptake that conspires with leaky RyR2 to deplete SR Ca2+. Two new therapeutic strategies for heart failure (HF) are now being tested in clinical trials: (a) fixing the leak in RyR2 channels with a novel class of Ca2+-release channel stabilizers called Rycals and (b) increasing expression of SERCA2a to improve SR Ca2+ reuptake with viral-mediated gene therapy. There are many potential opportunities for additional mechanism-based therapeutics involving the machinery that regulates Ca2+ cycling in the heart.
Excitation-contraction coupling
With each beat of the heart, Ca2+ is released from the sarcoplasmic reticulum (SR) via the ryanodine receptor 2, cardiac (RyR2), raising the cytosolic Ca2+ concentration about ten-fold (∼1 μM) and activating cardiac muscle contraction (Figure 1). The Ca2+ is then pumped back into the SR by the sarcoplasmic/endoplasmic reticulum Ca2+ ATPase 2a (SERCA2a), lowering the cytosolic Ca2+ concentration to baseline levels (∼100 nM) and causing relaxation. The Ca2+ release and reuptake cycle is initiated by the action potential, an electrical signal that depolarizes the plasma membrane and the specialized invagination called the transverse tubule (T tubule). Voltage-gated Ca2+ channels on the T tubule are activated by depolarization and allow a small amount of Ca2+ to run down its concentration gradient from mM external Ca2+ concentration to nM internal Ca2+ concentration. This entering Ca2+ binds to and activates RyR2 channels, which release Ca2+ stored at high concentration (in the millimolar range) in the SR. The Ca2+ binds to troponin C, allowing actin-myosin cross-bridging and the thick and thin filaments of the sarcomere to slide past each other, shortening the sarcomere and causing cardiac muscle contraction.
Figure 1. Ca2+ cycling in cardiomyocytes and regulation by PKA.
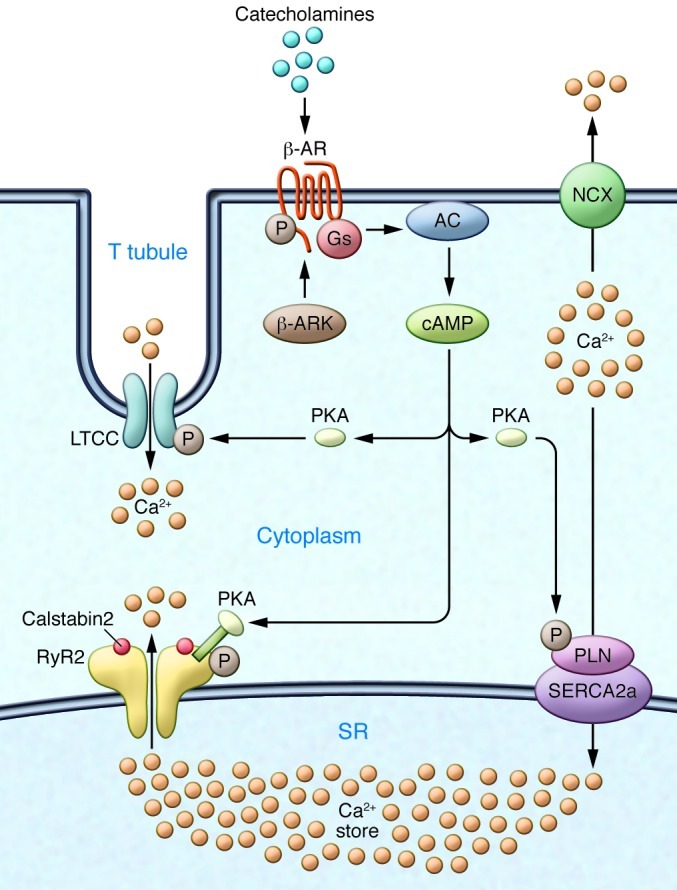
EC coupling in the heart starts with depolarization of the T tubule, which activates voltage-gated L-type Ca2+ channels (LTCCs) in the plasma membrane. Ca2+ influx via LTCCs triggers Ca2+ release from the SR via RyR2 (SR Ca2+ release channel). During systole, the free intracellular Ca2+ concentration increases ten-fold from ∼100 nM to ∼1 μM, which enables muscle contraction. The β-AR signaling pathway can increase the Ca2+ transient by activating the trigger (LTCC), release (RyR2), and uptake (SERCA/phospholamban [SERCA/PLN]) pathways. Catecholamine activation of β-ARs allows for the activation of adenylate cyclase (AC), mediated by specific G proteins (Gs), and the generation of cAMP, which in turn activates PKA. Relaxation occurs after intracellular Ca2+ is pumped out of the cytoplasm by SERCA2a, which is regulated by phospholamban. In addition, Ca2+ is extruded from the cell by the sarcolemmal NCX. RyR2 is a macromolecular complex comprised of four RyR2 monomers, PP1-spinophilin, PP2A-PR130, PKA-PDE4D3-mAKAP, calstabin2, CaMKII, and calmodulin. Calsequestrin regulates luminal SR free Ca2+, and junctin and triadin help maintain the integrity of the T tubule–SR junction. β-ARK, β-AR kinase.
Heart failure
Heart failure (HF) is the leading cause of mortality and morbidity in developed countries. The incidence of HF continues to increase after age 65, affecting nearly 1 in 100 individuals (1). This is despite substantial advances in the care of patients, brought about by coronary care units and the development of devices for the treatment of HF including biventricular pacing and left ventricular assist devices (LVADs) (2). The most common cause of HF in developed countries is atherosclerosis and concomitant ischemic heart disease. Other causes include hypertension (which leads to hypertrophy that can degenerate to dilated cardiomyopathy and HF), dilated non-ischemic cardiomyopathies, and much rarer genetic causes. While HF initially involves the myocardium, resulting in decreased cardiac performance, it rapidly affects multiple organs including, most prominently, the neurohormonal, circulatory, and renal systems. Indeed, patients with HF have chronic activation of the sympathetic nervous system, which results in a maladaptive attempt to improve cardiac function. Moreover, β-adrenergic agonists or phosphodiesterase (PDE) inhibitors do increase contractility by increasing cAMP and increasing Ca2+ release, but they also increase mortality. In fact, blocking neurohormonal pathways is the focus of current HF therapy, and while this improves survival, it is limited by side effects and the requirement to titrate drugs to physiological parameters such as heart rate (HR) and blood pressure (3). Additional current therapies are aimed at reducing the symptoms of HF (e.g., diuretics for pulmonary and peripheral congestion), but they do not inhibit HF progression.
The search for novel therapeutics for HF has led investigators to examine the mechanisms underlying HF with the hope that this approach will uncover potential therapeutic targets to slow HF progression, improve quality of life, and reduce mortality.
Much attention has been paid to understanding the role of defects in Ca2+ regulation in HF (4). This is because, as noted above, Ca2+ is the signal that regulates cardiac muscle contraction. Cardiac contractility is determined by the amplitude and kinetics of Ca2+ cycling, which in turn are regulated by phosphorylation and dephosphorylation of key proteins involved in excitation-contraction (EC) coupling by kinases and phosphatases. Stress-induced activation of G protein–coupled β-adrenergic receptors (β-ARs) activates adenylyl cyclase, which in turn leads to cAMP production and PKA activation. PKA phosphorylates many proteins in cardiac muscle, including those involved in EC coupling: the L-type Ca2+ channel, RyR2, and phospholamban (Figure 2), as well as troponin I, myosin binding protein C, and protein phosphatase inhibitor-1. PKA phosphorylation of these proteins (e.g., RyR2) results in increased HR as well as increased amplitude and velocity of Ca2+ release and reuptake and enhanced contractility (5). Whereas PKA is the “on switch” for cardiac HR and contractility, the off switches are the protein phosphatases, including PP1 and PP2A, as well as PDE4D3, which hydrolyzes cAMP (6).
Figure 2. Defective Ca2+ handling in failing hearts due to sympathetic overactivity.
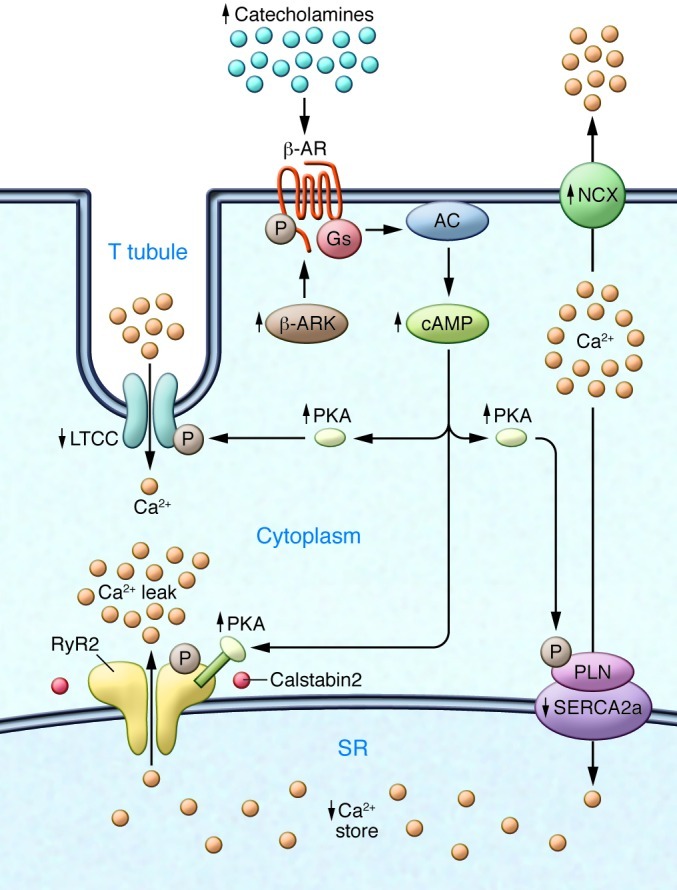
Chronic activity of the sympathetic nervous system leads to phosphorylation of the β-AR, activation of β-AR kinase, and desensitization of β-ARs. The LTCC is also phosphorylated, and NCX expression is upregulated. An important contributor to impaired Ca2+ handling in HF is PKA hyperphosphorylation of RyR2. This leads to a higher sensitivity to Ca2+-induced Ca2+ release at low cytoplasmic Ca2+ concentrations, resulting in increased RyR2 open probability at low Ca2+ concentrations and a diastolic SR Ca2+ leak. The long-term effect of the diastolic Ca2+ leak is depletion of SR Ca2+ stores. SERCA2a expression and activity are decreased in HF, which is linked to phospholamban hypophosphorylation. In contrast, NCX expression and activity are upregulated in HF. Arrows indicate increased or decreased expression or activity in HF.
It is now generally accepted that defective SR Ca2+ handling plays an important role in HF pathophysiology (7). This defective SR Ca2+ handling is characterized chiefly by leaky RyR2 channels, due to stress-induced dissociation of the stabilizing RyR2 subunit calstabin2 (also known as FKBP12.6) resulting in a diastolic SR Ca2+ leak, reduced SR Ca2+ content, and decreased Ca2+ transient (6–10). Compounding this problem is impaired SR Ca2+ uptake due to reduced activity of SERCA2a, as a consequence of both reduced SERCA2a expression and increased inhibition of the pump by phospholamban (11). Thus, these two major players in cardiac EC coupling, the SR Ca2+ release channel (RyR2) and the SR Ca2+ uptake pump (SERCA2a), conspire to deplete the SR of Ca2+ and lead to impaired cardiac contractility (4). Not surprisingly, therefore, both the RyR2 leak and the impaired uptake have been targeted with novel therapeutics, both of which are now undergoing clinical testing in HF patients.
Ca2+ signaling defects in HF: leaky RyR2 channels, decreased SR Ca2+ content, and reduced transients
Over 20 years ago, reports of reduced Ca2+ transient amplitude, increased Ca2+ transient duration, prolonged Ca2+ transient decay time, and more recently reduced SR Ca2+ load suggested that a decrease in systolic Ca2+ transient amplitude secondary to reduced SR Ca2+ stores was responsible for the decreased contractility and reduced cardiac output in HF (12). Moreover, aberrant diastolic release of SR Ca2+ can generate a transient inward current that causes delayed afterdepolarizations (DADs) (10, 13).
Impaired SERCA2a function and enhanced Na2+/Ca2+ exchanger (NCX) activity have been proposed as causes of reduced SR Ca2+ load in HF (14). However, over the past 12 years the role of a pathological diastolic SR Ca2+ leak via calstabin2-depleted leaky RyR2 has been recognized as an important contributor to altered Ca2+ handling in HF (5). In failing hearts, the RyR2-mediated Ca2+ leak is caused by the chronic hyperadrenergic state observed in patients with HF, which in turn induces chronic PKA hyperphosphorylation at Ser2808 and oxidation of RyR2, causing depletion of calstabin2 (5) from the channel complex that destabilizes the closed state of the channel (Figure 3). The term hyperphosphorylation describes RyR2 in which 3–4 of the four RyR2 monomers are chronically PKA phosphorylated. PKA-hyperphosphorylated/calstabin2-depleted channels are sensitized to cytosolic Ca2+, leading to inappropriate Ca2+ release during diastole, referred to as a diastolic SR Ca2+ leak. Ca2+ leak reduces SR Ca2+ stores and activates the transient inward current (15).
Figure 3. Dysfunctional RyR2 in failing hearts.

(A) The RyR2 macromolecular complex includes four identical RyR2 subunits (numerals 1–4 indicate the four monomers). Each RyR2 subunit binds one calstabin2 (also known as FKBP12.6) as well as mAKAP, to which PKA catalytic and regulatory subunits and PDE4D3 are bound; PP2A and its targeting protein PR130; and PP1 and its targeting protein sphinophilin (accessory molecules are only shown for one of the four RyR2 subunits, except calstabin2, which is shown for all four RyR2 subunits). The β-adrenergic signaling pathway can activate PKA through the second messenger cAMP. (B) In HF, PKA hyperphosphorylation of Ser2809, due to reduced PDE4D3, PP1, and PP2A levels in the RyR2 macromolecular complex, depletes calstabin2 from the RyR2 channel complex. The functional effect of these changes in the macromolecular composition of RyR2 is a pathological increase in Ca2+-dependent activation of RyR2 and depletion of SR Ca2+ stores, as well as functional uncoupling of RyR2 from their neighboring channels. Ca2+ leak due to abnormal RyR2 channel openings may be prevented by treatment with β-blockers (BBs), which interfere with the upstream β-AR signaling pathway, or with Rycals, which selectively increase the binding affinity of calstabin2 to PKA-phosphorylated and oxidized RyR2.
The RyR2 Ca2+ leak model of HF is supported by studies demonstrating that: (a) β-adrenergic stimulation causes depletion of calstabin2 from the RyR2 complex; (b) HF patients have PKA-hyperphosphorylated and calstabin2-depleted RyR2 (16–23); and (c) patients whose cardiac function has been normalized by treatment with LVADs have reduced levels of circulating catecholamines (24) and reduced phosphorylation of RyR2 at Ser2809 (5). Indeed, improved cardiac function in patients treated with LVAD is associated with restoration of calstabin2 binding to RyR2 (5).
The RyR2 leak is caused by stress-induced remodeling of the RyR2 macromolecular complex due to PKA hyperphosphorylation, nitrosylation, and oxidation of the channel that results in depletion of calstabin2, phosphatases (25), and PDE4D3 (6) from the RyR2 channel in HF (7). Depletion of PDE4D3 and phosphatases results in elevated levels of cAMP at RyR2 (6) and a decreased rate of dephosphorylation of a hyperphosphorylated channel, promoting further PKA hyperphosphorylation (7, 25). The depletion of the channel subunit calstabin2 from the channel results in destabilization of the channel closed state and the diastolic SR Ca2+ leak that reduce the SR Ca transient, resulting in impaired contractility and trigger fatal cardiac arrhythmias (5, 26). Importantly, the same stress-induced remodeling affects the skeletal muscle RyR1 complex and contributes to weakened skeletal muscle function and impaired exercise capacity in HF (27, 28).
The discovery of leaky RyR2 in failing hearts also provides a mechanism to explain the therapeutic efficacy of β-AR blockers in HF. Indeed, β-blockers inhibit PKA phosphorylation of Ser2808 in murine RyR2 (Ser2809 in human RyR2) by blocking β-ARs, indirectly prevent calstabin2 depletion from the RyR2 complex, and thereby reduce SR Ca2+ leak in HF patients, resulting in improved contractility and decreased arrhythmogenesis (7, 29, 30).
Multiple animal models have been used to support a role for PKA hyperphosphorylation of RyR2 in HF progression (31). Genetically altered mice harboring RyR2 that cannot be PKA phosphorylated (RyR2-S2808A) were protected against calstabin2 depletion from the RyR2 complex and HF progression 4 weeks after myocardial infarction (MI) (32). PDE4D3-deficient mice develop age-dependent cardiomyopathy and arrhythmias, RyR2 PKA hyperphosphorylation, and calstabin2 depletion. The crossing of PDE4D3-deficient mice and RyR2-S2808A mice has been shown to be protective against HF progression and arrhythmias (6). Transgenic mice expressing a mutant calstabin2-D37V, which remains bound to PKA-phosphorylated RyR2 channels, are also protected against post-MI HF (33). Calstabin2-deficient mice exhibit DADs and exercise-induced ventricular tachycardia (VT) (13), and RyR2s from calstabin2-deficient mice exhibit slightly increased open probability at baseline that increases substantially when the mice are exercised.
Arrhythmias: lessons from catecholaminergic polymorphic VT
Catecholaminergic polymorphic VT (CPVT) is a rare inherited form of exercise-induced sudden cardiac death (SCD) that occurs in individuals with structural normal hearts and normal ECGs. Mutations in RyR2 have been linked to CPVT (13, 34).
We originally reported that RyR2 CPVT mutations reduced the affinity for calstabin2, resulting in leaky channels during exercise (13). Treating mice with a Rycal (a novel class of drugs described below that prevent diastolic SR Ca2+ leak via RyR2 channels) has been shown to prevent exercise-induced depletion of calstabin2 from the RyR2 complex and reduced VT and SCD (9, 13). Calstabin2-deficient and haploinsufficient mice exhibit CPVT (13) but do not develop HF. Presumably the reason for the lack of cardiac dysfunction is that, in contrast to the post-MI model, in which calstabin2 depletion from the RyR2 complex has been shown to promote HF progression, the calstabin2-deficient mice have otherwise normal cardiac function and are able to compensate for the chronic diastolic SR Ca2+ leak in the absence of a compromised ventricle (e.g., no MI). However, PDE4D3-deficient mice that have chronic PKA hyperphosphorylation of RyR2 and RyR2-S2808D mice that mimic chronically PKA-hyperphosphorylated RyR2 both have calstabin2 depletion from the RyR2 complex, and both exhibit progressive cardiac dysfunction leading to a dilated cardiomyopathy in the absence of MI (6). This indicates that the combination of calstabin2 deficiency plus another insult (e.g., PKA hyperphosphorylation of RyR2) is sufficient to cause cardiac dysfunction in the absence of MI (7). However, the loss of calstabin2 alone, without another insult to the myocardium, can be compensated for at baseline, although this causes SCD with exercise due to leaky RyR2.
Fixing leaky RyR2 channels
Identification of the diastolic SR Ca2+ leak via RyR2 as a mechanism underlying HF progression and cardiac arrhythmias has led to novel therapeutic approaches. JTV-519 (K201), a 1,4-benzothiazepine, was noted to have effects on intracellular Ca2+ (35) and cardioprotective effects (36). However, the target was not known and there was no mechanism of action for JTV-519. Moreover, JTV-519 was found to inhibit Na+, Ca2+, and K+ currents (37, 38). Using a canine model of pacing-induced HF, Matsuzaki and colleagues reported that JTV-519 improved cardiac function (19).
Testing the drug in calstabin2-deficient mice showed that the ability of JTV-519 to prevent HF progression and fatal cardiac arrhythmias and improve skeletal muscle function requires stabilization of the closed state of RyR2 by calstabin2 (28, 39). Moreover, JTV-519 had no effect on the gating properties of normal RyR channels and no effects in healthy dogs and mice (9).
My laboratory generated many derivatives of the 1,4-benzothiazepine JTV-519 and have developed a novel class of Ca2+ release channel stabilizers known as Rycals. An orally available Rycal, S107, improves skeletal muscle force generation and exercise capacity, reduces arrhythmias, and improves muscle function in mice with Duchenne muscular dystrophy by reducing pathologic SR Ca2+ leak in cardiac and skeletal muscle (7, 9, 40–43). Rycals are protective against post-MI HF progression (7, 44) and have been shown to suppress VT/ventricular fibrillation (VT/VF) and SCD in murine models of human CPVT. S107 also raises the seizure threshold in mice with leaky neuronal RyR2 channels and improves exercise capacity in mouse models of sarcopenia (age-related loss of muscle function) (6, 9, 10, 34, 41).
Dantrolene, a drug used to prevent malignant hyperthermia in patients with mutations in RyR1 who have been exposed to volatile anesthetics, has been proposed to have therapeutic potential in heart disease (45). The Na+ channel antagonist flecainide prevents ventricular arrhythmias in patients with CPVT mutations (46). Based on single-channel data, flecainide has been proposed as an open channel blocker of RyR2. However, it is unlikely that this could be the mechanism of its anti-arrhythmic activity in CPVT, since blocking the open state of RyR2 would seriously impair cardiac contractility. Moreover, the leak that triggers the fatal ventricular arrhythmias in CPVT is a diastolic SR Ca2+ leak that occurs when the channel is supposed to be tightly shut, not open (8, 13, 39). Finally, it is more likely that some other activity of flecainide, such as its well-documented sodium channel blockade (47), is the mechanism underlying its anti-arrhythmic actions in CPVT.
Focus on the pump
SERCA activity and protein levels are decreased in failing hearts (11). Mice that are haploinsufficient for the SERCA2a gene (Atp2a2) develop accelerated HF progression compared with wild-type controls (48). However, patients with an ATP2A2 mutation and haploinsufficiency (Darier disease) do not have cardiac dysfunction (49). Nevertheless, restoration of normal levels of SERCA2a has been targeted as a novel therapeutic for HF (4). Phospholamban controls the affinity of SERCA2 for Ca2+. Unphosphorylated phospholamban inhibits SERCA2 Ca2+ affinity, and phosphorylation by PKA and Ca2+/calmodulin-dependent protein kinase II (CaMKII) relieves this inhibition and results in enhanced SR Ca2+ uptake. However, a human mutation in phospholamban results in severe dilated cardiomyopathy (50).
Increasing SR Ca2+ uptake
Both SERCA overexpression and phospholamban inhibition have been targeted as therapeutic strategies for HF. Cardiac-specific expression of SERCA2a was shown to improve contractility (51). However, results regarding arrhythmias are controversial. Chen et al. reported increased arrhythmias leading to mortality after MI in transgenic rats (52), whereas decreases in ventricular arrhythmias after postischemic injury were reported (53). Beneficial results observed in preclinical testing have led to clinical trials in patients with HF to enhance SR Ca2+ uptake, including a first-in-human gene therapy trial, Calcium Upregulation by Percutaneous administration of gene therapy In cardiac Disease (CUPID), using an adeno-associated type 1 vector carrying SERCA2a. In 39 patients with New York Heart Association class III/IV HF, treatment with the SERCA2a adenovirus resulted in improvement or stabilization in the New York Heart Association class, Minnesota Living With Heart Failure Questionnaire, 6-minute walk test, peak maximum oxygen consumption, N-terminal prohormone brain natriuretic peptide levels, and left ventricular end-systolic volume, as well as decreased frequency of cardiovascular events and duration of hospitalizations (54). Further trials are planned.
Controversies in the field
The diastolic SR Ca2+ leak model for HF and arrhythmias first proposed in 2000 (5) has generated great interest and has led to clinical trials for HF and arrhythmias as noted above. Indeed, many laboratories have examined aspects of our findings. While essentially all of our findings have been reproduced by others (16, 17, 19–23, 55–57), controversy surrounding our work has emerged over the years, with reports challenging our studies as summarized recently (58). I have previously addressed this controversy and pointed out that most of the differences are based either on opposite interpretations of the same findings or on profound differences in experimental approach (59).
We originally reported that RyR2s are PKA hyperphosphorylated in human and canine failing hearts (5) and defined PKA hyperphosphorylation as phosphorylation of 3–4 of the four PKA sites (Ser2808) in the RyR2 homotetramer (60, 61). We also reported that PKA hyperphosphorylation of RyR2 was associated with depletion of the stabilizing subunit calstabin2 from the channel (there are four calstabin2 bound to each RyR2 channel, again one per monomer; ref. 5) and that this renders the channel “leaky,” meaning that at diastolic cytosolic Ca2+ concentration (∼100 nM) when the RyR2 channel should be tightly closed, the PKA-hyperphosphorylated, calstabin2-depleted channels are not tightly closed; that is, HF RyR2 channels have increased single-channel open probability when recorded in planar lipid bilayers (5). This results in a diastolic SR Ca2+ leak that depletes SR Ca2+, contributing to impaired contractility and HF progression, and provides signals that lead to membrane depolarization and triggering of fatal ventricular arrhythmias in failing hearts (13, 34).
Alternative mechanisms have been proposed to explain SR Ca2+ leak in HF. CaMKIIδ levels are elevated in human HF (62), and there is an increase in CaMKII-dependent phosphorylation of RyR2 in HF. The observation that mice expressing the CaMKII inhibitory peptide AC3-I are protected against HF led to the proposal that CaMKII inhibitors may prevent HF progression (63). It has been reported that PKA hyperphosphorylation of RyR2 is involved in ischemic (post-MI) but not in non-ischemic (aortic banding) cardiomyopathy, and that CaMKII phosphorylation of RyR2 is the mechanism for the leak (64). The aortic banding model is a hypertrophy model in which cardiac function is initially increased and later decreases. It is possible that elevated CaMKII phosphorylation of RyR2 is associated with the hypertrophy.
Moreover, we developed genetically altered mice that harbor RyR2 that cannot be phosphorylated by CaMKII (RyR2-S2814A) and showed that they were not protected against post-MI HF progression (65). Instead, RyR2 CaMKII phosphorylation is required for the rate-related increase in contractility (65). CaMKII is Ca2+ activated and is exposed to more Ca2+ at higher HRs, resulting in increased CaMKII phosphorylation of RyR2, increased SR Ca2+ release, and increased contractility (known as the Bowditch phenomenon) (65). HR is typically elevated in HF due to increased levels of catecholamines, and thus CaMKII activation during HF is likely a result of increased HR.
We originally reported that PKA hyperphosphorylation of RyR2 causes depletion of calstabin2 from the RyR2 complex (5). We subsequently observed that oxidation and nitrosylation of RyR2 also cause depletion of calstabin2 from the RyR2 complex, and that the combination of all three deplete nearly all of the calstabin2 from the channel complex (7, 44). Some of the divergent results reported in the literature concerning the effects of PKA phosphorylation on calstabin binding to the channels could be due to variations in the state of oxidation of the channels. For example, we found that when RyR2-S2808D channels were expressed in HEK cells, there was progressive oxidation of the channel, such that the binding of calstabin2 to the channel decreased each day the transfected cells were in culture (7). This progressive oxidation is likely due to mitochondrial Ca2+ overload resulting from leaky RyR2 channels and ROS production, which oxidizes the channel (41).
Future directions: new and old pathways, additional therapeutic targets
It should be evident from Figure 1 that there are many potential therapeutic targets in the key pathways that regulate Ca2+ handling in cardiac muscle. For example, the cardiac RyR2 is a macromolecular signaling complex with multiple enzymes and targeting proteins (15), each of which should be explored as a potential cause of, and therapeutic target for, HF at the cellular, animal, and human genetic levels (8). Moreover, recent work on mechanisms of SR Ca2+ leak in different disease models has shown the critical role of nitrosylation of RyR1 in muscular dystrophy (43) and RyR2s (42) and of interactions between RyR1 and mitochondria in skeletal muscle, whereby oxidation of RyR1 causes Ca2+ overload of mitochondria and further oxidation of the channel, worsening the SR Ca2+ leak (41). These studies suggest that targeting molecules involved in oxidation/nitrosylation and mitochondria should be explored for future therapeutics.
Acknowledgments
Much of the work referred to in this Review was supported by the National Heart, Lung and Blood Institute, the Fondation Leducq, the Doris Duke Charitable Foundation, and the Ellison Foundation. The author thanks all members of the Marks laboratory past and present whose dedication and hard work have led to advances in our understanding of mechanisms and therapeutic opportunities in cardiovascular diseases.
Footnotes
Conflict of interest: Andrew R. Marks is a consultant for and owns shares in ARMGO Pharma Inc., a start-up company developing RyR-targeted therapeutics.
Citation for this article: J Clin Invest. 2013;123(1):46–52. doi:10.1172/JCI62834.
References
- 1.Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006;3(11):e442. doi: 10.1371/journal.pmed.0030442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kamouh A, Francis GS. Kamouh A, Francis GS. Contemporary management and research directions in advanced heart failure: where are we going? Congest Heart Fail. 2011;17(5):241–247. doi: 10.1111/j.1751-7133.2011.00243.x. [DOI] [PubMed] [Google Scholar]
- 3.Dube P, Weber KT. Congestive heart failure: pathophysiologic consequences of neurohormonal activation and the potential for recovery: part II. Am J Med Sci. 2011;342(6):503–506. doi: 10.1097/MAJ.0b013e3182327527. [DOI] [PubMed] [Google Scholar]
- 4.Lompre AM, Hajjar RJ, Harding SE, Kranias EG, Lohse MJ, Marks AR. Ca2+ cycling and new therapeutic approaches for heart failure. Circulation. 2010;121(6):822–830. doi: 10.1161/CIRCULATIONAHA.109.890954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marx SO, et al. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101(4):365–376. doi: 10.1016/S0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 6.Lehnart SE, et al. Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell. 2005;123(1):25–35. doi: 10.1016/j.cell.2005.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shan J, et al. Role of chronic ryanodine receptor phosphorylation in heart failure and beta-adrenergic receptor blockade in mice. J Clin Invest. 2010;120(12):4375–4387. doi: 10.1172/JCI37649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Andersson DC, Marks AR. Fixing ryanodine receptor Ca leak - a novel therapeutic strategy for contractile failure in heart and skeletal muscle. Drug Discov Today Dis Mech. 2010;7(2):e151–e157. doi: 10.1016/j.ddmec.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lehnart SE, et al. Leaky Ca2+ release channel/ryanodine receptor 2 causes seizures and sudden cardiac death in mice. J Clin Invest. 2008;118(6):2230–2245. doi: 10.1172/JCI35346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lehnart SE, et al. Stabilization of cardiac ryanodine receptor prevents intracellular calcium leak and arrhythmias. Proc Natl Acad Sci U S A. 2006;103(20):7906–7910. doi: 10.1073/pnas.0602133103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arai M, Alpert NR, MacLennan DH, Barton P, Periasamy M. Alterations in sarcoplasmic reticulum gene expression in human heart failure. A possible mechanism for alterations in systolic and diastolic properties of the failing myocardium. Circ Res. 1993;72(2):463–469. doi: 10.1161/01.RES.72.2.463. [DOI] [PubMed] [Google Scholar]
- 12.Morgan JP, Erny RE, Allen PD, Grossman W, Gwathmey JK. Abnormal intracellular calcium handling, a major cause of systolic and diastolic dysfunction in ventricular myocardium from patients with heart failure. Circulation. 1990;81(2 suppl):III21–III32. [PubMed] [Google Scholar]
- 13.Wehrens XH, et al. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell. 2003;113(7):829–840. doi: 10.1016/S0092-8674(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 14.Hobai IA, O’Rourke B. Decreased sarcoplasmic reticulum calcium content is responsible for defective excitation-contraction coupling in canine heart failure. Circulation. 2001;103(11):1577–1584. doi: 10.1161/01.CIR.103.11.1577. [DOI] [PubMed] [Google Scholar]
- 15.Kushnir A, Marks AR. The ryanodine receptor in cardiac physiology and disease. Adv Pharmacol. 2010;59:1–30. doi: 10.1016/S1054-3589(10)59001-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ogrodnik J, Niggli E. Increased Ca(2+) leak and spatiotemporal coherence of Ca(2+) release in cardiomyocytes during beta-adrenergic stimulation. J Physiol. 2010;588(pt 1):225–242. doi: 10.1113/jphysiol.2009.181800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morimoto S, et al. Protein kinase A-dependent phosphorylation of ryanodine receptors increases Ca2+ leak in mouse heart. Biochem Biophys Res Commun. 2009;390(1):87–92. doi: 10.1016/j.bbrc.2009.09.071. [DOI] [PubMed] [Google Scholar]
- 18.Blayney LM, Jones JL, Griffiths J, Lai FA. A mechanism of ryanodine receptor modulation by FKBP12/12.6, protein kinase A, and K201. Cardiovasc Res. 2010;85(1):68–78. doi: 10.1093/cvr/cvp273. [DOI] [PubMed] [Google Scholar]
- 19.Yano M, et al. FKBP12.6-mediated stabilization of calcium-release channel (ryanodine receptor) as a novel therapeutic strategy against heart failure. Circulation. 2003;107(3):477–484. doi: 10.1161/01.CIR.0000044917.74408.BE. [DOI] [PubMed] [Google Scholar]
- 20.George CH, Higgs GV, Lai FA. Ryanodine receptor mutations associated with stress-induced ventricular tachycardia mediate increased calcium release in stimulated cardiomyocytes. Circ Res. 2003;93(6):531–540. doi: 10.1161/01.RES.0000091335.07574.86. [DOI] [PubMed] [Google Scholar]
- 21.Yano M, et al. Altered stoichiometry of FKBP12.6 versus ryanodine receptor as a cause of abnormal Ca(2+) leak through ryanodine receptor in heart failure. Circulation. 2000;102(17):2131–2136. doi: 10.1161/01.CIR.102.17.2131. [DOI] [PubMed] [Google Scholar]
- 22.Ono K, et al. Altered interaction of FKBP12.6 with ryanodine receptor as a cause of abnormal Ca(2+) release in heart failure. Cardiovasc Res. 2000;48(2):323–331. doi: 10.1016/S0008-6363(00)00191-7. [DOI] [PubMed] [Google Scholar]
- 23.Doi M, et al. Propranolol prevents the development of heart failure by restoring FKBP12.6-mediated stabilization of ryanodine receptor. Circulation. 2002;105(11):1374–1379. doi: 10.1161/hc1102.105270. [DOI] [PubMed] [Google Scholar]
- 24.Estrada-Quintero T, Uretsky BF, Murali S, Griffith BP, Kormos RL. Neurohormonal activation and exercise function in patients with severe heart failure and patients with left ventricular assist system. A comparative study. Chest. 1995;107(6):1499–1503. doi: 10.1378/chest.107.6.1499. [DOI] [PubMed] [Google Scholar]
- 25.Reiken S, et al. Protein kinase A phosphorylation of the cardiac calcium release channel (ryanodine receptor) in normal and failing hearts. Role of phosphatases and response to isoproterenol. J Biol Chem. 2003;278(1):444–453. doi: 10.1074/jbc.M207028200. [DOI] [PubMed] [Google Scholar]
- 26.Brillantes AB, et al. Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein. Cell. 1994;77(4):513–523. doi: 10.1016/0092-8674(94)90214-3. [DOI] [PubMed] [Google Scholar]
- 27.Reiken S, et al. PKA phosphorylation activates the calcium release channel (ryanodine receptor) in skeletal muscle: defective regulation in heart failure. J Cell Biol. 2003;160(6):919–928. doi: 10.1083/jcb.200211012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wehrens XH, et al. Enhancing calstabin binding to ryanodine receptors improves cardiac and skeletal muscle function in heart failure. Proc Natl Acad Sci U S A. 2005;102(27):9607–9612. doi: 10.1073/pnas.0500353102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reiken S, et al. Beta-blockers restore calcium release channel function and improve cardiac muscle performance in human heart failure. Circulation. 2003;107(19):2459–2466. doi: 10.1161/01.CIR.0000068316.53218.49. [DOI] [PubMed] [Google Scholar]
- 30.Reiken S, et al. beta-adrenergic receptor blockers restore cardiac calcium release channel (ryanodine receptor) structure and function in heart failure. Circulation. 2001;104(23):2843–2848. doi: 10.1161/hc4701.099578. [DOI] [PubMed] [Google Scholar]
- 31.Kushnir A, Betzenhauser MJ, Marks AR. Ryanodine receptor studies using genetically engineered mice. FEBS Lett. 2010;584(10):1956–1965. doi: 10.1016/j.febslet.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wehrens XH, Lehnart SE, Reiken S, Vest JA, Wronska A, Marks AR. Ryanodine receptor/calcium release channel PKA phosphorylation: a critical mediator of heart failure progression. Proc Natl Acad Sci U S A. 2006;103(3):511–518. doi: 10.1073/pnas.0510113103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang F, Shan J, Reiken S, Wehrens XH, Marks AR. Analysis of calstabin2 (FKBP12.6)-ryanodine receptor interactions: rescue of heart failure by calstabin2 in mice. Proc Natl Acad Sci U S A. 2006;103(9):3456–3461. doi: 10.1073/pnas.0511282103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lehnart SE, et al. Sudden death in familial polymorphic ventricular tachycardia associated with calcium release channel (ryanodine receptor) leak. Circulation. 2004;109(25):3208–3214. doi: 10.1161/01.CIR.0000132472.98675.EC. [DOI] [PubMed] [Google Scholar]
- 35.Kaneko N, Matsuda R, Toda M, Shimamoto K. Inhibition of annexin V-dependent Ca2+ movement in large unilamellar vesicles by K201, a new 1,4-benzothiazepine derivative. Biochim Biophys Acta. 1997;1330(1):1–7. doi: 10.1016/s0005-2736(97)00132-6. [DOI] [PubMed] [Google Scholar]
- 36.Ito K, Shigematsu S, Sato T, Abe T, Li Y, Arita M. JTV-519, a novel cardioprotective agent, improves the contractile recovery after ischaemia-reperfusion in coronary perfused guinea-pig ventricular muscles. Br J Pharmacol. 2000;130(4):767–776. doi: 10.1038/sj.bjp.0703373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kiriyama K, Kiyosue T, Wang JC, Dohi K, Arita M. Effects of JTV-519, a novel anti-ischaemic drug, on the delayed rectifier K+ current in guinea-pig ventricular myocytes. Naunyn Schmiedebergs Arch Pharmacol. 2000;361(6):646–653. doi: 10.1007/s002100000230. [DOI] [PubMed] [Google Scholar]
- 38.Kimura J, Kawahara M, Sakai E, Yatabe J, Nakanishi H. Effects of a novel cardioprotective drug, JTV-519, on membrane currents of guinea pig ventricular myocytes. Jpn J Pharmacol. 1999;79(3):275–281. doi: 10.1254/jjp.79.275. [DOI] [PubMed] [Google Scholar]
- 39.Wehrens XH, et al. Protection from cardiac arrhythmia through ryanodine receptor-stabilizing protein calstabin2. Science. 2004;304(5668):292–296. doi: 10.1126/science.1094301. [DOI] [PubMed] [Google Scholar]
- 40.Fauconnier J, et al. Ryanodine receptor leak mediated by caspase-8 activation leads to left ventricular injury after myocardial ischemia-reperfusion. Proc Natl Acad Sci U S A. 2011;108(32):13258–13263. doi: 10.1073/pnas.1100286108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Andersson DC, et al. Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging. Cell Metab. 2011;14(2):196–207. doi: 10.1016/j.cmet.2011.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fauconnier J, et al. Leaky RyR2 trigger ventricular arrhythmias in Duchenne muscular dystrophy. Proc Natl Acad Sci U S A. 2010;107(4):1559–1564. doi: 10.1073/pnas.0908540107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bellinger AM, et al. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat Med. 2009;15(3):325–330. doi: 10.1038/nm.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shan J, et al. Phosphorylation of the ryanodine receptor mediates the cardiac fight or flight response in mice. J Clin Invest. 2010;120(12):4388–4398. doi: 10.1172/JCI32726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kobayashi S, et al. Dantrolene, a therapeutic agent for malignant hyperthermia, inhibits catecholaminergic polymorphic ventricular tachycardia in a RyR2(R2474S/+) knock-in mouse model. Circ J. 2010;74(12):2579–2584. doi: 10.1253/circj.CJ-10-0680. [DOI] [PubMed] [Google Scholar]
- 46.van der Werf C, et al. Flecainide therapy reduces exercise-induced ventricular arrhythmias in patients with catecholaminergic polymorphic ventricular tachycardia. J Am Coll Cardiol. 2011;57(22):2244–2254. doi: 10.1016/j.jacc.2011.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sheets MF, Fozzard HA, Lipkind GM, Hanck DA. Sodium channel molecular conformations and antiarrhythmic drug affinity. Trends Cardiovasc Med. 2010;20(1):16–21. doi: 10.1016/j.tcm.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schultz Jel J, et al. Accelerated onset of heart failure in mice during pressure overload with chronically decreased SERCA2 calcium pump activity. Am J Physiol Heart Circ Physiol. 2004;286(3):H1146–H1153. doi: 10.1152/ajpheart.00720.2003. [DOI] [PubMed] [Google Scholar]
- 49.Tavadia S, Authi KS, Hodgins MB, Munro CS. Expression of the sarco/endoplasmic reticulum calcium ATPase type 2 and 3 isoforms in normal skin and Darier’s disease. Br J Dermatol. 2004;151(2):440–445. doi: 10.1111/j.1365-2133.2004.06130.x. [DOI] [PubMed] [Google Scholar]
- 50.Haghighi K, et al. Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J Clin Invest. 2003;111(6):869–876. doi: 10.1172/JCI17892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baker DL, et al. Targeted overexpression of the sarcoplasmic reticulum Ca2+-ATPase increases cardiac contractility in transgenic mouse hearts. Circ Res. 1998;83(12):1205–1214. doi: 10.1161/01.RES.83.12.1205. [DOI] [PubMed] [Google Scholar]
- 52.Chen Y, et al. Constitutive cardiac overexpression of sarcoplasmic/endoplasmic reticulum Ca2+-ATPase delays myocardial failure after myocardial infarction in rats at a cost of increased acute arrhythmias. Circulation. 2004;109(15):1898–1903. doi: 10.1161/01.CIR.0000124230.60028.42. [DOI] [PubMed] [Google Scholar]
- 53.del Monte F, et al. Abrogation of ventricular arrhythmias in a model of ischemia and reperfusion by targeting myocardial calcium cycling. Proc Natl Acad Sci U S A. 2004;101(15):5622–5627. doi: 10.1073/pnas.0305778101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jessup M, et al. Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID): a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation. 2011;124(3):304–313. doi: 10.1161/CIRCULATIONAHA.111.022889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kohno M, et al. A new cardioprotective agent, JTV519, improves defective channel gating of ryanodine receptor in heart failure. Am J Physiol Heart Circ Physiol. 2003;284(3):H1035–H1042. doi: 10.1152/ajpheart.00722.2002. [DOI] [PubMed] [Google Scholar]
- 56.Antos CL, et al. Dilated cardiomyopathy and sudden death resulting from constitutive activation of protein kinase a. Circ Res. 2001;89(11):997–1004. doi: 10.1161/hh2301.100003. [DOI] [PubMed] [Google Scholar]
- 57.George CH, Higgs GV, Mackrill JJ, Lai FA. Dysregulated ryanodine receptors mediate cellular toxicity: restoration of normal phenotype by FKBP12.6. J Biol Chem. 2003;278(31):28856–28864. doi: 10.1074/jbc.M212440200. [DOI] [PubMed] [Google Scholar]
- 58.Bers DM. Ryanodine receptor s2808 phosphorylation in heart failure: smoking gun or red herring. Circ Res. 2012;110(6):796–799. doi: 10.1161/CIRCRESAHA.112.265579. [DOI] [PubMed] [Google Scholar]
- 59.Marks AR. A guide for the perplexed: towards an understanding of the molecular basis of heart failure. Circulation. 2003;107(11):1456–1459. doi: 10.1161/01.CIR.0000059745.95643.83. [DOI] [PubMed] [Google Scholar]
- 60.Timerman AP, et al. Selective binding of FKBP12.6 by the cardiac ryanodine receptor. J Biol Chem. 1996;271(34):20385–20391. doi: 10.1074/jbc.271.34.20385. [DOI] [PubMed] [Google Scholar]
- 61.Timerman AP, Jayaraman T, Wiederrecht G, Onoue H, Marks AR, Fleischer S. The ryanodine receptor from canine heart sarcoplasmic reticulum is associated with a novel FK-506 binding protein. Biochem Biophys Res Commun. 1994;198(2):701–706. doi: 10.1006/bbrc.1994.1101. [DOI] [PubMed] [Google Scholar]
- 62.Hoch B, et al. Differentiation-dependent expression of cardiac delta-CaMKII isoforms. J Cell Biochem. 1998;68(2):259–268. doi: 10.1002/(SICI)1097-4644(19980201)68:2<259::AID-JCB12>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 63.Ling H, et al. Requirement for Ca2+/calmodulin-dependent kinase II in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. J Clin Invest. 2009;119(5):1230–1240. doi: 10.1172/JCI38022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Respress JL, et al. Role of RyR2 phosphorylation at S2814 during heart failure progression. Circ Res. 2012;110(11):1474–1483. doi: 10.1161/CIRCRESAHA.112.268094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kushnir A, Shan J, Betzenhauser MJ, Reiken S, Marks AR. Role of CaMKIIdelta phosphorylation of the cardiac ryanodine receptor in the force frequency relationship and heart failure. Proc Natl Acad Sci U S A. 2010;107(22):10274–10279. doi: 10.1073/pnas.1005843107. [DOI] [PMC free article] [PubMed] [Google Scholar]