

Abstract
The discovery of the genetic basis of inherited arrhythmias has paved the way for an improved understanding of arrhythmogenesis in a wide spectrum of life-threatening conditions. In vitro expression of mutations and transgenic animal models have been instrumental in enhancing this understanding, but the applicability of results to the human heart remains unknown. The ability to differentiate induced pluripotent stem cells (iPSs) into cardiomyocytes enables the potential to generate patient-specific myocytes, which could be used to recapitulate the features of inherited arrhythmias in the context of the patient’s genetic background. Few studies have been reported on iPS-derived myocytes obtained from patients with heritable arrhythmias, but they have demonstrated the applicability of this innovative approach to the study of inherited arrhythmias. Here we review the results achieved by iPS investigations in arrhythmogenic syndromes and discuss the existing challenges to be addressed before the use of iPS-derived myocytes can become a part of personalized management of inherited arrhythmias.
Introduction
The discovery of genes responsible for inherited arrhythmogenic syndromes has provided major insights in the pathophysiology of cardiac arrhythmias (1). In the last 20 years, the electrophysiological mechanisms leading to sudden cardiac death in diseases such as long QT syndrome (LQTS), Brugada syndrome, and catecholaminergic polymorphic ventricular tachycardia (CPVT) have been dissected, thanks to the in vitro characterization of human mutations in heterologous systems and the development of animal models (2, 3). However, animal models of inherited arrhythmias only approximate human mutations because of species differences in the electrophysiological properties of the heart, thus limiting our understanding of pathogenesis and the development of targeted therapies. The heart rate of a mouse is around 600 beats per minute, ten times faster than that of human beings; as a consequence, the duration of the action potential is much shorter and ion channels have different properties. Although mutations inserted into the genome of heart cells of animals recapitulate some of the properties seen in affected patients (4, 5), not all observations in these models are applicable to humans. Though access to affected human cardiac tissue or cells would be ideal, until recently, deriving human myocytes from heart biopsies has been a very complex exercise.
One challenging aspect in understanding inherited arrhythmias is the evidence that the same genetic mutations may give rise to remarkably different clinical phenotypes in different family members. Usually referred to as “variable penetrance of the disease,” this observation has been attributed to largely undefined genetic modifiers (6, 7). Studies of induced pluripotent stem cell–derived (iPS-derived) cells might shed light on the elusive role of genetic modifiers in arrhythmias (discussed in detail below).
Studies of patient-derived cardiomyocytes from differentiated pluripotent stem cells could potentially provide a fascinating opportunity to make a quantum leap in the field (8). In the near future, skin cells from an individual affected by an inherited arrhythmic disease might be differentiated into cardiac cells that carry the genetic defect of the patient. The disease model thus obtained may be used to gain insight into the pathophysiology of the disease and identify targeted therapies for the individual patient, which is the ideal goal of personalized medicine.
Despite the appeal and technical feasibility of this concept, initial data have shown that the unique complexity of cardiac excitability provides several challenges, and technical advances are required before the methods can be applied to the clinical setting.
In this review, we present the state of the art in using iPSs for the study of cardiac electrophysiology, highlighting the results achieved thus far and envisioning the promises and challenges of the field.
iPSs
Pluripotent stem cells are characterized by self-renewal and the capacity for differentiation into all somatic cell lineages (9). ES cells (ESCs), obtained from the inner cell mass of mouse embryos, were the first pluripotent cells isolated (10). ESCs have also been obtained from human embryos (11).
In addition to embryonic cells, differentiated somatic cells can be pluripotent with adequate reprogramming. As early as 1962, Gurdon demonstrated that nuclear transplantation of somatic nuclei into eggs could generate animals (12). More recently, Takahashi and Yamanaka found that mouse somatic cells could be reprogrammed into a pluripotent state through the overexpression of four transcription factors, OCT4, SOX2, KLF4, and MYC; the cells obtained in this way were termed iPSs (ref. 13 and Figure 1). The four pluripotency factors initiate poorly defined epigenetic events that reactivate the endogenous pluripotency genes Oct4, Nanog, and Sox2 and promote an autoregulatory loop that maintains the pluripotent state independent of the transgenes (13). iPSs were later generated from adult human fibroblasts by defined factors (14).
Figure 1. iPS generation, characterization, and derivation of beating cardiomyocytes.

(A) iPS generation strategy. Skin fibroblasts obtained from patients are expanded and transduced with a combination of embryonic genes (Oct4, Sox2, Klf4, Nanog, Linn28, and Myc). Transduced fibroblasts are maintained in an embryonic-specific medium supplemented with bFGF until iPS colonies appear. Clones are isolated manually based on their ESC-like morphology and expanded for further characterization. (B) iPS characterization. Pluripotency marker expression (left) and developmental competence (right) should be evaluated to prove pluripotency of derived iPS lines. Analysis of the expression of typical markers of pluripotency in iPS progenitors includes expression of the “stemness” markers alkaline phosphatase, SSEA4, TRA-1-80, and OCT4 using FACS and immunofluorescence. Developmental competence is assessed both in vivo and in vitro. For in vivo analysis, teratoma formation assay is used: bona fide iPSs, when injected subcutaneously into immunodeficient mice, can give rise to a teratoma containing the three germ layers (ectoderm, mesoderm, and endoderm). In vitro, iPSs are induced to differentiate through EB aggregation and give rise to cells of all three germ layers. Neuronal (β3-tubulin positive) and intestinal (AFP positive) markers indicate differentiation to ectodermal and endodermal lineages, respectively. (C) Generation of iPS-derived beating cardiomyocytes. Cardiac differentiation is induced by aggregation of EBs. The inductive process is enhanced in the presence of specific medium and ascorbic acid. Appearance of beating areas within the EBs is indicative of cardiomyocyte differentiation; cells are dissociated into single cells, which are analyzed for cardiac markers and electrophysiology.
Differentiation of pluripotent cells toward cardiomyogenic lineages
The differentiation of cardiac cells from ESCs mimics embryonic processes specifying the cardiovascular lineage and involves a transition through a sequence of increasingly restricted progenitor cells. In vitro, differentiation can be induced by suspending ESCs in medium with high levels of fetal calf serum to obtain three-dimensional aggregates known as embryoid bodies (EBs) (15). Differentiated cell types of all three germ layers can be obtained by allowing the EBs to adhere to the culture substrate. However, this method is complex, time consuming, and above all can be poorly cardiogenic, with rhythmically contracting outgrowths in the 5%–15% range (16).
One serum-free, EB-based suspension technique has proven particularly effective for analyzing growth factor variables and timing during cardiac differentiation. In this protocol, EBs are formed in the presence of a low level of bone morphogenetic protein 4 (BMP4) and then exposed to optimized levels of BMP4, FGF2, and activin A between days 1 and 4; VEGFA and dickkopf 1 (DKK1) from days 4 to 8; and VEGF and FGF2 from day 8 onward (17)
A simpler, monolayer-based method has also been developed, in which human pluripotent stem cells are cultured to a high density and then treated with a high dose of activin A, followed by 4 days of exposure to BMP4 (18). Contracting cardiomyocytes can be seen at day 12, with a purity of approximately 30%.
These reports indicate that correct dosing and timing of the right combination of growth factors has the utmost importance in inducing an efficient differentiation process. Much work is therefore focused on transferring developmental biology knowledge to approaches to derive cardiomyocytes from pluripotent cells and to study the differentiated cells produced.
Interestingly, recent data show that it is possible to knock out pathogenic genes in patient-derived cells after genetic modification. This application of gene editing allows for the development of “isogenic controls” that may be used to define the effect of a mutation in the specific genetic environment of a given patient. Pioneers of this field have been Soldner et al. (19) and Liu et al. (20), who successfully restored the wild-type coding sequence in mutant patient-derived cells to correct phenotypes of Parkinson’s disease and progeria, respectively. Applying this technology to inherited arrhythmias may be particularly promising, allowing for a comparison of the phenotype of disease-carrying iPS-derived myocytes and their isogenic control myocytes.
Do iPS-derived myocytes reliably model the complexity of the human heart?
The electrical impulse is generated and propagated through a heterogeneous pool of cells in the heart. Each cell type (e.g., pacemaker cell, atrioventricular nodal cell, Purkinje fiber, atrial myocyte, ventricular myocyte) is characterized by a distinctive expression pattern of genes encoding a variety of ion channels that determine their specific electrophysiological profile. These electrophysiological properties of the heart develop during embryogenesis and further mature postnatally; accordingly, embryonic and fetal cells do not exhibit the same electrical behavior seen in adult myocytes (21).
The architecture of the electrical structure of the heart is highly sophisticated; even within a specific cell type, such as the ventricular cells, regional differences exist in the cellular electrophysiological profile. Transmural sections of the ventricular wall are composed of different types of cells, such as epicardial, mid-myocardial, and endocardial, that have well-documented differences in electrical properties, as seen by the variable shape and duration of their action potentials (22). Similar differences exist between cells in the apex and the base of the heart and in the right versus the left ventricles (23). Even within the right ventricle, distinct electrical properties are present. Compared with other ventricular regions, the outflow tract presents a delayed expression of proteins during development that are responsible for rapid conduction of the cardiac impulse, such as connexin 43 (Cx43) and Cx40 (24). During postnatal maturation, this region of the right heart retains some slow conduction properties that may facilitate arrhythmogenesis (21, 24). This observation suggests that mutations in inherited arrhythmias may differentially affect cellular electrophysiology in the various heart cell types. It also raises the question whether iPS-derived myocytes are appropriate to study region-specific genetic conditions such as Brugada syndrome, in which the RV outflow tract is the preferred arrhythmogenic site. Consequently, for iPS-derived myocytes to reliably model patient arrhythmogenesis, the differentiated cells must replicate the complexity of electrical heterogeneity in the human heart.
Action potential morphologies in iPS-derived myocytes
Electrical characterization of iPS-derived myocytes showed that the differentiation process of cardiac cells reflects, at least to some extent, the heterogeneity seen in the heart. Zhang et al. (25) performed a systematic study of the electrical properties of iPS-derived cardiomyocytes using microelectrode recordings from spontaneously beating cells at 56–70 days after the formation of the EBs. These experiments identified three types of cardiac cells resembling atrial, ventricular, and nodal cells (i.e., cells from the AV node). As expected, the distinguishing feature of atrial cells was the reduced duration of the action potential and the lack of a plateau phase, whereas nodal cells were recognized by their pronounced phase 4 depolarization preceding the onset of the action potential; ventricular cells presented the typical plateau phase and, therefore, had the longest action potential duration (APD). A striking feature of the published electrophysiology data on iPS-derived myocytes is the impressive heterogeneity in the electrical parameters, such as the duration and amplitude of the action potential and the resting membrane potential. This heterogeneity suggests that cells obtained by different laboratories may not be similar and raises the question whether results are reproducible across research groups. A comparison of the electrophysiological properties of iPS-derived cardiomyocytes from published articles is summarized in Table 1. In these published studies, APDs at 50% and 90% of the amplitude (APD50 and APD90, respectively), for example, are very different even when the pacing rate is identical. These inconsistencies may be related to the different times elapsed between EB formation and the electrophysiological evaluation (Table 1). Differentiation protocols may also account for the development of cells at variable stage of maturation. Malan et al. circumvent the problem of APD variability by studying the effect of pacing or drugs within a single cell in iPS-derived myocytes from genetically modified mice (26); this longitudinal approach could also be tested in human studies.
Table 1.
Summary of studies of iPS-derived cardiomyocytes from patients with inherited arrhythmias
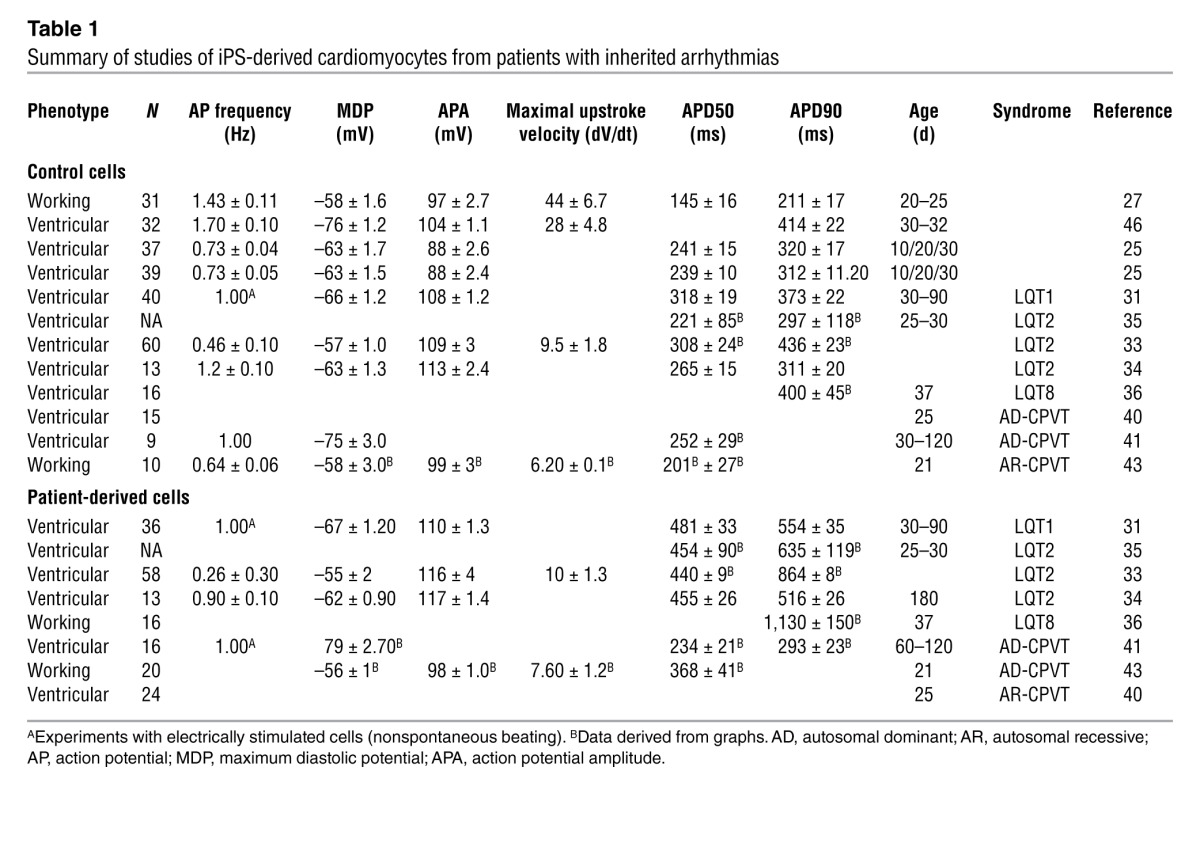
The key question that emerges is whether differentiated myocytes obtained with available protocols provide a reproducible model for the adult human heart and, therefore, whether this model is applicable to the clinical setting. Based on the published data, iPS-derived myocytes appear to lack the distinction between epicardial and endocardial cells that occurs in adult mammalian ventricular myocytes. According to some authors, even the distinction between atrial-like and ventricular-like myocytes is blurred by the existence of cells with an intermediate phenotype (27). Thus, some authors prefer to use the phrase “working myocardial cells” to cluster atrial-like and ventricular-like cells (see Table 1) and distinguish them from nodal cells (i.e., cells from the AV node). These observations point to the evidence that iPS-derived myocytes are at an early stage of differentiation with a continuous spectrum of electrical properties, resulting in heterogeneity. As discussed below, this observation is further supported by morphological evaluation of iPS-derived cardiac cells.
Ultrastructural properties of iPS-derived myocytes
An interesting study by Gherghiceanu et al. (28) reports important information on the ultrastructural characteristics of iPS-derived myocytes and compares them to human ESC-derived cardiomyocytes. Interestingly, both cell types presented features of immature cells: large glycogen deposits, lipid droplets, large endoplasmic reticulum, rough endoplasmic reticulum, and an immature sarcoplasmic reticulum (SR). The latter feature is more accentuated in iPS-derived myocytes. Remarkably, iPS-derived myocytes lack gap junctions, intercalated disks, and T tubules (despite the presence of caveolae, considered precursors to T tubules) (28). However, studies in cardiomyocytes derived from human ESCs have demonstrated the presence of gap junctions (29). In examining the entire EB, a progression of cell maturation was found — embryonic myocytes were present in the center of the EB, whereas cells located at the periphery of the EB were similar to fetal myocytes (29).
Modeling LQTS with iPS-derived myocytes
Although we have highlighted that iPS-derived myocytes obtained with the currently available differentiation protocols do not resemble adult myocytes, several studies have already characterized iPS-derived myocytes from patients with inherited arrhythmias, providing intriguing results (Table 1).
LQTS is an inherited disease characterized by genetically determined prolongation of cardiac repolarization that is quantified as the duration of the QT interval on the ECG (30). The disease predisposes the heart to malignant ventricular tachyarrhythmia that may lead to sudden death. Most of the arrhythmic events in patients are precipitated by stress, emotion, or physical activity. Patients are usually treated with antiadrenergic therapy (β-blockers). The genes that cause LQTS encode either for subunits that form cardiac ion channels or for proteins that regulate the assembly and the localization of ion channels. The common functional consequence of mutations found in LQTS-related genes is the prolongation of cardiac action potential and induction of early afterdepolarizations (EADs) that result in the development of a specific type of ventricular tachycardia called Torsade des pointes (Figure 1).
Moretti and coworkers (31) characterized for the first time iPS-derived myocytes from an LQTS patient carrying the R190W mutation in KCNQ1, which encodes the potassium channel that conducts the slow component of the delayed rectifier current (IKs) (Figure 2). Electrophysiological recording in iPS-derived myocytes has shown that (a) the R190W mutation prolongs the APD, (b) the IKs current in R190W myocytes is dramatically reduced, possibly because of a trafficking defect, and (c) adrenergic stimulation facilitates development of EADs in R190W myocytes, whereas β-blockers revert the arrhythmogenic effect of β-adrenergic stimulation. These observations showed that iPS-derived cells replicate the clinical features of the disease (QTc prolongation, proarrhythmic effect of adrenergic activation, and protective action of β-blockers) and reproduce results of functional characterization of the R190W mutation in heterologous systems (reduction the IKs current and trafficking defect) (32).
Figure 2. Common mutations in LQTS.

(A and B) Predicted transmembrane topology of KCNH2 and KCNQ1 proteins and the three LQT2 mutations (A) and one LQT1 mutation (B) studied in cardiomyocytes differentiated from iPSs obtained from tissues of affected patients. (C–E) Example of EADs and prolonged action potential (C), underlying QT prolongation with T wave morphological abnormalities (D), and polymorphic ventricular tachycardia (Torsade de Pointes) (E) observed in patients with LQTS.
Three additional reports investigated the properties of iPS-derived myocytes of LQTS patients carrying a mutation in the KCNH2 gene, which encodes for the channel that conducts the rapid component of the delayed rectifier current (IKr) (33–35). Studies by Itzhaki et al. (33) and Matsa et al. (35) characterized cells derived from carriers of mutations localized in the pore region of the HERG channel (A561T and A614V), known to cause a severe form of LQTS. As expected, both studies showed that the iPS-derived myocytes from affected individuals have prolonged APD and a decreased IKr current. Moreover, pharmacological challenges showed that the IKr blocking compound E4031 further prolonged repolarization in iPSs from LQTS patients and induced or worsened the EADs. In a related study, Lahti et al. (34) characterized iPS-derived myocytes from a carrier of a mild missense mutation in the KCNH2 gene. The mutation causes the replacement of an arginine residue at position 176, corresponding to the cytoplasmic N terminus of the channel (R176W). The patient that donated a skin biopsy for the research protocol was asymptomatic and presented a normal QT interval (QTc = 437 ms). Surprisingly, however, the myocytes derived from the patient fibroblasts presented a marked prolongation of the APD90 that was approximately 170% that of the APD of cells derived from a control individual (Table 1; APD90 in the R176W carrier, 516 ± 26 ms; APD90 in a control individual, 310.5 ± 19.6 ms; data at 1 Hz pacing; P < 0.01). The discrepancy between the normal QTc of the patient and the marked prolongation of APD in iPS-derived myocytes is hard to explain and questions the ability of iPS-derived myocytes to predict QT prolongation in patients. Even more intriguing (and unexplained) is that in cells from the carrier of the R176W mutation, despite an APD90 prolongation (166% of control) comparable to that observed in myocytes from carriers of the A614V (200%) and the A561T (188%) mutations, the occurrence of EADs in R176W myocytes was much lower. Unfortunately, it is not possible to determine whether the iPS-derived myocytes were truly able to predict symptoms or simply were unable to develop EADs because of lack of proper regulation of intracellular Ca2+ (iCa2+), because the authors did not study myocytes from asymptomatic family members who carried the R176W mutation.
Matsa et al. (35) obtained iPS-derived myocytes from two family members carrying the A561T mutation. In this study, the authors compared the cells derived from an asymptomatic mother carrier of the A561T mutation to cells derived from her highly symptomatic daughter. Consistent with the more severe clinical manifestations, the QTc was longer in the daughter (571 ms) than in the mother (514 ms). Interestingly, the APD90 was longer in the cells obtained from the daughter (the APD90 recorded from iPS-derived myocytes from the daughter was 188% of the APD90 of control cells; in myocytes derived from the mother, APD90 was 150% of the value measured in control myocytes). Even more intriguingly, myocytes derived from maternal tissue failed to develop EADs, whereas afterpotentials developed in the cells from the symptomatic daughter.
As speculated by the authors, the lack of EADs in the iPS-derived myocytes obtained from the asymptomatic mother may reflect the effect of unrecognized genetic modifiers that may protect her cells from development of arrhythmias. Unfortunately, since Matsa et al. (35) did not demonstrate the presence of genetic or epigenetic factors that silence the effects of the mutation in the mother, the finding remains without mechanistic explanation.
The last characterization of iPS-derived myocytes from LQTS patients was performed by Yazawa et al. in two individuals affected by Timothy syndrome (LQT8) (36). LQT8 is caused by mutations in the gene encoding the L-type calcium channel CaV1.2 (CACNA1C) (37). This form of LQTS is a malignant multisystem disorder that, in addition to causing cardiac electrical vulnerability, induces cardiac malformations, syndactyly of the hands and feet, delayed social development, and autism. The majority of LQT8 patients carry the G406R missense mutation, which increases the inward calcium current by altering voltage-dependent inactivation of the channel, leading to QT interval prolongation. This comprehensive study reported the electrophysiological properties of LQT8 iPS-derived myocytes, documenting marked prolongation of APD and development of EADs and triggered beats. In addition, in iPS-derived myocytes from LQT8 patients, calcium transients presented irregular timing and irregular amplitude as compared with transients recorded in control iPS-derived myocytes. Finally, the authors tested the hypothesis that roscovitine, a compound that increases the voltage-dependent inactivation of the cardiac L-type calcium channels, may restore the electrical properties and Ca2+ signaling in cardiomyocytes from LQT8 patients. Roscovitine successfully abbreviated the action potential and abolished arrhythmogenic activity in myocytes from LQT8 patients. Although intriguing, the observation is not sufficient to support the notion that iPS-derived myocytes may help identify novel therapies for arrhythmogenic diseases because the efficacy of this therapeutic approach in humans has not yet been confirmed.
Modeling CPVT in iPS-derived myocytes
CPVT is an inherited arrhythmogenic disease (38) that occurs in patients with an unremarkable resting ECG and structurally intact heart. Adrenergic stimulation related to stress, emotion, or physical activity induces a peculiar form of life-threatening ventricular tachycardia called “bidirectional tachycardia.” Similar to LQTS patients, individuals with CPVT are treated with β-blockers to prevent arrhythmias. An autosomal dominant form of CPVT has been associated with mutations on RYR2, which encodes the cardiac ryanodine receptor located in the SR of cardiomyocytes. The ryanodine receptor regulates calcium release from the SR to the cytosol to initiate cardiac contraction.
An autosomal recessive form of CPVT has been linked to mutations in CASQ2, which encodes cardiac calsequestrin, a high-capacity, low-affinity Ca2+-binding glycoprotein located inside the SR. Mutations in RYR2 and in CASQ2 lead to spontaneous diastolic calcium release, inducing delayed afterdepolarizations (DADs) and initiating arrhythmias (refs. 5, 39, and Figure 3).
Figure 3. CPVT.

(A) Subcellular localization of the calcium-handling proteins involved in the pathogenesis of CPVT. The pathophysiology of both ryanodine receptor (RyR2) and calsequestrin (CASQ2) has been studied in cardiomyocytes differentiated from patient-derived iPSs. (B–D) The cellular and clinical phenotype of CPVT is shown, with an example of DADs and triggered activity recorded in cardiomyocytes isolated from the heart of a RyR2-R4496C knockin mouse (B), a resting electrocardiogram (C), and bidirectional ventricular tachycardia in a patient with CPVT (D).
Two studies characterized cardiomyocytes from iPSs obtained from patients with the dominant form of CPVT (40, 41). Fatima et al. (40) studied iPS-derived myocytes from a carrier of the F2483I mutation, located in the FKBP12.6 domain of RyR2. The mutation was identified in an adult female patient that presented typical bidirectional ventricular tachycardia during an exercise stress test. The authors differentiated iPS-derived myocytes from fibroblasts of the CPVT patient and demonstrated that cells presented the typical morphologies of atrial, ventricular, and nodal cells. Unfortunately, no data for APD were reported in the study, and the ventricular cells presented some features that are atypical of CPVT: upon administration of isoproterenol, control cells showed a physiological increase in the spontaneous beating rate, but myocytes derived from the CPVT patient had a negative chronotropic response. This observation is inconsistent with the clinical response of CPVT patients to β-adrenergic stimulation. Patients usually show an acceleration of sinus rhythm and development of arrhythmias. Myocytes isolated from the heart of knockin mice for CPVT mutations also present acceleration of triggered rhythm during adrenergic stimulation (39). Another puzzling aspect of the study is the unusual morphology of the humps that were classified as DADs. These irregularly irregular wavelets did not present the typical morphology of DADs. Calcium imaging studies showed that, in agreement with a CPVT phenotype, iPS-derived myocytes from the CPVT patient presented abnormal spontaneous calcium transients characterized by higher amplitude and longer duration compared with healthy cardiomyocytes.
Jung et al. (41) reported data from iPS-derived CPVT myocytes carrying the S406L RyR2 mutation. Action potential recording experiments were performed, and it is not clear why the APD90 of iPS-derived ventricular myocytes from control patients was longer than APD90 recorded in myocytes from the CPVT patients (Table 1). This finding differs from clinical evidence that patients with CPVT have a slightly prolonged QT interval. This study clearly showed the presence of the frequency-dependent development of DADs (41). The authors also characterized calcium sparks in iPS -derived myocytes from control and CPVT patients. Under basal conditions, Ca2+ spark frequency did not differ between control and CPVT myocytes, but Ca2+ spark amplitude was higher in diseased cells. During exposure of cells to catecholamines, Ca2+ spark frequency considerably increased in CPVT cells compared with control cells.
Interestingly, Jung et al. (41) tested the ability of dantrolene to abolish triggered activity in iPS-derived myocytes. The demonstration that dantrolene is able to prevent arrhythmias in an knock-in mouse model of CPVT was previously reported (42), therefore Jung et al. tested whether the drug would be effective also in iPS-derived myocytes obtained from a CPVT patient carrier of the S406L mutation. Dantrolene was indeed able to abolish DADs and suggesting that the drug may be effective also to treat CPVT in humans.
Novak et al. (43) studied iPS-derived myocytes obtained from patients with the D307H mutation in the gene encoding for cardiac calsequestrin. This mutation has been extensively characterized, and it is associated with a very severe form of CPVT (44). The study adds to data from Jung et al. (41) and Fatima et al. (40), highlighting once more the susceptibility of iPS-derived CPVT myocytes to the development of DADs and triggered activity. Perhaps most importantly, data obtained with electron microscopy showed that cells derived from CPVT patients present an immature morphology with less-organized myofibrils, enlarged SR cisternae, and reduced number of caveolae. These data confirm data derived from knockout and knockin Casq2 mice showing that defective calsequestrin leads to ultrastructural abnormalities in the SR (5, 45). Unfortunately, the level of CASQ2 in iPS-derived myocytes was not quantified to determine whether the protein is markedly decreased, as observed in the studies performed in the transgenic mouse model carrier of the same mutation. In addition, the authors did not discuss how the absence of T tubules affected calcium handling physiology. Considering that iPS-derived cells have an immature calcium-handling apparatus, the model may not be suitable to confirm observations made in animal models of CPVT.
Concluding remarks
A substantial amount of knowledge on the electrophysiological properties of iPS-derived myocytes has been collected. Data show that differentiation of stem cells into cardiomyocytes leads to the generation of immature cardiac cells that resemble fetal myocytes. According to most studies, iPS-derived myocytes have a depolarized resting membrane potential and a decreased Vmax (Table 1). Interestingly, these myocytes lack gap junctions, intercalated disks, and T tubules. In order for the field to progress toward the clinical use of iPS-derived myocytes in predicting susceptibility to arrhythmias and response to therapy, the development of new differentiation protocols to produce iPS-derived myocytes with the electrophysiological properties of adult cardiomyocytes and with a fully functional calcium-handling apparatus will clearly be of paramount importance. Patch clamp studies and microelectrode recordings are technically challenging in iPS-derived myocytes and, therefore, a thorough characterization of the major ionic currents is still lacking. Potentially, the introduction of automatic high-throughput electrophysiological analysis may overcome these limitations.
Studies published until now have successfully demonstrated that iPS-derived myocytes obtained from skin biopsies of affected individuals can recapitulate the key features of the disease. These results are important but do not support the claim that this technology is ready to enter the clinic. While it is clear that the predictive value of the study of iPS-derived myocytes for individualized management of patients is still far from proven, it is reasonable to expect that this new model will provide additional insight in the biology of inherited arrhythmias in the short term, now that the critical proof-of-principle studies have been done.
These comments are not intended to shed a pessimistic view on the value of iPS-derived myocytes for the understanding of inherited arrhythmias, but rather reflect a realistic assessment about where we stand and what needs to be improved, so that the field may mature toward its clinical potential.
Acknowledgments
The authors’ work was supported by Telethon grant GGP11141 (to S.G. Priori), CARIPLO grant pr.2008.2275 (to S.G. Priori), Fondation Leducq Award 08CVD01 (to S.G. Priori), a Fondazione Veronesi Award on inherited arrhythmogenic diseases (to S.G. Priori), and Ministero Salute grant GR2010_2315996 (to S.G. Priori, G. Condorelli, and E. Di Pasquale).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(1):84–91. doi:10.1172/JCI62838.
References
- 1.Cerrone M, Napolitano C, Priori SG. Genetics of ion-channel disorders. Curr Opin Cardiol. 2012;27(3):242–252. doi: 10.1097/HCO.0b013e328352429d. [DOI] [PubMed] [Google Scholar]
- 2.Liu GX, et al. Differential conditions for early after-depolarizations and triggered activity in cardiomyocytes derived from transgenic LQT1 and LQT2 rabbits. J Physiol. 2012;590(pt 5):1171–1180. doi: 10.1113/jphysiol.2011.218164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tamargo J, Caballero R, Núñez L, Gómez R, Vaquero M, Delpón E. Genetically engineered mice as a model for studying cardiac arrhythmias. Front Biosci. 2007;12:22–38. doi: 10.2741/2046. [DOI] [PubMed] [Google Scholar]
- 4.Cerrone M, et al. Bidirectional ventricular tachycardia and fibrillation elicited in a knock-in mouse model carrier of a mutation in the cardiac ryanodine receptor. Circ Res. 2005;96(10):e77–e82. doi: 10.1161/01.RES.0000169067.51055.72. [DOI] [PubMed] [Google Scholar]
- 5.Rizzi N, et al. Unexpected structural and functional consequences of the R33Q homozygous mutation in cardiac calsequestrin: a complex arrhythmogenic cascade in a knock in mouse model. Circ Res. 2008;103(3):298–306. doi: 10.1161/CIRCRESAHA.108.171660. [DOI] [PubMed] [Google Scholar]
- 6.Tomas M, et al. Polymorphisms in the NOS1AP gene modulate QT interval duration and risk of arrhythmias in the Long QT Syndrome. J Am Coll Cardiol. 2010;55(24):2745–2752. doi: 10.1016/j.jacc.2009.12.065. [DOI] [PubMed] [Google Scholar]
- 7.Priori SG, Napolitano C, Schwartz PJ. Low penetrance in the long-QT syndrome: clinical impact. Circulation. 1999;99(4):529–533. doi: 10.1161/01.CIR.99.4.529. [DOI] [PubMed] [Google Scholar]
- 8.Davis RP, van den Berg CW, Casini S, Braam SR, Mummery CL. Pluripotent stem cell models of cardiac disease and their implication for drug discovery and development. Trends Mol Med. 2011;17(9):475–484. doi: 10.1016/j.molmed.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 9.Jaenisch R, Young R. Stem cells, the molecular circuitry of pluripotency and nuclear reprogramming. Cell. 2008;132(4):567–582. doi: 10.1016/j.cell.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Evans MJ, Kaufman MH. Establishment in culture of pluripotential cells from mouse embryos. Nature. 1981;292(5819):154–156. doi: 10.1038/292154a0. [DOI] [PubMed] [Google Scholar]
- 11.Thomson JA, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282(5391):1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- 12.Gurdon JB. The transplantation of nuclei between two species of Xenopus. Dev Biol. 1962;5:68–83. doi: 10.1016/0012-1606(62)90004-0. [DOI] [PubMed] [Google Scholar]
- 13.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 14.Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 15.Kehat I, et al. Human embryonic stem cells can differentiate into myocytes with structural and functional properties of cardiomyocytes. J Clin Invest. 2001;108(3):407–414. doi: 10.1172/JCI12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zwi L, et al. Cardiomyocyte differentiation of human induced pluripotent stem cells. Circulation. 2009;120(15):1513–1523. doi: 10.1161/CIRCULATIONAHA.109.868885. [DOI] [PubMed] [Google Scholar]
- 17.Yang L, et al. Human cardiovascular progenitor cells develop from a KDR+ embryonic-stem-cell-derived population. Nature. 2008;453(7194):524–528. doi: 10.1038/nature06894. [DOI] [PubMed] [Google Scholar]
- 18.Laflamme MA, et al. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat Biotechnol. 2007;25(9):1015–1024. doi: 10.1038/nbt1327. [DOI] [PubMed] [Google Scholar]
- 19.Soldner F, et al. Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell. 2011;146(2):318–331. doi: 10.1016/j.cell.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu GH, et al. Targeted gene correction of laminopathy-associated LMNA mutations in patient-specific iPSCs. Cell Stem Cell. 2011;8(6):688–694. doi: 10.1016/j.stem.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boukens BJ, Christoffels VM, Coronel R, Moorman AF. Developmental basis for electrophysiological heterogeneity in the ventricular and outflow tract myocardium as a substrate for life-threatening ventricular arrhythmias. Circ Res. 2009;104(1):19–31. doi: 10.1161/CIRCRESAHA.108.188698. [DOI] [PubMed] [Google Scholar]
- 22.Antzelevitch C. Transmural dispersion of repolarization and the T wave. Cardiovasc Res. 2001;50(3):426–431. doi: 10.1016/S0008-6363(01)00285-1. [DOI] [PubMed] [Google Scholar]
- 23.Janse MJ, Coronel R, Opthof T, Sosunov EA, Anyukhovsky EP, Rosen MR. Repolarization gradients in the intact heart: Transmural or apico-basal? Prog Biophys Mol Biol. 2012;109(1–2):6–15. doi: 10.1016/j.pbiomolbio.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 24.de Jong F, et al. Persisting zones of slow impulse conduction in developing chicken hearts. Circ Res. 1992;71(2):240–250. doi: 10.1161/01.RES.71.2.240. [DOI] [PubMed] [Google Scholar]
- 25.Zhang J, et al. Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ Res. 2009;104(4):e30–e41. doi: 10.1161/CIRCRESAHA.108.192237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Malan D, Friedrichs S, Fleischmann BK, Sasse P. Cardiomyocytes obtained from induced pluripotent stem cells with long-QT syndrome 3 recapitulate typical disease-specific features in vitro. Circ Res. 2011;109(8):841–847. doi: 10.1161/CIRCRESAHA.111.243139. [DOI] [PubMed] [Google Scholar]
- 27.Zhu WZ, Xie Y, Moyes KW, Gold JD, Askari B, Laflamme MA. Neuregulin/ErbB signaling regulates cardiac subtype specification in differentiating human embryonic stem cells. Circ Res. 2010;107(6):776–786. doi: 10.1161/CIRCRESAHA.110.223917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gherghiceanu M, et al. Cardiomyocytes derived from human embryonic and induced pluripotent stem cells: comparative ultrastructure. J Cell Mol Med. 2011;15(11):2539–2551. doi: 10.1111/j.1582-4934.2011.01417.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kehat I, Gepstein A, Spira A, Itskovitz-Eldor J, Gepstein L. High-resolution electrophysiological assessment of human embryonic stem cell-derived cardiomyocytes: a novel in vitro model for the study of conduction. Circ Res. 2002;91(8):659–661. doi: 10.1161/01.RES.0000039084.30342.9B. [DOI] [PubMed] [Google Scholar]
- 30.Napolitano C. Genetic testing of inherited arrhythmias. Pediatr Cardiol. 2012;33(6):980–987. doi: 10.1007/s00246-012-0287-7. [DOI] [PubMed] [Google Scholar]
- 31.Moretti A, et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N Engl J Med. 2010;363(15):1397–1409. doi: 10.1056/NEJMoa0908679. [DOI] [PubMed] [Google Scholar]
- 32.Marban E. Cardiac channelopathies. Nature. 2002;415(6868):213–218. doi: 10.1038/415213a. [DOI] [PubMed] [Google Scholar]
- 33.Itzhaki I, et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature. 2011;471(7337):225–229. doi: 10.1038/nature09747. [DOI] [PubMed] [Google Scholar]
- 34.Lahti AL, et al. Model for long QT syndrome type 2 using human iPS cells demonstrates arrhythmogenic characteristics in cell culture. Dis Model Mech. 2011;5(2):220–230. doi: 10.1242/dmm.008409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matsa E, et al. Drug evaluation in cardiomyocytes derived from human induced pluripotent stem cells carrying a long QT syndrome type 2 mutation. Eur Heart J. 2011;32(8):952–962. doi: 10.1093/eurheartj/ehr073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yazawa M, et al. Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature. 2011;471(7337):230–234. doi: 10.1038/nature09855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Splawski I, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119(1):19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 38.Priori SG, et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2002;106(1):69–74. doi: 10.1161/01.CIR.0000020013.73106.D8. [DOI] [PubMed] [Google Scholar]
- 39.Liu N, et al. Arrhythmogenesis in catecholaminergic polymorphic ventricular tachycardia: insights from a RyR2 R4496C knock-in mouse model. Circ Res. 2006;99(3):292–298. doi: 10.1161/01.RES.0000235869.50747.e1. [DOI] [PubMed] [Google Scholar]
- 40.Fatima A, et al. In vitro modeling of ryanodine receptor 2 dysfunction using human induced pluripotent stem cells. Cell Physiol Biochem. 2011;28(4):579–592. doi: 10.1159/000335753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jung CB, et al. Dantrolene rescues arrhythmogenic RYR2 defect in a patient-specific stem cell model of catecholaminergic polymorphic ventricular tachycardia. EMBO Mol Med. 2012;4(3):180–191. doi: 10.1002/emmm.201100194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kobayashi S, et al. Dantrolene, a therapeutic agent for malignant hyperthermia, inhibits catecholaminergic polymorphic ventricular tachycardia in a RyR2(R2474S/+) knock-in mouse model. Circ J. 2010;74(12):2579–2584. doi: 10.1253/circj.CJ-10-0680. [DOI] [PubMed] [Google Scholar]
- 43.Novak A, et al. Cardiomyocytes generated from CPVTD307H patients are arrhythmogenic in response to beta-adrenergic stimulation. J Cell Mol Med. 2012;16(3):468–482. doi: 10.1111/j.1582-4934.2011.01476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lahat H, et al. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am J Hum Genet. 2001;69(6):1378–1384. doi: 10.1086/324565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Knollmann BC, et al. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J Clin Invest. 2006;116(9):2510–2520. doi: 10.1172/JCI29128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ma J, et al. High purity human-induced pluripotent stem cell-derived cardiomyocytes: electrophysiological properties of action potentials and ionic currents. Am J Physiol Heart Circ Physiol. 2011;301(5):H2006–H2017. doi: 10.1152/ajpheart.00694.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]