

Abstract
Many remarkable advances have improved our understanding of the cellular and molecular events in the pathogenesis of atherosclerosis. Chief among these is the accumulating knowledge of how the immune system contributes to all phases of atherogenesis, including well-known inflammatory reactions consequent to intimal trapping and oxidation of LDL. Advances in our understanding of the innate and adaptive responses to these events have helped to clarify the role of inflammation in atherogenesis and suggested new diagnostic modalities and novel therapeutic targets. Here we focus on recent advances in understanding how adaptive immunity affects atherogenesis.
Introduction: innate immunity informs adaptive responses in atherosclerosis
There have been remarkable advances in understanding immune contributions to the pathogenesis of atherosclerosis, and especially the inflammatory reactions consequent to generation of oxidized LDL (oxLDL) within the intima (1–6). Because of the explosion of new knowledge and the brevity of this Review, it is not possible to discuss or cite much of the relevant literature, for which we apologize. We mainly focus on recent advances in understanding how adaptive immunity affects atherogenesis. However, because innate recognition of disease-specific antigens is a prerequisite for adaptive immune responses to occur, we begin by briefly introducing atherosclerosis-relevant antigens recognized by innate immunity.
Innate immunity plays a fundamental role in initiating and modulating atherosclerosis, as reviewed in depth elsewhere (3, 5, 7). It recognizes signature molecules, either pathogen-associated molecular patterns (PAMPs) of microbial origin or danger- or damage-associated molecular patterns (DAMPs), which are “self” molecules that become accessible to the immune system following cell injury or death, or are “altered-self” molecules that generate neoepitopes. The innate immune system utilizes germ line-encoded pattern recognition receptors (PRRs) to recognize PAMPs and DAMPs, effecting responses that are usually protective, such as killing of the inciting microbe or maintenance of homeostasis by stimulating removal of damaged or dead tissue. Importantly, these initial responses are accompanied by secretion of chemokines and cytokines that recruit and activate lymphocytes, and by presentation of antigens, which together initiate definitive adaptive responses. The major type of response to innate immune recognition is inflammation.
Identifying the antigens to which innate immune responses occur is central to understanding the role of immunity in atherogenesis. The possibility that infectious agents provoke relevant immune responses has been considered, but little evidence supports a primary role, though contributory roles are possible (reviewed in refs. 8, 9). Similarly, DAMPs generated as a result of tissue injury, such as heat shock proteins (HSPs) (10) or cholesterol crystals (11), require pre-existing injury, and although they may contribute to chronic inflammation, they are not likely to initiate the primary inflammatory cascade.
Although other antigens may also be important, much evidence suggests that major atherosclerosis-relevant antigens consist of neoepitopes generated as a consequence of oxidative reactions, as occur when oxLDL is formed or when cells undergo apoptosis (1, 7). Innate immunity has apparently evolved multiple mechanisms to mediate removal of these oxidatively modified molecules, cells, and debris, which would otherwise be proinflammatory and immunogenic (2, 7, 12). Analogous to recognition of PAMPs on pathogens, recognition of oxidation-damaged molecular complexes occurs via the detection of oxidation-specific epitopes (OSEs), which constitute common motifs of oxidative damage that are ligands for a common set of innate PRRs, including macrophage scavenger receptors, natural Abs (NAbs), and innate plasma proteins. Maintenance of homeostasis against OSEs has led to evolutionary pressure for PRRs against such epitopes, and consequently, OSEs are a major target of innate immunity (reviewed in ref. 7). This not only provides a conceptual framework for the involvement of innate immunity in atherogenesis, but because innate responses are prerequisites for adaptive responses, it explains the compelling data that OSEs are also immunodominant, disease-specific antigens that activate adaptive responses in atherogenesis.
Understanding adaptive immunity in atherosclerosis
Adaptive responses occur following recognition of an antigen by membrane Ig on B cells and TCRs on T cells. Unlike PRRs of innate immunity, the genes encoding Ig receptors and TCRs are formed by somatic recombination, which generates tremendous diversity of receptor specificities, each unique to a single lymphocyte clone. Antigen recognition drives lymphocyte proliferation and differentiation into effector cells with a variety of pro-inflammatory properties that protect from infection, but which can also cause tissue damage and disease, especially when the inciting antigens are persistent or when intrinsic defects occur in lymphocyte regulation. Because adaptive immunity can randomly generate lymphocytes that recognize self-molecules as well as harmless foreign molecules, various tolerance mechanisms exist to prevent the survival or activation of these dangerous B and T cells. One essential tolerogenic mechanism is that T cells must see molecules called costimulators, which are induced by innate immune responses to PAMPs or DAMPs, in addition to seeing antigen, in order to become activated. Thus, naive T cells specific for normal self-antigens will not be costimulated, and tolerance, rather than effector responses, will be induced
Adaptive immunity exerts diverse and profound effects on atherosclerosis (3, 5, 13). In humans, the evidence is largely correlative data from analysis of blood and atherosclerotic lesions obtained surgically or at autopsy. Plasma Ab titers against OSEs of oxLDL and immune complexes with oxLDL in human lesions were demonstrated more than two decades ago (14, 15), and Ab titers to HSP60 correlate with cardiovascular disease (CVD) (10). Studies have found correlations between acute coronary syndromes (ACSs) and the numbers of various subsets of T cells in the blood and changes in Ab titers to oxLDL. The strikingly elevated risk for atherosclerotic disease in patients with systemic autoimmune diseases (16–18) also supports the hypothesis that adaptive immune responses promote atherosclerosis.
Studies of atherosclerosis in hypercholesterolemic Apoe–/– or Ldlr–/– mice in which adaptive immunity is deleted (e.g., Rag-null mice or mice on a SCID background) demonstrate that while adaptive immunity is not a prerequisite for atherogenesis, its presence profoundly affects lesion formation and on balance is proatherogenic (19–23). However, a total absence of adaptive immunity leads to a lack of both protective and proatherogenic influences. Atherosclerosis is the result of a complex outcome of the balance between subsets of immune cells and their products, between T effector cells and Tregs, between costimulatory and coinhibitory molecules, and between B-1 cells and B-2 cells and their respective IgM and IgG Abs, as well as between proinflammatory and antiinflammatory subsets of macrophages. Much progress has been made in understanding this balance through selective interventions and genetic manipulations of murine models.
T cells in atherosclerosis
Beginning with seminal studies in the 1980s that demonstrated activated CD4+ and CD8+ T cells in human atheromata (5, 24), extensive data now support that T cells mediate proinflammatory and proatherogenic immune responses. A key unresolved question is the identity of the relevant activating antigens. Candidate antigens include oxLDL and native LDL (25) as well as modified or even native fragments of apoB-100 (the major protein of LDL) (26, 27) and HSP60 (10). The mechanisms of loss of T cell tolerance in the case of self-proteins may arise due to strong innate responses to these same molecules and/or the oxidatively driven covalent modification of apoB-100 to form OSE neoepitopes. Effector T cell responses within arterial lesions are likely initiated by antigen activation of naive T cell precursors in secondary lymphoid organs (see Figure 1). Many of the putative driving antigens can be present systemically, such as lipoproteins that may be in excess and oxidized in dyslipidemic individuals, and may activate lymphocytes within lymph nodes or the spleen. Therefore, proatherogenic T cell responses can be considered systemic in nature. Below we review the major T cell subsets involved. We refer the reader to recent reviews for discussions of minor subsets such as iNKT cells (28) or CD4+CD28null effector T cells preferentially found during chronic inflammatory diseases (29, 30), for which less information is known.
Figure 1. Theoretical events in initiation and effector phases of a proatherogenic T cell response.
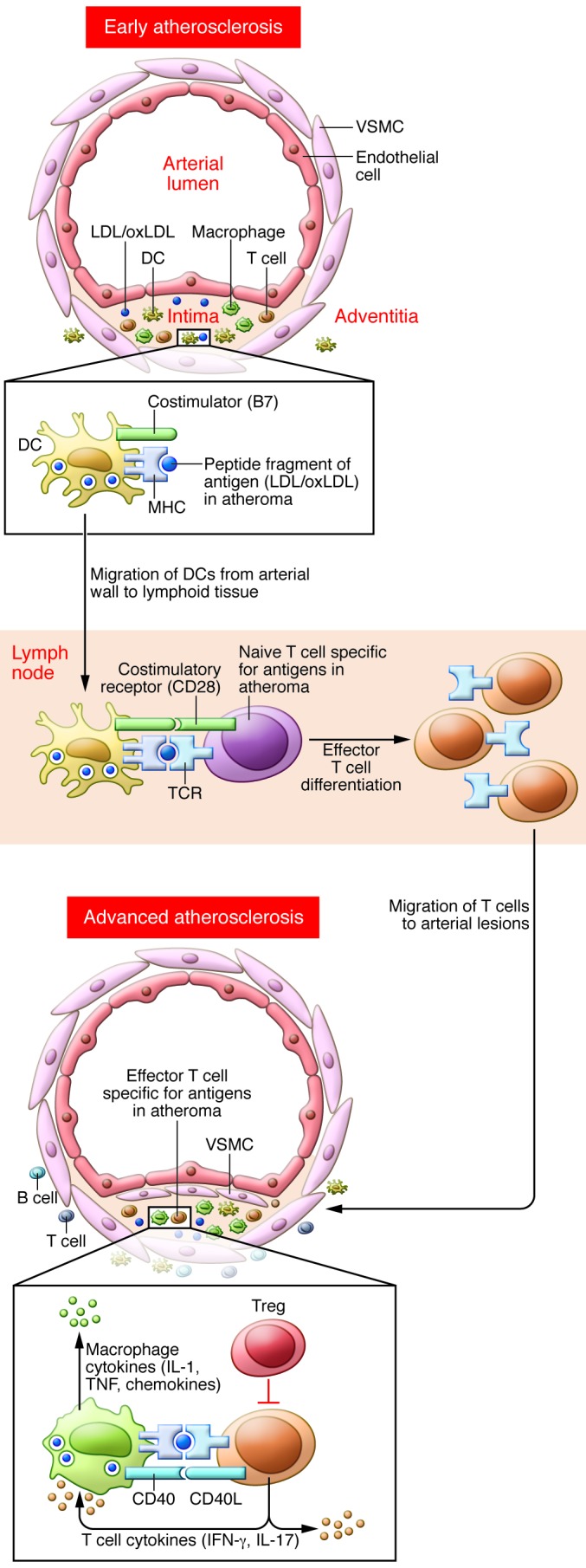
Modified self-proteins generated in early atherosclerotic lesions (or systemically), such as the oxidatively modified apoB-100 component of LDL, are processed by DCs and presented as peptide/MHC complexes to naive T cells in secondary lymphoid tissues, leading to T cell clonal expansion and differentiation into effector T cells, such as Th1 or Th17 cells. The effector T cells migrate into arterial lesions, where resident macrophages or DCs present the same peptide-MHC antigens, leading to effector T cell activation and expression of pro-inflammatory effector molecules, such as secreted IFN-γ and IL-17 and membrane-bound CD40 ligand. These molecules promote lesion growth and/or destabilization.
CD4+ Th cells.
In response to combined stimulation with antigen, costimulators, and particular cytokines, naive CD4+ Th cells differentiate into distinct effector or Th subsets distinguished from one another by the cytokines they produce. The three best-characterized Th are Th1, which secretes IFN-γ, Th2, which secretes IL-4, IL-5, and IL-13, and Th17, which secretes IL-17 and IL-22. Chronic or repeated antigen exposure, which likely occurs in human atherosclerosis, often results in the emergence of a dominant polarized Th subset. Identification of the Th subsets that enhance atherosclerosis provides a therapeutic opportunity to block either the cytokines produced or factors driving specific subset differentiation.
Compelling data implicate Th1 cells and IFN-γ in promoting atherogenesis and inflammation. These data include identification of IFN-γ in human lesions and the expression of IFN-γ by T cells cloned from human plaques (5, 25). In addition, genetic ablation of the IFN-γ receptor or its receptor (31, 32) or the Th1 lineage–defining transcription factor T-bet (33) reduced atherogenesis in murine models, while exogenous IFN-γ enhanced lesion development (34). IFN-γ has a variety of biological effects predicted to promote lesion development and destabilize established lesions (reviewed in ref. 35). Of importance, oxLDL and LDL activated human plaque T cells to secrete IFN-γ in a class II MHC-restricted manner (25), documenting Th1 activation by these antigens. Other studies revealed a proatherogenic role for IL-12 (36), which drives Th1 differentiation, and for IL-18, which enhances IFN-γ production by T cells (37). These may be of relevance for humans, as monoclonal Abs to the p40 subunit receptor for IL-12/IL-23 are used for psoriasis, and may be paradoxically associated with elevated IL-12 levels, and concerns have been raised about a possible increase in CVD (38, 39).
Both murine and human studies demonstrate that both Th17 and Th1 cells contribute to pathogenesis of many of the same diseases (40). However, reports of the role of Th17 cells or IL-17 in atherogenesis are inconsistent, likely reflecting poorly understood relationships between Th1 and Th17 responses (41). Although IL-17 has been detected in human lesions (42, 43), IL-17–expressing T cells appear to be rare (44). Data on the frequency of Th17 cells or IL-17 in the blood of patients with ACS or acute myocardial infarction also provide an unclear message (45–50). Several murine studies support a proinflammatory, proatherogenic role for the IL-17/IL-17 receptor axis (51–56), but others failed to find IL-17 to be proatherogenic, and some even found it to be atheroprotective (43, 57–59). Manipulations of mice that indirectly enhanced Th17 differentiation, including genetic deletion of suppressor of cytokine signaling 3 (Socs3) or anti-CD20–mediated B cell depletion, were atheroprotective (43, 60). The inconsistencies in the data may relate to the existence of IL-17–producing CD4+ T cells that also produce IFN-γ (61, 62), or to plasticity of Th17 cells, which can redifferentiate into Th1 cells (63). In light of the open questions about IL-17 and atherogenesis, it will be important to determine the impact on CVD in autoimmune patients treated with anti–IL-17 Abs (64–66).
There are limited and inconsistent data on the influence of Th2 cells on atherosclerosis. Ldlr–/– mice that lack the Th1-driving transcription factor T-bet have enhanced systemic Th2 responses and decreased lesion development (33). However, in severely hypercholesterolemic mice, advancing atherosclerosis is associated with a switch to Th2 cells (67). The Th2 cytokine IL-4 has been reported to be proatherogenic (36, 68) or have no effect (69). On the other hand, IL-13 was recently reported to be atheroprotective (70), as was IL-5 (71). Because inhibition of IL-5 activity in humans is under study for a variety of conditions associated with eosinophilia (72), long-term follow up for the impact on CVD should be informative.
CTLs.
CD8+ CTLs, which are generally less numerous than CD4+ T cells in human lesions, may nevertheless constitute up to 50% of the T cells in advanced lesions (73, 74). In murine atherosclerosis, few lesional CD8+ T cells are present, except when immunoregulatory molecules are also deficient (75), although systemic CD8+ T cell responses to hypercholesterolemia are detectable (76). Genetic deficiencies in either CD8 or TAP-1, required for class I MHC antigen presentation, did not significantly affect lesion development (77, 78). Although not apparently contributing to early lesion development, CTLs are activated in the context of hypercholesterolemia and within lesions can promote plaque inflammation and instability.
B cells and Ab responses in atherosclerosis
B-1 cells and IgM NAbs.
In mice, B-1a cells are more properly regarded as an arc of innate immunity. They respond to T cell–independent antigens by secretion of IgM and IgA NAbs, whose repertoire results from natural selection. Importantly, OSEs such as those found on oxLDL and apoptotic cells are a major target of NAbs in both mice and humans (reviewed in ref. 7). Considerable evidence supports an atheroprotective role for IgM specific for OSEs in mice. Immunization of mice with heat-killed S. pneumoniae, which shares molecular identity with oxidized phospholipids of oxLDL, leads to marked increases in IgM specific for oxLDL, which blocks oxLDL uptake by macrophages and inhibits atherosclerosis (79). T-bet deficiency and CD74 deficiency, both associated with reduced atherosclerosis, result in marked increases in IgM NAbs specific for oxLDL (33, 80). Ldlr–/– mice deficient in the ability to secrete IgM (sIgM–/– mice) have dramatically increased atherosclerosis (81). Splenectomy was shown to enhance atherosclerosis (82), and recent studies have shown that this can be rescued by transfer of B-1 cells but not B-2 cells (83). Furthermore, transfer of B-1 cells from sIgM–/– mice were incapable of such rescue, demonstrating that the IgMs were atheroprotective. In humans, IgM titers to epitopes of oxLDL are inversely associated with CVD in univariate analysis, also consistent with an atheroprotective role (84). The recent identification in humans of CD20+CD27+CD43+ B cells that spontaneously secrete IgM NAbs suggests an equivalent human B-1 cell population (85). As with B-2 cells, B-1 cells express CD20, which is currently being targeted with anti-CD20 monoclonal Abs in autoimmune diseases and therefore could be depleted.
B-2 cells and IgG-adaptive Abs.
Conventional, adaptive B-2 (or follicular B) cells respond to T cell–dependent antigens by secretion of IgG isotype Abs. IgG Abs specific for OSEs are found in blood and lesions of both normal and atherosclerotic patients, and in experimental animals (reviewed in refs. 7, 86, 87). In murine models, IgG titers to oxLDL epitopes increase and decrease in parallel with lesion progression and regression, respectively (88). Many IgG Abs to oxLDL inhibit the uptake of oxLDL by macrophages, at least in culture, suggesting that they should inhibit atherogenesis. Indeed, the seminal observation more than two decades ago that immunization of animal models with MDA-LDL, a model epitope of oxLDL, could inhibit atherosclerosis was an important stimulus to the study of adaptive immunity in atherogenesis (89). However, such immunizations increase both IgG1 from B-2 cells and IgM from B-1 cells, the latter resulting from an induced Th2 response with IL-5 release, which is a known B-1 cell stimulant (71). Subsequent studies have shown that elevations of titers of OSE Abs in murine models, whether by immunization, passive infusions, or adenoviral-mediated expression, can inhibit atherogenesis (7, 86, 90, 91). This includes use of fragment, antigen-binding (Fab) or even single-chain variable fragment (scFv) anti-oxLDL Ab fragments that have no effector functions (92), demonstrating the inherent ability of such OSE Abs to inhibit foam cell formation. These data suggest the possibility that passively or therapeutically raising the titers of such OSE Abs in humans may be beneficial.
In humans, IgG titers to oxLDL are much more heterogeneous than in the murine models but in general correlate positively with CVD manifestations in univariate analyses (84, 87). This has led some investigators to suggest that such IgGs are proatherogenic. However, these associations are lost when adjusted for common covariates such as age, likely reflecting the fact that the ambient IgG titers present in disease are biomarkers rather than disease modifiers.
Studies to determine the role of B-2 cells in atherosclerosis are complicated by potential roles of the B-2 cells themselves versus the roles of the Abs they secrete. Bone marrow deficiency of B cells supported an atheroprotective role for B cells (93). On the other hand, use of anti-CD20 to deplete B-2 cells in mice, which spared B-1 cells in the peritoneum, reduced atherosclerosis, as well as activation and proliferation of DCs and CD4+ T cells, suggesting that B-2 cells were proatherogenic by several mechanisms (60, 94). Furthermore B cell–activating factor receptor (BAFF-R) deficiency, which causes a reduction in B-2 but not B-1 cells, also reduced lesion development and macrophage and T cell infiltration (95, 96). However, recent data demonstrated a role for the Id3-CCR6 pathway in mediating aortic B cell homing, and demonstrated that resident adventitial B cells mediated protection from early atherosclerosis in part through inhibiting intimal macrophage accumulation (97). Indeed subsets of regulatory B cells, including B cells that secrete IL-10, are well known (98). These data indicate a complex role for B cells and B cell subsets in atherogenesis, suggesting that they affect lesion formation both systemically via secretion of Abs and locally within the aorta, by affecting macrophage and T cell biology.
FDA-approved anti-CD20 and anti-BAFF Ab drugs are being used in patients with autoimmune disease. Both selectively target B-2 cells in mice, but their impact on the newly described B-1 cell population in humans is unknown and clearly should be an important topic of study. This is particularly important because of a recently described human CD20+ B-1 subset that secretes IL-10 and regulates T cell activity (99).
In response to T cell help and germinal center reactions, B-2 cells undergo isotype switching and differentiation into Ab-secreting cells that produce IgG, IgA, or IgE Abs. The effector functions of some IgG subtypes and IgE Abs depend in part on isotype-specific Fc receptors expressed on various cell types. Genetic ablation studies in mice indicate that CD16 (FcγRIII) (100) or the common γ chain of activating Fcγ receptors (101) are proatherogenic, while the inhibitory FcγRIIb receptor decreases atherosclerosis (102, 103). Because IgG isotypes bind to these Fcγ receptors with different affinities, they may differentially affect atherogenesis through this mechanism, independent of other effector functions. Furthermore, loss of the IgE receptor FcεR1α reduced atherosclerotic plaque development and inflammation in Apoe–/– mice (104). Some IgG isoforms also activate the classical complement cascade. Although complement activation occurs in murine atherosclerotic lesions, the impact of this is controversial (reviewed in ref. 105), and in particular, the influence of Ab-dependent complement activation on atherosclerosis is unknown.
Regulation of proatherogenic adaptive immune responses
Adaptive immune responses are highly regulated by multiple mechanisms. One can view atherosclerosis as a disease in which there is inadequate regulation of adaptive immune responses to normal or altered components of lipoproteins that accumulate in lymphoid organs and in the intima of arteries. In this discussion, we focus mainly on two interrelated mechanisms of adaptive immune regulation: Tregs and costimulatory and coinhibitory molecules.
Tregs.
T cell subsets whose major functions are to inhibit immune responses, rather than to promote protective immunity against pathogens, are called Tregs (106). Most studies of rodent and human Tregs focus on CD4+ αβ TCR+ CD25+ cells that also express the transcription factor Foxp3. These Foxp3+ Tregs include natural Tregs, which develop in the thymus and constitute the majority of circulating Tregs in human blood, and induced Tregs (also known as adaptive or peripheral Tregs), which differentiate from conventional CD4+ T cells in peripheral tissues in response to antigens, cytokines, and other cues. Tregs suppress immune responses by multiple mechanisms including CD25-mediated competition for IL-2 (107), CTLA-4–mediated competitive binding (and perhaps removal) of B7-1 and B7-2 costimulators on APCs (refs. 108, 109, and see Figure 2), and secretion of the suppressive cytokines IL-10, IL-35, IL-9, and TGF-β (110). The targets of suppression of Tregs include naive and effector CD4+ and CD8+ T cells, DCs, and even endothelial cells.
Figure 2. Costimulatory and coinhibitory molecules and their receptors.

Costimulatory molecules, expressed on APCs, engage receptors on T cells concurrent with antigen recognition and induce signals that are required for naive T cell activation or that enhance effector/memory T cell responses. Coinhibitory receptors (CTLA-4 and PD-1) are expressed on activated T cells and, upon binding ligands on APCs, inhibit T cell responses. The major costimulatory and coinhibitory molecules, which belong to the B7/CD28 families or TNF/TNFR superfamilies, are shown in the table.
An atheroprotective role for Tregs in murine atherosclerosis has been supported by studies in which Tregs have been depleted, induced, or injected, leading to diminished disease (13). Impaired Treg development or function due to hematopoietic deficiency of B7-1 and B7-2, CD28, or inducible T cell costimulator (ICOS) also results in enhanced lesion development (13, 111). Furthermore, Treg numbers decrease and effector T cells increase as atherosclerotic lesions progress in mice (112), which is consistent with the ultimate failure of Tregs to keep up with proatherogenic effector T cells in human disease. Many publications have reported that various manipulations that result in reduced lesion size or inflammation (113–116) are also associated with increased Treg numbers.
Tregs are detectable within human arterial lesions, and changes in the numbers of circulating Tregs correlate with changes in disease activity and/or with effective therapy. Only low numbers of CD4+Foxp3+ T cells have been identified in human atherosclerotic plaques at various stages of development (117), but increased numbers of carotid plaque Foxp3+ CD3+ T cells have been associated with symptomatic disease (118). The enumeration of Foxp3+CD4+ T cells in the blood reveals a generalized trend toward decreased numbers of blood Tregs in patients with clinically active CVD compared with controls (48, 119, 120) and increased Treg numbers in the blood or lesions in response to various therapeutic interventions including statins (121–123). The effects of statins on Treg development may contribute to the drugs’ well-documented pleiotropic anti-inflammatory effects and are in need of further study (124). Overall, these studies suggest that pharmacologically increasing Tregs may be a feasible goal for patients with vulnerable plaques.
APCs and costimulatory and coinhibitory molecules.
T lymphocyte activation at the initiation and effector phases of adaptive immune responses requires the presentation of antigenic peptide-MHC complexes displayed on the cell surface by APCs, which also provide costimulatory molecules (described below) and cytokines required for T cell activation. DCs are the most important and perhaps only type of APC that can efficiently activate naive T cells, while effector and memory CD4+ T cells can be activated by other APCs that express class II MHC, including macrophages and B cells (125). CTLs can be activated by almost any cell type that displays the right class I MHC-associated peptide.
CD11chi myeloid DCs are detectable in the arterial intima in atherosclerosis-prone areas (126) and in the adventitia (127), even in arteries without lesions, and DCs increase in number in both sites as lesions grow (128, 129). Although proatherogenic T cell responses could plausibly occur in any secondary lymphoid tissue, effector T cells must migrate into the arterial wall and be locally activated by APCs to influence lesion development. Both DCs and lesional macrophages, including cholesterol-loaded foam cells (130, 131), likely participate in the local activation of effector CD4+ and CD8+ T cells. In addition, stimulation of plasmacytoid DCs also plays an atherogenic role (132).
Costimulatory and coinhibitory molecules expressed on the surface of APCs engage receptors on T cells at the same time that the TCR recognizes an antigen (see Figure 2), triggering signaling that enhances or inhibits T cell activation, respectively. Naive T cell activation requires both antigen and costimulation, and activation of effector and memory T cells is enhanced by costimulation. Costimulatory and coinhibitory pathways have significant effects on atherosclerotic lesion development, as recently reviewed (133). The costimulators CD80, CD86, CD275, and CD252 can be detected on lesional macrophages in human and mouse lesions (134–138), as can their receptors on lesional T cells. A proatherogenic effect has been demonstrated for CD80, CD86, and CD252 (136, 137, 139) as well as for CD137, a costimulatory receptor expressed on T lymphocytes (138, 140). Human CD4+CD28null T cells, which are associated with ACS, express high levels of the costimulatory receptors CD134 and CD137 compared with conventional CD4+CD28+ T cells, and in vitro studies show that the ligands for these receptors can costimulate these T cells (141).
The coinhibitor programmed cell death ligand 1 (PDL1) is expressed on macrophages and DCs within mouse aortic lesions (142). Combined deficiency of PDL1 and PDL2 or of their receptor PD-1 in Ldlr–/– mice results in increased atherosclerosis and lesional CD4+ and CD8+ T cells (75, 142). Blood T cells and myeloid DCs from patients with coronary artery disease express less PD-1 and PDL1 compared with cells from healthy individuals (143).
B7 CD28 family–mediated costimulation is not only required for effector T cell differentiation, but also for the development of Tregs (144). Furthermore, coinhibitory molecules expressed by Tregs may in part mediate Treg function. In mice, transplantation of Cd80–/–Cd86–/– or Cd28–/– or Icos–/– bone marrow into Ldlr–/– recipients resulted in Treg deficiency and increased atherosclerosis with increased inflammation in lesions (111, 145). Therefore, the net effect of costimulatory or coinhibitory deficiencies in experimental models, and perhaps in patients treated with costimulatory blockers, may be complicated by the simultaneous loss of both effector and regulatory responses.
Induction of tolerance to treat atherosclerosis.
Investigators have explored methods to induce peripheral tolerance for the treatment and prevention of autoimmune disease and allograft rejection. In preclinical models, nasal or oral administration of HSP65 reduced atherosclerosis in Ldlr–/– mice (146, 147). Similar findings are reported for nasal administration of oxLDL (148, 149) or β2-glycoprotein I (150). Another approach is to deliver the relevant antigens via tolerogenic DCs that have suppressed costimulatory functions but maintain antigen-presenting functions. Atherosclerosis was attenuated and Tregs induced by treatment of hypercholesterolemic Ldlr–/– mice transgenic for human apoB-100 with DCs that had been pulsed with apoB-100 in combination with the immunosuppressive cytokine IL-10 (151). Alternatively, an immunogenic peptide fragment of human apoB, which when conjugated to human albumin and given subcutaneously, induced Tregs and reduced atherosclerosis in Apoe–/– mice (152). A major challenge to developing effective tolerance-inducing therapies in humans will be the identification of the relevant antigens provoking inflammatory responses.
Translational aspects of new understandings
The intense study of immunological mechanisms in atherogenesis has yielded not only novel insights into its pathogenesis, but novel reagents useful as biomarkers, novel imaging agents, and even therapeutics. For example, as shown in Figure 3, Abs to OSEs have been used to develop biomarkers that provide independent prognostic value of CVD outcomes, to noninvasively image atherosclerotic lesions, or — when expressed in vivo in high titers — to inhibit atherogenesis. Another more widely applicable approach to enhancing titers of such Abs is a vaccine approach (7, 90). This is attractive because if the relevant antigen could be identified to produce high-titered oxLDL-neutralizing Ab titers, it could be widely applied in a cost-effective manner. Various OSEs of oxLDL have been shown to work in murine models, such as oxidized phospholipid epitopes and MDA, although whether these would be equally immunogenic in humans is unknown.
Figure 3. Translational applications of OSE Abs.

(A) E06 to oxidized phospholipid (oxPL) can be used to measure oxPL/apoB in human plasma as a biomarker of CVD. Because most oxPL is bound by Lp(A) lipoproteins, the assay primarily reflects the oxPL content on the most atherogenic Lp(A) particles (154). The 10-year predictive value of oxPL/apoB in death, myocardial infarction (MI), and stroke in the prospectively followed Bruneck population, which represents a general community, is shown (155). Groups 1, 2, and 3 represent the top, middle, and bottom tertiles, respectively. (B) Molecular imaging of murine atherosclerosis using MDA2, a malondialdehyde-specific monoclonal Ab. Nanoparticles consisting of micelles containing manganese (Mn) and MDA2 can increase relaxivity through binding to extracellular oxLDL, internalization into macrophages, and intracellular release as free Mn, thus becoming an indirect OSE-dependent macrophage-targeting agent. Noninvasive magnetic resonance imaging of OSEs is accomplished by injection of these nanoparticles and imaging of the abdominal aorta in cholesterol-fed Apoe–/– mice. Note the lack of signal at the preinjection scan and the strong signal (white contrast) in the 48-hour scan (156). Arrow indicates lesions in the abdominal aorta. (C) Therapeutic use of the human OSE Ab IK17 (as a scFv) to inhibit atherosclerosis. High-plasma titers of IK17 scFv were achieved in cholesterol-fed Ldlr–/–Rag1–/– mice via adenoviral-mediated hepatic expression, leading to a 46% reduction in en face atherosclerosis, compared with that in control mice treated with an adenovirus–enhanced green fluorescent protein vector (92). Importantly, peritoneal macrophages isolated from Adv-IK17-scFv–treated mice had decreased lipid accumulation, consistent with the ability of IK17 to inhibit oxLDL uptake by macrophages. Images reprinted with permission from Biomarkers in Medicine (154), Arteriosclerosis, Thrombosis, and Vascular Biology (155), and Journal of the American College of Cardiology (156).
Targeted blockage of immune cytokines and receptors, such as the use of Abs to TNF-α, the p40 subunit of IL-12/IL-23, or IL-17, are ongoing for inflammatory diseases, and prospective observation of these treated patients for the impact of the interventions on CVD outcomes may provide important insights for future therapy. Similarly, modulation of costimulatory and coinhibitory pathways is currently in use or in advanced stages of development for the treatment of autoimmune disease and cancer. CTLA-4 Ig is a CD80/CD87 blocker, which is approved for the treatment of rheumatoid arthritis. Blocking Abs specific for CTLA-4 or PD-1 are both being used to enhance T cell–mediated antitumor responses in cancer patients (153). Therapeutic blockade of TNF/TNFR superfamily members such as the OX40 ligand/OX40 pathway may be effective for inhibiting CD4+CD28– T cells in the setting of ACS (141). In addition, B cell–depletion strategies are being investigated as well, and their impact on B-2 and B-1 cells and on CVD needs to be determined. Finally, we await with great interest the outcome of an ongoing trial of the ability of canakinumab, a human monoclonal antibody that neutralizes IL-1b, to reduce CVD in high-risk patients with existing CVD. This therapy has minimal effects on lipoprotein levels but does inhibit CRP levels, indicative of a generalized ability to inhibit inflammation (157). This placebo-controlled study in approximately 17,000 patients should be a key test of the hypothesis that inhibition of inflammation will be an important new strategy to reduce the burden of CVD (158).
The recognition that both innate and adaptive immunity due intimately involved in atherogenesis has provided novel targets to reduce the proinflammatory milieu of the developing atherosclerotic lesion, as discussed throughout this Review. Above all, one should not forget that hypercholesterolemia per se appears to be the primary mechanism driving the entire inflammatory cascade, and numerous experimental and clinical studies document that lowering plasma cholesterol effectively reduces inflammation. Because such hypolipidemic therapy most often only begins in midlife in the developed world, when atherosclerotic lesions are almost universal and already established as chronic inflammatory lesions, direct interventions to inhibit inflammation will be needed to provide the maximal opportunity to reduce progression and even effect regression. Thus, targeting one or more of the immunological mechanisms that control inflammation may be of great value. However, it is important to recognize the complexity of immunological control, and any immunotherapeutic strategies utilized for the specific purpose of reducing atherogenesis, be it therapeutic use of Abs, vaccination, or targeted inhibition of adaptive proinflammatory responses, should be approached with great care. The new opportunities to examine the impact of interventions aimed at autoimmune diseases should be utilized to examine biomarkers, imaging modalities of CVD, and CVD outcomes. Such knowledge may provide important clues to the role of adaptive immunity in human atherosclerosis.
Acknowledgments
We thank our many colleagues with whom we work for their helpful suggestions.
Footnotes
Conflict of interest: Joseph L. Witztum and Sotirios Tsimikas have patents and patent applications for the commercial use of antibodies specific for oxidation-specific epitopes that are assigned to the UCSD.
Citation for this article: J Clin Invest. 2013;123(1):27–36. doi:10.1172/JCI63108.
References
- 1.Steinberg D, Witztum JL. Oxidized low-density lipoprotein and atherosclerosis. Arterioscler Thromb Vasc Biol. 2010;30(12):2311–2316. doi: 10.1161/ATVBAHA.108.179697. [DOI] [PubMed] [Google Scholar]
- 2.Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145(3):341–355. doi: 10.1016/j.cell.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Packard RR, Lichtman AH, Libby P. Innate and adaptive immunity in atherosclerosis. Semin Immunopathol. 2009;31(1):5–22. doi: 10.1007/s00281-009-0153-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weber C, Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med. 2011;17(11):1410–1422. doi: 10.1038/nm.2538. [DOI] [PubMed] [Google Scholar]
- 5.Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. 2011;12(3):204–212. doi: 10.1038/ni.2001. [DOI] [PubMed] [Google Scholar]
- 6.Libby P. Counterregulation rules in atherothrombosis. J Am Coll Cardiol. 2012;59(16):1438–1440. doi: 10.1016/j.jacc.2012.01.023. [DOI] [PubMed] [Google Scholar]
- 7.Miller YI, et al. Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ Res. 2011;108(2):235–248. doi: 10.1161/CIRCRESAHA.110.223875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosenfeld ME, Campbell LA. Pathogens and atherosclerosis: update on the potential contribution of multiple infectious organisms to the pathogenesis of atherosclerosis. Thromb Haemost. 2011;106(5):858–867. doi: 10.1160/TH11-06-0392. [DOI] [PubMed] [Google Scholar]
- 9.Tufano A, et al. The infectious burden in atherothrombosis. Semin Thromb Hemost. 2012;38(5):515–523. doi: 10.1055/s-0032-1315759. [DOI] [PubMed] [Google Scholar]
- 10.Wick G, Knoflach M, Xu Q. Autoimmune and inflammatory mechanisms in atherosclerosis. Annu Rev Immunol. 2004;22:361–403. doi: 10.1146/annurev.immunol.22.012703.104644. [DOI] [PubMed] [Google Scholar]
- 11.Duewell P, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464(7293):1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang MK, et al. Apoptotic cells with oxidation-specific epitopes are immunogenic and proinflammatory. J Exp Med. 2004;200(11):1359–1370. doi: 10.1084/jem.20031763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mallat Z, Taleb S, Ait-Oufella H, Tedgui A. The role of adaptive T cell immunity in atherosclerosis. J Lipid Res. 2009;50(suppl):S364–S369. doi: 10.1194/jlr.R800092-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Palinski W, et al. Low density lipoprotein undergoes oxidative modification in vivo. Proc Natl Acad Sci U S A. 1989;86(4):1372–1376. doi: 10.1073/pnas.86.4.1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ylä-Herttuala S, Palinski W, Butler SW, Picard S, Steinberg D, Witztum JL. Rabbit and human atherosclerotic lesions contain IgG that recognizes epitopes of oxidized LDL. Arterioscler Thromb. 1994;14(1):32–40. doi: 10.1161/01.ATV.14.1.32. [DOI] [PubMed] [Google Scholar]
- 16.Wade NS, Major AS. The problem of accelerated atherosclerosis in systemic lupus erythematosus: insights into a complex co-morbidity. Thromb Haemost. 2011;106(5):849–857. doi: 10.1160/TH11-05-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kitas GD, Gabriel SE. Cardiovascular disease in rheumatoid arthritis: state of the art and future perspectives. Ann Rheum Dis. 2011;70(1):8–14. doi: 10.1136/ard.2010.142133. [DOI] [PubMed] [Google Scholar]
- 18.Patel RV, Shelling ML, Prodanovich S, Federman DG, Kirsner RS. Psoriasis and vascular disease-risk factors and outcomes: a systematic review of the literature. J Gen Intern Med. 2011;26(9):1036–1049. doi: 10.1007/s11606-011-1698-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dansky HM, Charlton SA, Harper MM, Smith JD. T and B lymphocytes play a minor role in atherosclerotic plaque formation in the apolipoprotein E-deficient mouse. Proc Natl Acad Sci U S A. 1997;94(9):4642–4646. doi: 10.1073/pnas.94.9.4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daugherty A. The effects of total lymphocyte deficiency on the extent of atherosclerosis in apolipoprotein E–/– mice. . J Clin Invest. 1997;100(6):1575–1580. doi: 10.1172/JCI119681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou X, Nicoletti A, Elhage R, Hansson GK. Transfer of CD4+ T cells aggravates atherosclerosis in immunodeficient apolipoprotein E knockout mice. Circulation. 2000;102(24):2919–2922. doi: 10.1161/01.CIR.102.24.2919. [DOI] [PubMed] [Google Scholar]
- 22.Song L, Leung C, Schindler C. Lymphocytes are important in early atherosclerosis. J Clin Invest. 2001;108(2):251–259. doi: 10.1172/JCI11380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reardon CA, et al. Effect of immune deficiency on lipoproteins and atherosclerosis in male apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2001;21(6):1011–1016. doi: 10.1161/01.ATV.21.6.1011. [DOI] [PubMed] [Google Scholar]
- 24.Jonasson L, Holm J, Skalli O, Bondjers G, Hansson GK. Regional accumulations of T cells, macrophages, and smooth muscle cells in the human atherosclerotic plaque. Arteriosclerosis. 1986;6(2):131–138. doi: 10.1161/01.atv.6.2.131. [DOI] [PubMed] [Google Scholar]
- 25.Stemme S, Faber B, Holm J, Wiklund O, Witztum JL, Hansson GK. T lymphocytes from human atherosclerotic plaques recognize oxidized low density lipoprotein. Proc Natl Acad Sci U S A. 1995;92(9):3893–3897. doi: 10.1073/pnas.92.9.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hermansson A, et al. Inhibition of T cell response to native low-density lipoprotein reduces atherosclerosis. J Exp Med. 2010;207(5):1081–1093. doi: 10.1084/jem.20092243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ketelhuth DF, Hansson GK. Cellular immunity, low-density lipoprotein and atherosclerosis: break of tolerance in the artery wall. Thromb Haemost. 2011;106(5):779–786. doi: 10.1160/TH11-05-0321. [DOI] [PubMed] [Google Scholar]
- 28.Getz GS, Vanderlaan PA, Reardon CA. Natural killer T cells in lipoprotein metabolism and atherosclerosis. Thromb Haemost. 2011;106(5):814–819. doi: 10.1160/TH11-05-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Betjes MG, Meijers RW, de Wit LE, Litjens NH. A killer on the road: circulating CD4(+)CD28null T cells as cardiovascular risk factor in ESRD patients. J Nephrol. 2012;25(2):183–191. doi: 10.5301/jn.5000057. [DOI] [PubMed] [Google Scholar]
- 30.Olofsson PS. Targeting T cell costimulation to prevent atherothrombosis. Circ Res. 2012;110(6):800–801. doi: 10.1161/CIRCRESAHA.112.265108. [DOI] [PubMed] [Google Scholar]
- 31.Buono C, Come CE, Stavrakis G, Maguire GF, Connelly PW, Lichtman AH. Influence of interferon-gamma on the extent and phenotype of diet-induced atherosclerosis in the LDLR-deficient mouse. Arterioscler Thromb Vasc Biol. 2003;23(3):454–460. doi: 10.1161/01.ATV.0000059419.11002.6E. [DOI] [PubMed] [Google Scholar]
- 32.Gupta S, Pablo AM, Jiang X, Wang N, Tall AR, Schindler C. IFN-gamma potentiates atherosclerosis in ApoE knock-out mice. J Clin Invest. 1997;99(11):2752–2761. doi: 10.1172/JCI119465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Buono C, Binder CJ, Stavrakis G, Witztum JL, Glimcher LH, Lichtman AH. T-bet deficiency reduces atherosclerosis and alters plaque antigen-specific immune responses. Proc Natl Acad Sci U S A. 2005;102(5):1596–1601. doi: 10.1073/pnas.0409015102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Whitman SC, Ravisankar P, Elam H, Daugherty A. Exogenous interferon-[gamma] enhances atherosclerosis in apolipoprotein E–/– mice. . Am J Pathol. 2000;157(6):1819–1824. doi: 10.1016/S0002-9440(10)64820-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ait-Oufella H, Taleb S, Mallat Z, Tedgui A. Recent advances on the role of cytokines in atherosclerosis. Arterioscler Thromb Vasc Biol. 2011;31(5):969–979. doi: 10.1161/ATVBAHA.110.207415. [DOI] [PubMed] [Google Scholar]
- 36.Davenport P, Tipping PG. The role of interleukin-4 and interleukin-12 in the progression of atherosclerosis in apolipoprotein E-deficient mice. Am J Pathol. 2003;163(3):1117–1125. doi: 10.1016/S0002-9440(10)63471-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Whitman SC, Ravisankar P, Daugherty A. Interleukin-18 enhances atherosclerosis in apolipoprotein E(–/–) mice through release of interferon-gamma. Circ Res. 2002;90(2):E34–E38. doi: 10.1161/hh0202.105292. [DOI] [PubMed] [Google Scholar]
- 38.Ryan C, et al. Association between biologic therapies for chronic plaque psoriasis and cardiovascular events: a meta-analysis of randomized controlled trials. JAMA. 2011;306(8):864–871. doi: 10.1001/jama.2011.1211. [DOI] [PubMed] [Google Scholar]
- 39.Tzellos T, Kyrgidis A, Zouboulis CC. J Eur Acad Dermatol Venereol. Re-evaluation of the risk for major adverse cardiovascular events in patients treated with anti-IL-12/23 biological agents for chronic plaque psoriasis: a meta-analysis of randomized controlled trials [published online ahead of print March 8, 2012]. doi: 10.1111/j.1468-3083.2012.04500.x . [DOI] [PubMed] [Google Scholar]
- 40.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 41.Taleb S, Tedgui A, Mallat Z. Interleukin-17: friend or foe in atherosclerosis? Curr Opin Lipidol. 2010;21(5):404–408. doi: 10.1097/MOL.0b013e32833dc7f9. [DOI] [PubMed] [Google Scholar]
- 42.de Boer OJ, et al. Differential expression of interleukin-17 family cytokines in intact and complicated human atherosclerotic plaques. J Pathol. 2010;220(4):499–508. doi: 10.1002/path.2667. [DOI] [PubMed] [Google Scholar]
- 43.Taleb S, et al. Loss of SOCS3 expression in T cells reveals a regulatory role for interleukin-17 in atherosclerosis. J Exp Med. 2009;206(10):2067–2077. doi: 10.1084/jem.20090545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eid RE, et al. Interleukin-17 and interferon-gamma are produced concomitantly by human coronary artery-infiltrating T cells and act synergistically on vascular smooth muscle cells. Circulation. 2009;119(10):1424–1432. doi: 10.1161/CIRCULATIONAHA.108.827618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang X, et al. Interleukin-17A gene variants and risk of coronary artery disease: a large angiography-based study. Clin Chim Acta. 2011;412(3–4):327–331. doi: 10.1016/j.cca.2010.10.027. [DOI] [PubMed] [Google Scholar]
- 46.Wang Z, et al. Increased Th17 cells in coronary artery disease are associated with neutrophilic inflammation. Scand Cardiovasc J. 2011;45(1):54–61. doi: 10.3109/14017431.2010.491123. [DOI] [PubMed] [Google Scholar]
- 47.Hashmi S, Zeng QT. Role of interleukin-17 and interleukin-17-induced cytokines interleukin-6 and interleukin-8 in unstable coronary artery disease. Coron Artery Dis. 2006;17(8):699–706. doi: 10.1097/01.mca.0000236288.94553.b4. [DOI] [PubMed] [Google Scholar]
- 48.Cheng X, et al. The Th17/Treg imbalance in patients with acute coronary syndrome. Clin Immunol. 2008;127(1):89–97. doi: 10.1016/j.clim.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 49.Zhao Z, et al. Activation of Th17/Th1 and Th1, but not Th17, is associated with the acute cardiac event in patients with acute coronary syndrome. Atherosclerosis. 2011;217(2):518–524. doi: 10.1016/j.atherosclerosis.2011.03.043. [DOI] [PubMed] [Google Scholar]
- 50.Patel KD, et al. Interleukin 17: an unlikely marker of acute coronary syndrome? Atherosclerosis. 2009;205(1):33–34. doi: 10.1016/j.atherosclerosis.2008.11.022. [DOI] [PubMed] [Google Scholar]
- 51.Erbel C. Inhibition of IL-17A attenuates atherosclerotic lesion development in apoE-deficient mice. J Immunol. 2009;183(12):8167–8175. doi: 10.4049/jimmunol.0901126. [DOI] [PubMed] [Google Scholar]
- 52.Gao Q, et al. A critical function of Th17 proinflammatory cells in the development of atherosclerotic plaque in mice. J Immunol. 2010;185(10):5820–5827. doi: 10.4049/jimmunol.1000116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Usui F, et al. Interleukin-17 deficiency reduced vascular inflammation and development of atherosclerosis in Western diet-induced apoE-deficient mice. Biochem Biophys Res Commun. 2012;420(1):72–77. doi: 10.1016/j.bbrc.2012.02.117. [DOI] [PubMed] [Google Scholar]
- 54.Butcher MJ, Gjurich BN, Phillips T, Galkina EV. The IL-17A/IL-17RA axis plays a proatherogenic role via the regulation of aortic myeloid cell recruitment. Circ Res. 2012;110(5):675–687. doi: 10.1161/CIRCRESAHA.111.261784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.van Es T. Attenuated atherosclerosis upon IL-17R signaling disruption in LDLr deficient mice. Biochem Biophys Res Commun. 2009;388(2):261–265. doi: 10.1016/j.bbrc.2009.07.152. [DOI] [PubMed] [Google Scholar]
- 56.Chen S, Shimada K, Zhang W, Huang G, Crother TR, Arditi M. IL-17A is proatherogenic in high-fat diet-induced and chlamydia pneumoniae infection-accelerated atherosclerosis in mice. J Immunol. 2010;185(9):5619–5627. doi: 10.4049/jimmunol.1001879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Danzaki K, et al. Interleukin-17A deficiency accelerates unstable atherosclerotic plaque formation in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2012;32(2):273–280. doi: 10.1161/ATVBAHA.111.229997. [DOI] [PubMed] [Google Scholar]
- 58.Cheng X, et al. Inhibition of IL-17A in atherosclerosis. Atherosclerosis. 2011;215(2):471–474. doi: 10.1016/j.atherosclerosis.2010.12.034. [DOI] [PubMed] [Google Scholar]
- 59.Madhur MS, et al. Role of interleukin 17 in Inflammation, atherosclerosis, and vascular function in apolipoprotein E–deficient mice. Arterioscler Thromb Vasc Biol. 2011;31(7):1565–1572. doi: 10.1161/ATVBAHA.111.227629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ait-Oufella H, et al. B cell depletion reduces the development of atherosclerosis in mice. J Exp Med. 2010;207(8):1579–1587. doi: 10.1084/jem.20100155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wilke CM, Bishop K, Fox D, Zou W. Deciphering the role of Th17 cells in human disease. Trends Immunol. 2011;32(12):603–611. doi: 10.1016/j.it.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Eid RE, et al. Interleukin-17 and interferon-[gamma] are produced concomitantly by human coronary artery-infiltrating T cells and act synergistically on vascular smooth muscle cells. Circulation. 2009;119(10):1424–1432. doi: 10.1161/CIRCULATIONAHA.108.827618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee YK, et al. Late developmental plasticity in the T helper 17 lineage. Immunity. 2009;30(1):92–107. doi: 10.1016/j.immuni.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Leonardi C, et al. Anti-interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N Engl J Med. 2012;366(13):1190–1199. doi: 10.1056/NEJMoa1109997. [DOI] [PubMed] [Google Scholar]
- 65.Papp KA, et al. Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. N Engl J Med. 2012;366(13):1181–1189. doi: 10.1056/NEJMoa1109017. [DOI] [PubMed] [Google Scholar]
- 66.Waisman A. To be 17 again—anti-interleukin-17 treatment for psoriasis. N Engl J Med. 2012;366(13):1251–1252. doi: 10.1056/NEJMe1201071. [DOI] [PubMed] [Google Scholar]
- 67.Zhou X, Paulsson G, Stemme S, Hansson GK. Hypercholesterolemia is associated with a T helper (Th) 1/Th2 switch of the autoimmune response in atherosclerotic apo E-knockout mice. J Clin Invest. 1998;101(8):1717–1725. doi: 10.1172/JCI1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.King VL, Szilvassy SJ, Daugherty A. Interleukin-4 deficiency decreases atherosclerotic lesion formation in a site-specific manner in female LDL receptor–/– mice. . Arterioscler Thromb Vasc Biol. 2002;22(3):456–461. doi: 10.1161/hq0302.104905. [DOI] [PubMed] [Google Scholar]
- 69.King VL, Cassis LA, Daugherty A. Interleukin-4 does not influence development of hypercholesterolemia or angiotensin II-induced atherosclerotic lesions in mice. Am J Pathol. 2007;171(6):2040–2047. doi: 10.2353/ajpath.2007.060857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cardilo-Reis L, et al. Interleukin-13 protects from atherosclerosis and modulates plaque composition by skewing the macrophage phenotype. EMBO Mol Med. 2012;4(10):1072–1086. doi: 10.1002/emmm.201201374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Binder CJ, et al. IL-5 links adaptive and natural immunity specific for epitopes of oxidized LDL and protects from atherosclerosis. J Clin Invest. 2004;114(3):427–437. doi: 10.1172/JCI20479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Corren J. Inhibition of interleukin-5 for the treatment of eosinophilic diseases. Discov Med. 2012;13(71):305–312. [PubMed] [Google Scholar]
- 73.Kleindienst R, Xu Q, Willeit J, Waldenberger FR, Weimann S, Wick G. Immunology of atherosclerosis. Demonstration of heat shock protein 60 expression and T lymphocytes bearing alpha/beta or gamma/delta receptor in human atherosclerotic lesions. Am J Pathol. 1993;142(6):1927–1937. [PMC free article] [PubMed] [Google Scholar]
- 74.van der Wal AC, Das PK, Bentz van de Berg D, van der Loos CM, Becker AE. Atherosclerotic lesions in humans. In situ immunophenotypic analysis suggesting an immune mediated response. Lab Invest. 1989;61(2):166–170. [PubMed] [Google Scholar]
- 75.Bu DX, et al. Impairment of the programmed cell death-1 pathway increases atherosclerotic lesion development and inflammation. Arterioscler Thromb Vasc Biol. 2011;31(5):1100–1107. doi: 10.1161/ATVBAHA.111.224709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kolbus D, et al. CD8+ T cell activation predominate early immune responses to hypercholesterolemia in Apoe–/– mice. . BMC Immunol. 2010;11:58. doi: 10.1186/1471-2172-11-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Elhage R, et al. Deleting TCR alpha beta+ or CD4+ T lymphocytes leads to opposite effects on site-specific atherosclerosis in female apolipoprotein E-deficient mice. Am J Pathol. 2004;165(6):2013–2018. doi: 10.1016/S0002-9440(10)63252-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kolbus D, et al. TAP1-deficiency does not alter atherosclerosis development in Apoe–/– mice. . PLoS One. 2012;7(3):e33932. doi: 10.1371/journal.pone.0033932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Binder CJ, et al. Pneumococcal vaccination decreases atherosclerotic lesion formation: molecular mimicry between Streptococcus pneumoniae and oxidized LDL. Nat Med. 2003;9(6):736–743. doi: 10.1038/nm876. [DOI] [PubMed] [Google Scholar]
- 80.Sun J, et al. Deficiency of antigen-presenting cell invariant chain reduces atherosclerosis in mice. Circulation. 2010;122(8):808–820. doi: 10.1161/CIRCULATIONAHA.109.891887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lewis MJ, Malik TH, Ehrenstein MR, Boyle JJ, Botto M, Haskard DO. Immunoglobulin M is required for protection against atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation. 2009;120(5):417–426. doi: 10.1161/CIRCULATIONAHA.109.868158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Caligiuri G, Nicoletti A, Poirier B, Hansson GK. Protective immunity against atherosclerosis carried by B cells of hypercholesterolemic mice. J Clin Invest. 2002;109(6):745–753. doi: 10.1172/JCI07272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kyaw T, et al. B1a B lymphocytes are atheroprotective by secreting natural IgM that increases IgM deposits and reduces necrotic cores in atherosclerotic lesions. Circ Res. 2011;109(8):830–840. doi: 10.1161/CIRCRESAHA.111.248542. [DOI] [PubMed] [Google Scholar]
- 84.Ravandi A, et al. Relationship of IgG and IgM autoantibodies and immune complexes to oxidized LDL with markers of oxidation and inflammation and cardiovascular events: results from the EPIC-Norfolk Study. J Lipid Res. 2011;52(10):1829–1836. doi: 10.1194/jlr.M015776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Griffin DO, Holodick NE, Rothstein TL. Human B1 cells in umbilical cord and adult peripheral blood express the novel phenotype CD20+ CD27+ CD43+ CD70. J Exp Med. 2011;208(1):67–80. doi: 10.1084/jem.20101499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Binder CJ, et al. Innate and acquired immunity in atherogenesis. Nat Med. 2002;8(11):1218–1226. doi: 10.1038/nm1102-1218. [DOI] [PubMed] [Google Scholar]
- 87.Gounopoulos P, Merki E, Hansen LF, Choi SH, Tsimikas S. Antibodies to oxidized low density lipoprotein: epidemiological studies and potential clinical applications in cardiovascular disease. Minerva Cardioangiol. 2007;55(6):821–837. [PubMed] [Google Scholar]
- 88.Tsimikas S, Palinski W, Witztum JL. Circulating autoantibodies to oxidized LDL correlate with arterial accumulation and depletion of oxidized LDL in LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2001;21(1):95–100. doi: 10.1161/01.ATV.21.1.95. [DOI] [PubMed] [Google Scholar]
- 89.Palinski W, Miller E, Witztum JL. Immunization of low density lipoprotein (LDL) receptor-deficient rabbits with homologous malondialdehyde-modified LDL reduces atherogenesis. Proc Natl Acad Sci U S A. 1995;92(3):821–825. doi: 10.1073/pnas.92.3.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hansson GK, Nilsson J. Vaccination against atherosclerosis? Induction of atheroprotective immunity. Semin Immunopathol. 2009;31(1):95–101. doi: 10.1007/s00281-009-0151-x. [DOI] [PubMed] [Google Scholar]
- 91.Nilsson J, Björkbacka H, Fredrikson GN. Apolipoprotein B100 autoimmunity and atherosclerosis — disease mechanisms and therapeutic potential. Curr Opin Lipidol. 2012;23(5):422–428. doi: 10.1097/MOL.0b013e328356ec7c. [DOI] [PubMed] [Google Scholar]
- 92.Tsimikas S, et al. Human oxidation-specific antibodies reduce foam cell formation and atherosclerosis progression. J Am Coll Cardiol. 2011;58(16):1715–1727. doi: 10.1016/j.jacc.2011.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Major AS, Fazio S, Linton MF. B-lymphocyte deficiency increases atherosclerosis in LDL receptor-null mice. Arterioscler Thromb Vasc Biol. 2002;22(11):1892–1898. doi: 10.1161/01.ATV.0000039169.47943.EE. [DOI] [PubMed] [Google Scholar]
- 94.Kyaw T, et al. Conventional B2 B cell depletion ameliorates whereas its adoptive transfer aggravates atherosclerosis. J Immunol. 2010;185(7):4410–4419. doi: 10.4049/jimmunol.1000033. [DOI] [PubMed] [Google Scholar]
- 95.Kyaw T, et al. Depletion of B2 but not B1a B cells in BAFF receptor-deficient ApoE mice attenuates atherosclerosis by potently ameliorating arterial inflammation. PLoS One. 2012;7(1):e29371. doi: 10.1371/journal.pone.0029371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sage AP, et al. BAFF receptor deficiency reduces the development of atherosclerosis in mice—brief report. Arterioscler Thromb Vasc Biol. 2012;32(7):1573–1576. doi: 10.1161/ATVBAHA.111.244731. [DOI] [PubMed] [Google Scholar]
- 97.Doran AC, et al. B-cell aortic homing and atheroprotection depend on Id3. Circ Res. 2012;110(1):e1–e12. doi: 10.1161/CIRCRESAHA.111.256438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mauri C, Bosma A. Immune regulatory function of B cells. Annu Rev Immunol. 2012;30:221–241. doi: 10.1146/annurev-immunol-020711-074934. [DOI] [PubMed] [Google Scholar]
- 99.Griffin DO, Rothstein TL. Human “orchestrator” CD11b(+) B1 cells spontaneously secrete IL-10 and regulate T cell activity. Mol Med. 2012;18(9):1003–1008. doi: 10.2119/molmed.2012.00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kelly JA, et al. Inhibition of arterial lesion progression in CD16-deficient mice: evidence for altered immunity and the role of IL-10. Cardiovasc Res. 2010;85(1):224–231. doi: 10.1093/cvr/cvp300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hernandez-Vargas P, et al. Fcgamma receptor deficiency confers protection against atherosclerosis in apolipoprotein E knockout mice. Circ Res. 2006;99(11):1188–1196. doi: 10.1161/01.RES.0000250556.07796.6c. [DOI] [PubMed] [Google Scholar]
- 102.Zhao M, et al. FcgammaRIIB inhibits the development of atherosclerosis in low-density lipoprotein receptor-deficient mice. J Immunol. 2010;184(5):2253–2260. doi: 10.4049/jimmunol.0902654. [DOI] [PubMed] [Google Scholar]
- 103.Mendez-Fernandez YV, et al. The inhibitory FcgammaRIIb modulates the inflammatory response and influences atherosclerosis in male apoE(–/–) mice. Atherosclerosis. 2011;214(1):73–80. doi: 10.1016/j.atherosclerosis.2010.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang J, et al. IgE stimulates human and mouse arterial cell apoptosis and cytokine expression and promotes atherogenesis in Apoe–/– mice. . J Clin Invest. 2011;121(9):3564–3577. doi: 10.1172/JCI46028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Speidl WS, Kastl SP, Huber K, Wojta J. Complement in atherosclerosis: friend or foe? J Thromb Haemost. 2011;9(3):428–440. doi: 10.1111/j.1538-7836.2010.04172.x. [DOI] [PubMed] [Google Scholar]
- 106.Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. 2012;30:531–564. doi: 10.1146/annurev.immunol.25.022106.141623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat Immunol. 2007;8(12):1353–1362. doi: 10.1038/ni1536. [DOI] [PubMed] [Google Scholar]
- 108.Wing K, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322(5899):271–275. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- 109.Qureshi OS, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science. 2011;332(6029):600–603. doi: 10.1126/science.1202947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.von Boehmer H. Mechanisms of suppression by suppressor T cells. Nat Immunol. 2005;6(4):338–344. doi: 10.1038/ni1180. [DOI] [PubMed] [Google Scholar]
- 111.Gotsman I, et al. Impaired regulatory T-cell response and enhanced atherosclerosis in the absence of inducible costimulatory molecule. Circulation. 2006;114(19):2047–2055. doi: 10.1161/CIRCULATIONAHA.106.633263. [DOI] [PubMed] [Google Scholar]
- 112.Maganto-Garcia E, Tarrio ML, Grabie N, Bu DX, Lichtman AH. Dynamic changes in regulatory T cells are linked to levels of diet-induced hypercholesterolemia. Circulation. 2011;124(2):185–195. doi: 10.1161/CIRCULATIONAHA.110.006411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ng HP, Burris RL, Nagarajan S. Attenuated atherosclerotic lesions in apoE-Fcγ-chain-deficient hyperlipidemic mouse model is associated with inhibition of Th17 cells and promotion of regulatory T cells. J Immunol. 2011;187(11):6082–6093. doi: 10.4049/jimmunol.1004133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Engel D, et al. Caveolin-1 deficiency decreases atherosclerosis by hampering leukocyte influx into the arterial wall and generating a regulatory T-cell response. FASEB J. 2011;25(11):3838–3848. doi: 10.1096/fj.11-183350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Weber C, et al. CCL17-expressing dendritic cells drive atherosclerosis by restraining regulatory T cell homeostasis in mice. J Clin Invest. 2011;121(7):2898–2910. doi: 10.1172/JCI44925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Taleb S, et al. Defective leptin/leptin receptor signaling improves regulatory T cell immune response and protects mice from atherosclerosis. Arterioscler Thromb Vasc Biol. 2007;27(12):2691–2698. doi: 10.1161/ATVBAHA.107.149567. [DOI] [PubMed] [Google Scholar]
- 117.de Boer OJ, van der Meer JJ, Teeling P, van der Loos CM, van der Wal AC. Low numbers of FOXP3 positive regulatory T cells are present in all developmental stages of human atherosclerotic lesions. PLoS One. 2007;2(8):e779. doi: 10.1371/journal.pone.0000779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Patel S, Chung SH, White G, Bao S, Celermajer DS. The “atheroprotective” mediators apolipoproteinA-I and Foxp3 are over-abundant in unstable carotid plaques. Int J Cardiol. 2010;145(2):183–187. doi: 10.1016/j.ijcard.2009.05.024. [DOI] [PubMed] [Google Scholar]
- 119.Kofler S, Sisic Z, Shvets N, Lohse P, Weis M. Expression of circulatory dendritic cells and regulatory T-cells in patients with different subsets of coronary artery disease. J Cardiovasc Pharmacol. 2011;57(5):542–549. doi: 10.1097/FJC.0b013e3182124c53. [DOI] [PubMed] [Google Scholar]
- 120.Potekhina AV, et al. CD4(+)CD25(high)CD127(low) regulatory T cells in patients with stable angina and their dynamics after intracoronary sirolimus-eluting stent implantation. Hum Immunol. 2011;72(7):553–557. doi: 10.1016/j.humimm.2011.03.015. [DOI] [PubMed] [Google Scholar]
- 121.Meng X, et al. Statins induce the accumulation of regulatory T cells in atherosclerotic plaque. Mol Med. 2012;18(1):598–605. doi: 10.2119/molmed.2011.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhang D, et al. Effect of oral atorvastatin on CD4+CD25+ regulatory T cells, FoxP3 expression, and prognosis in patients with ST-segment elevated myocardial infarction before primary percutaneous coronary intervention. J Cardiovasc Pharmacol. 2011;57(5):536–541. doi: 10.1097/FJC.0b013e318211d016. [DOI] [PubMed] [Google Scholar]
- 123.Mausner-Fainberg K, et al. The effect of HMG-CoA reductase inhibitors on naturally occurring CD4+CD25+ T cells. Atherosclerosis. 2008;197(2):829–839. doi: 10.1016/j.atherosclerosis.2007.07.031. [DOI] [PubMed] [Google Scholar]
- 124.Bu DX, Griffin G, Lichtman AH. Mechanisms for the anti-inflammatory effects of statins. Curr Opin Lipidol. 2011;22(3):165–170. doi: 10.1097/MOL.0b013e3283453e41. [DOI] [PubMed] [Google Scholar]
- 125.Itano AA, Jenkins MK. Antigen presentation to naive CD4 T cells in the lymph node. Nat Immunol. 2003;4(8):733–739. doi: 10.1038/ni957. [DOI] [PubMed] [Google Scholar]
- 126.Paulson KE, Zhu S-N, Chen M, Nurmohamed S, Jongstra-Bilen J, Cybulsky MI. Resident intimal dendritic cells accumulate lipid and contribute to the initiation of atherosclerosis. Circ Res. 2010;106(2):383–390. doi: 10.1161/CIRCRESAHA.109.210781. [DOI] [PubMed] [Google Scholar]
- 127.Ma-Krupa W, Jeon M-S, Spoerl S, Tedder TF, Goronzy JJ, Weyand CM. Activation of arterial wall dendritic cells and breakdown of self-tolerance in giant cell arteritis. J Exp Med. 2004;199(2):173–183. doi: 10.1084/jem.20030850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Koltsova EK, Ley K. How dendritic cells shape atherosclerosis. Trends Immunol. 2011;32(11):540–547. doi: 10.1016/j.it.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Butcher MJ, Galkina EV. Phenotypic and functional heterogeneity of macrophages and dendritic cell subsets in the healthy and atherosclerosis-prone aorta. Front Physiol. 2012;3:44. doi: 10.3389/fphys.2012.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Packard RR, Maganto-Garcia E, Gotsman I, Tabas I, Libby P, Lichtman AH. CD11c(+) dendritic cells maintain antigen processing, presentation capabilities, and CD4(+) T-cell priming efficacy under hypercholesterolemic conditions associated with atherosclerosis. Circ Res. 2008;103(9):965–973. doi: 10.1161/CIRCRESAHA.108.185793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Koltsova EK, et al. Dynamic T cell-APC interactions sustain chronic inflammation in atherosclerosis. J Clin Invest. 2012;122(9):3114–3126. doi: 10.1172/JCI61758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Doring Y, et al. Auto-antigenic protein-DNA complexes stimulate plasmacytoid dendritic cells to promote atherosclerosis. Circulation. 2012;125(13):1673–1683. doi: 10.1161/CIRCULATIONAHA.111.046755. [DOI] [PubMed] [Google Scholar]
- 133.Lichtman AH. T cell costimulatory and coinhibitory pathways in vascular inflammatory diseases. Front Physiol. 2012;3:18. doi: 10.3389/fphys.2012.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.de Boer OJ, Hirsch F, van der Wal AC, van der Loos CM, Das PK, Becker AE. Costimulatory molecules in human atherosclerotic plaques: an indication of antigen specific T lymphocyte activation. Atherosclerosis. 1997;133(2):227–234. doi: 10.1016/S0021-9150(97)00135-4. [DOI] [PubMed] [Google Scholar]
- 135.Afek A, Harats D, Roth A, Keren G, George J. A functional role for inducible costimulator (ICOS) in atherosclerosis. Atherosclerosis. 2005;183(1):57–63. doi: 10.1016/j.atherosclerosis.2005.03.040. [DOI] [PubMed] [Google Scholar]
- 136.Buono C, Pang H, Uchida Y, Libby P, Sharpe AH, Lichtman AH. B7-1/B7-2 costimulation regulates plaque antigen-specific T-cell responses and atherogenesis in low-density lipoprotein receptor-deficient mice. Circulation. 2004;109(16):2009–2015. doi: 10.1161/01.CIR.0000127121.16815.F1. [DOI] [PubMed] [Google Scholar]
- 137.Wang X, et al. Positional identification of TNFSF4, encoding OX40 ligand, as a gene that influences atherosclerosis susceptibility. Nat Genet. 2005;37(4):365–372. doi: 10.1038/ng1524. [DOI] [PubMed] [Google Scholar]
- 138.Olofsson PS, et al. CD137 is expressed in human atherosclerosis and promotes development of plaque inflammation in hypercholesterolemic mice. Circulation. 2008;117(10):1292–1301. doi: 10.1161/CIRCULATIONAHA.107.699173. [DOI] [PubMed] [Google Scholar]
- 139.van Wanrooij EJ, van Puijvelde GH, de Vos P, Yagita H, van Berkel TJ, Kuiper J. Interruption of the Tnfrsf4/Tnfsf4 (OX40/OX40L) pathway attenuates atherogenesis in low-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2007;27(1):204–210. doi: 10.1161/01.ATV.0000251007.07648.81. [DOI] [PubMed] [Google Scholar]
- 140.Jeon HJ, et al. CD137 (4-1BB) deficiency reduces atherosclerosis in hyperlipidemic mice. Circulation. 2010;121(9):1124–1133. doi: 10.1161/CIRCULATIONAHA.109.882704. [DOI] [PubMed] [Google Scholar]
- 141.Dumitriu IE, et al. High levels of costimulatory receptors OX40 and 4-1BB characterize CD4+CD28null T cells in patients with acute coronary syndrome. Circ Res. 2012;110(6):857–869. doi: 10.1161/CIRCRESAHA.111.261933. [DOI] [PubMed] [Google Scholar]
- 142.Gotsman I, Grabie N, Dacosta R, Sukhova G, Sharpe A, Lichtman AH. Proatherogenic immune responses are regulated by the PD-1/PD-L pathway in mice. J Clin Invest. 2007;117(10):2974–2982. doi: 10.1172/JCI31344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lee J, et al. Contributions of PD-1/PD-L1 pathway to interactions of myeloid DCs with T cells in atherosclerosis. J Mol Cell Cardiol. 2009;46(2):169–176. doi: 10.1016/j.yjmcc.2008.10.028. [DOI] [PubMed] [Google Scholar]
- 144.Bour-Jordan H, Salomon BL, Thompson HL, Szot GL, Bernhard MR, Bluestone JA. Costimulation controls diabetes by altering the balance of pathogenic and regulatory T cells. J Clin Invest. 2004;114(7):979–987. doi: 10.1172/JCI20483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ait-Oufella H, et al. Natural regulatory T cells control the development of atherosclerosis in mice. Nat Med. 2006;12(2):178–180. doi: 10.1038/nm1343. [DOI] [PubMed] [Google Scholar]
- 146.Maron R, et al. Mucosal administration of heat shock protein-65 decreases atherosclerosis and inflammation in aortic arch of low-density lipoprotein receptor-deficient mice. Circulation. 2002;106(13):1708–1715. doi: 10.1161/01.CIR.0000029750.99462.30. [DOI] [PubMed] [Google Scholar]
- 147.van Puijvelde GH, et al. Induction of oral tolerance to HSP60 or an HSP60-peptide activates T cell regulation and reduces atherosclerosis. Arterioscler Thromb Vasc Biol. 2007;27(12):2677–2683. doi: 10.1161/ATVBAHA.107.151274. [DOI] [PubMed] [Google Scholar]
- 148.Zhong Y, et al. CD4+LAP+ and CD4+CD25 +Foxp3+ regulatory T cells induced by nasal oxidized low-density lipoprotein suppress effector T cells response and attenuate atherosclerosis in ApoE–/– mice. . J Clin Immunol. 2012;32(5):1104–1117. doi: 10.1007/s10875-012-9699-7. [DOI] [PubMed] [Google Scholar]
- 149.van Puijvelde GH, et al. Induction of oral tolerance to oxidized low-density lipoprotein ameliorates atherosclerosis. Circulation. 2006;114(18):1968–1976. doi: 10.1161/CIRCULATIONAHA.106.615609. [DOI] [PubMed] [Google Scholar]
- 150.George J, et al. Suppression of early atherosclerosis in LDL-receptor deficient mice by oral tolerance with beta 2-glycoprotein I. Cardiovasc Res. 2004;62(3):603–609. doi: 10.1016/j.cardiores.2004.01.028. [DOI] [PubMed] [Google Scholar]
- 151.Hermansson A, Johansson DK, Ketelhuth DF, Andersson J, Zhou X, Hansson GK. Immunotherapy with tolerogenic apolipoprotein B-100-loaded dendritic cells attenuates atherosclerosis in hypercholesterolemic mice. Circulation. 2011;123(10):1083–1091. doi: 10.1161/CIRCULATIONAHA.110.973222. [DOI] [PubMed] [Google Scholar]
- 152.Herbin O, et al. Regulatory T-cell response to apolipoprotein B100-derived peptides reduces the development and progression of atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2012;32(3):605–612. doi: 10.1161/ATVBAHA.111.242800. [DOI] [PubMed] [Google Scholar]
- 153.Topalian SL, Weiner GJ, Pardoll DM. Cancer immunotherapy comes of age. J Clin Oncol. 2011;29(36):4828–4836. doi: 10.1200/JCO.2011.38.0899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Taleb A, Witztum JL, Tsimikas S. Oxidized phospholipids on apoB-100-containing lipoproteins: a biomarker predicting cardiovascular disease and cardiovascular events. Biomark Med. 2011;5(5):673–694. doi: 10.2217/bmm.11.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kiechl S, et al. Oxidized phospholipids, lipoprotein(a), lipoprotein-associated phospholipase A2 activity, and 10-year cardiovascular outcomes: prospective results from the Bruneck study. Arterioscler Thromb Vasc Biol. 2007;27(8):1788–1795. doi: 10.1161/ATVBAHA.107.145805. [DOI] [PubMed] [Google Scholar]
- 156.Briley-Saebo KC, et al. In vivo detection of oxidation-specific epitopes in atherosclerotic lesions using biocompatible manganese molecular magnetic imaging probes. J Am Coll Cardiol. 2012;59(6):616–626. doi: 10.1016/j.jacc.2011.10.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ridker P, et al. Effects of interleukin-1b inhibition with canakinumab on hemoglobin A1c, lipids, C-reactive protein, interleukin-6, and fibrinogen: A Phase IIb Randomized, Placebo-Controlled Trial. Circulation. 2012;126(23):2739–2748. doi: 10.1161/CIRCULATIONAHA.112.122556. [DOI] [PubMed] [Google Scholar]
- 158.Ridker PM, Thuren T, Zalewski A, Libby P. Interleukin-1b inhibition and the prevention of recurrent cardiovascular events: rationale and design of the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS). Am Heart J. 2011;162(4):597–605. doi: 10.1016/j.ahj.2011.06.012. [DOI] [PubMed] [Google Scholar]