

Abstract
Low-grade chronic inflammation is a major characteristic of obesity and results from deregulated white adipose tissue function. Consequently, there is interest in identifying the underlying regulatory mechanisms and components that drive adipocyte inflammation. Here, we report that expression of the transcriptional corepressor complex subunits GPS2 and SMRT was significantly reduced in obese adipose tissue, inversely correlated to inflammatory status, and was restored upon gastric bypass surgery–induced weight loss in morbid obesity. These alterations correlated with reduced occupancy of the corepressor complex at inflammatory promoters, providing a mechanistic explanation for elevated inflammatory transcription. In support of these correlations, RNAi-mediated depletion of GPS2 and SMRT from cultured human adipocytes promoted derepression of inflammatory transcription and elevation of obesity-associated inflammatory markers, such as IL-6 and MCP-1. Furthermore, we identified a regulatory cascade containing PPARγ and TWIST1 that controlled the expression of GPS2 and SMRT in human adipocytes. These findings were clinically relevant, because treatment of diabetic obese patients with pioglitazone, an antidiabetic and antiinflammatory PPARγ agonist, restored expression of TWIST1, GPS2, and SMRT in adipose tissue. Collectively, our findings identify alterations in a regulatory transcriptional network in adipocytes involving the dysregulation of a specific corepressor complex as among the initiating events promoting adipose tissue inflammation in human obesity.
Introduction
Obesity is today understood to be a “metaflammatory” condition, characterized by both metabolic and inflammatory deteriorations. Inflammation is commonly known as a defense mechanism that protects the host from infection or trauma. Coordinated regulation of the inflammatory response, in particular its resolution, is essential to remain healthy and to maintain homeostasis. Failure to resolve inflammatory conditions leads to a broad spectrum of common diseases (1). While this has been long recognized in the case of classic inflammatory diseases, it is only recently that conditions of chronic low-grade inflammation associated with metabolic diseases have received attention, particularly in relation to obesity and related comorbidities (2). Obese subjects display moderate but chronically elevated circulating levels of inflammatory mediators such as IL-6 and TNF-α, and this chronic inflammatory status is usually associated with development of metabolic complications such as insulin resistance and liver steatosis (3–5).
Obesity complications are thought to be caused at least in part by adipose tissue–derived inflammatory mediators that trigger low-grade inflammation, linking them to inflammatory alterations. Recent efforts in the field are aimed at understanding how these apparently separate aspects are interconnected, raising substantial interest in identifying the regulatory pathways and components that are dysregulated in obese adipose tissue (6). At present, adipose tissue is considered to be a major endocrine gland that synthesizes and secretes a growing list of hormones, inflammatory mediators, and immune system effectors into the systemic circulation, including adiponectin and leptin (7–11). Among the candidates secreted by enlarged adipocytes, the increase in IL-6, IL-8, and monocyte chemotactic protein–1 (MCP-1; also known as CCL2) and the decrease in adiponectin are considered to be particularly important (12).
Obesity is an evolutionary process that goes through different phases, from an initial phase, characterized by a gradual increase in body weight and fat mass in response to energy imbalance, to a chronic phase, characterized by the occurrence of complications that increase morbidity and mortality (13–15). During these different phases, adipose tissue undergoes morphological modifications and inflammatory phenotype changes. During the weight gain phase, expansion of adipose tissue induces increased adipocyte size and then promotes immune cell infiltration. This adipocyte hypertrophy has been associated with its capability to produce inflammatory mediators. Adipocyte size, for example, determines the production of cytokines, such as IL-6 and IL-8, and key mediators of immune cell infiltration, such as MCP-1 (16). As a general model, “lean” adipocytes secrete factors, such as IL-13, that promote alternative activation of macrophages (i.e., the M2 subtype) and insulin-sensitizing factors, such as adiponectin. Alternatively activated M2 macrophages secrete antiinflammatory mediators, such as IL-10. In contrast, in “obese” adipocytes, overnutrition causes an increase in lipolysis and the release of proinflammatory nonesterified fatty acids (NEFAs) as well as factors that recruit and activate locally macrophages, such as MCP-1, IL-6, and IL-8. By secreting a large panel of chemoattractant molecules, both preadipocytes and mature adipocytes may play a role in the macrophage infiltration process (13).
An important step toward dissecting the molecular events that trigger inflammation of adipose tissue was the recognition that adipocytes display marked changes in gene expression of inflammatory mediators. Consistently, the NF-κB pathway, implicated in the direct transcriptional regulation of inflammatory responses in multiple cell types, appears to be activated in the majority of preadipocytes and mature adipocytes in obesity (17, 18). Bacterial LPS and saturated NEFAs, which are increased in obesity, are known to be strong inducers of NF-κB pathways. In addition, TLR4 is overexpressed in obese adipocytes (19). It was also reported that macrophage-derived TNF-α activates the TNF-α receptor–1 (TNFR-1) cascade in adipocytes, which could contribute to the costimulation of NF-κB, JNK, and AP-1 (also known as c-Jun) pathways (20).
The activation of these inflammatory transcription factors generally requires corepressor/coactivator exchange, making the coregulator matrix an important regulatory checkpoint of inflammatory gene expression (21, 22). Several coregulators appear to play major roles in adipocyte differentiation and metabolism, although their involvement in inflammatory gene expression or in obesity-mediated alterations in humans has not yet been demonstrated. For example, receptor interacting protein 140 (RIP140; also known as NRIP1) is expressed in white adipose tissue, where it regulates expression of genes involved in lipid metabolism and glucose homeostasis (23). RIP140-knockout mice exhibit elevated fatty acid oxidation, but surprisingly had reduced fat mass and body weight. 2 other coregulators, TLE3 and PGC1-α, have been proposed to contribute to adipogenesis and insulin sensitivity processes by influencing the activity of the nuclear receptor PPARγ (24). Based on current knowledge derived by recent studies, the primary transcription factors and the coregulators that control adipocyte differentiation appear to be distinct from those that control adipocyte inflammation (25).
A fundamental aspect of inflammatory transcription is the strict maintenance of gene repression in the absence of inflammatory signaling. Numerous studies emphasize the crucial involvement of transcriptional corepressor complexes linked to histone deacetylation in inflammatory gene regulation (20, 26, 27). Among various candidates, complexes assembled by 2 related corepressors, silencing mediator of retinoid and thyroid receptor (SMRT) and nuclear receptor corepressor 1 (NCOR1), have emerged as key players in the repression of inflammatory gene transcription, at least in macrophages (28) and in hepatocytes (29). A few studies have begun to address the biological function of these corepressors in murine adipocytes. Analysis of dominant-negative knockin mutants of SMRT in mice revealed a role during adipogenesis via direct repression of PPARγ (30). The first reported adipocyte-specific mouse model suggests that loss of NCOR1 phenocopies the effects of systemic PPARγ agonist treatment by increased adiposity, enhanced insulin sensitivity, and improved glucose tolerance (31). However, adipocyte inflammation was not elevated upon NCOR1 removal in mice, leaving open the question of whether these corepressors play roles in controlling adipocyte inflammation. These prior results, together with concerns about differences between mouse and human adipocyte pathways, emphasize the need to investigate this issue in the context of human adipose tissue.
In the present study, we aimed to identify those transcriptional alterations in human adipocytes that may be directly linked to the elevated inflammation status of obese adipose tissue. We found that 2 specific corepressor complex subunits, SMRT and G protein pathway suppressor 2 (GPS2), were differentially expressed and regulated in adipocytes of obese subjects, and investigated the functional consequences on inflammatory gene expression as well as the underlying regulatory mechanisms. Our work provides the first evidence to our knowledge that a specific transcriptional corepressor complex is dysregulated in human obesity and describes a transcriptional and potentially epigenetic reprogramming pathway that could be causally linked to metaflammation, i.e., the chronic inflammatory status of obese adipose tissue.
Results
SMRT and GPS2 expression negatively correlates with IL-6 expression in adipose tissue and in isolated adipocytes of obese subjects.
To identify obesity-associated transcriptional alterations, we revisited results from a previously reported microarray study (32) for biological pathways that were differentially regulated in subcutaneous adipose tissue of obese (BMI >30 kg/m2) compared with lean (BMI <28 kg/m2) subjects. Gene Ontology (GO) analysis revealed that, in addition to the known upregulation of inflammatory pathways, multiple transcriptional pathway components were dysregulated in obese adipose tissue. We specifically examined whether genes linked to the adipose tissue coregulator matrix were differentially regulated in this set of experiments. The microarray data revealed significantly reduced expression of the 2 corepressor complex subunits SMRT and GPS2 in obese adipose tissue. In agreement with previous studies (21–23), additional coactivators or corepressors, such as PGC1-α, TLE3, and RIP140, were also deregulated in obese adipose tissue (Supplemental Figure 1; supplemental material available online with this article; doi: 10.1172/JCI64052DS1).
To confirm the microarray results, we analyzed mRNA expression of all known corepressor complex subunits in subcutaneous adipose tissue of lean (n = 14) and obese (n = 36) subjects by quantitative real-time RT-PCR (qPCR; Table 1, Figure 1, and Supplemental Figure 2). GPS2 and SMRT expression was significantly decreased in adipose tissue of obese subjects (Figure 1, A and B), whereas other complex subunits, including the SMRT-related corepressor NCOR1, were not differentially expressed (Figure 1C and Supplemental Figure 2, D and E). As expected, gene expression of adiponectin was significantly reduced in the subcutaneous white adipose tissue of the obese group, whereas expression of leptin and IL-6 was significantly increased in those subjects (Figure 1, D–F). We also analyzed the expression profiles of GPS2, SMRT, and NCOR1 in relation to gender, but did not observe any gender-specific association in adipose tissue of obese subjects (Supplemental Figure 2, A–C).
Table 1.
Clinical parameters of lean and obese study populations
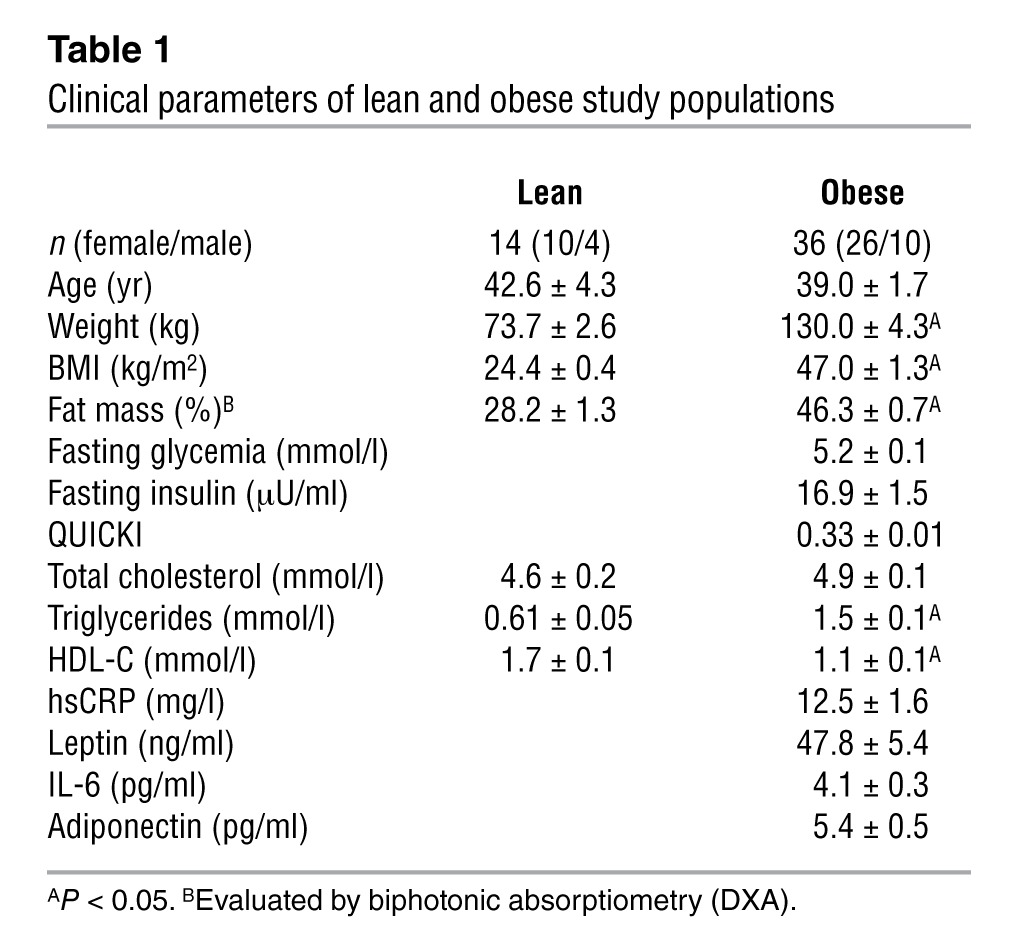
Figure 1. Expression of GPS2 and SMRT is negatively correlated with adipose tissue inflammation in human obesity.

qPCR analysis of mRNA expression of (A–C) the corepressor complex subunits SMRT, GPS2, and NCOR1; of (D and E) adiponectin and leptin; and of (F) IL-6 in subcutaneous adipose tissue of lean (n = 14) and obese (n = 36) male and female subjects. *P < 0.05. (G–I) Correlations were analyzed using Spearman statistical test.
To determine the association between expression of GPS2 and SMRT with inflammatory genes being deregulated in obesity, we conducted correlation studies among these different genes. We observed a significant inverse correlation between GPS2 and SMRT expression and between that of GPS2 and IL-6 (Figure 1, G and H). GPS2 gene expression was also positively correlated with adiponectin expression (Figure 1I). No significant correlations were identified between NCOR1 and IL-6 expression (Supplemental Figure 2, F–H). Adipose tissue is mainly composed of adipocytes, preadipocytes, and adipose tissue–associated immune cells. Previous studies have reported that adipose tissue inflammation is promoted in the first step of obesity by inflamed and hypertrophic adipocytes and preadipocytes (33). Therefore, we performed expression profiling in mature adipocytes of lean (n = 8) and obese (n = 12) subjects isolated from the subcutaneous adipose tissue. We confirmed that expression of SMRT and GPS2, but not that of NCOR1, was significantly reduced in isolated adipocytes of obese individuals (Figure 2, A–C). As was observed in obese adipose tissue, IL-6 expression was increased in the isolated adipocytes, whereas adiponectin expression was reduced (Figure 2, D and E). Correlation studies confirmed the association of GPS2 expression with SMRT and IL-6 levels (Figure 2, F and G, and Supplemental Figure 3A) and the positive correlation with adiponectin expression (Figure 2H), whereas no significant correlation was seen for NCOR1 (Supplemental Figure 3B). Finally, downregulation of GPS2 and SMRT, but not of NCOR1, in obese adipocytes was confirmed at the protein level by quantitative Western blot analysis (Figure 2I and Supplemental Figure 3C).
Figure 2. Adipocyte inflammation is associated with clearance of the SMRT-GPS2 complex from the IL-6 promoter.

(A–E) qPCR analysis of mRNA expression of SMRT, GPS2, NCOR1, adiponectin, and IL-6 in isolated human adipocytes from subcutaneous adipose tissue of lean (n = 8) and obese (n = 12) subjects. *P < 0.05. (F–H) Gene expression correlations were analyzed using Spearman statistical test. (I) Western blot analysis of protein levels of GPS2, SMRT, and NCOR1 in isolated human mature adipocytes from subcutaneous adipose tissue of lean and obese subjects (n = 3). (J) ChIP assays were performed to measure recruitment of GPS2, SMRT, NCOR1, HDAC3, and POL2 onto the IL-6 promoter, in conjunction with histone modifications (me2H3K9, indicative of repression, and me2H3K4 and acH3, indicative of activation), in isolated human adipocytes from subcutaneous adipose tissue of lean (n = 6) and obese (n = 5) subjects. IgG served as nonspecific control antibody.
Clearance of the SMRT-GPS2 complex from the IL-6 promoter triggers derepression of inflammatory gene expression in obese adipocytes.
To investigate the functional consequences of altered corepressor expression at the transcriptional level, we next studied the recruitment of GPS2, SMRT, and NCOR1 onto the IL-6 promoter in human adipocytes of lean and obese subjects using ChIP assays. In adipocytes of lean subjects, GPS2 and SMRT occupied the IL-6 promoter, while only low levels of RNA polymerase II (POL2) could be detected. In contrast, in adipocytes of obese subjects, GPS2 and SMRT recruitment was strongly reduced, and significantly increased recruitment of POL2 was observed (Figure 2J). The increased recruitment of POL2 was associated with changes of specific histone 3 marks linked to transcription activation (increased H3K4 methylation [me2H3K4] and H3 acetylation [acH3] and decreased H3K9 demethylation [me2H3K9]). Notably, the recruitment of HDAC3, the main histone deacetylase of the corepressor complex, mirrored expression of GPS2 and SMRT, while the subunit NCOR1 was not recruited onto the IL-6 promoter. These results support the concept that a corepressor complex containing SMRT, GPS2, and HDAC3 represses IL-6 expression in lean adipocytes, whereas IL-6 expression becomes derepressed due to corepressor complex clearance in obesity conditions.
Depletion of GPS2 and SMRT promotes adipocyte inflammation by increased expression of proinflammatory cytokines and chemoattractant molecules.
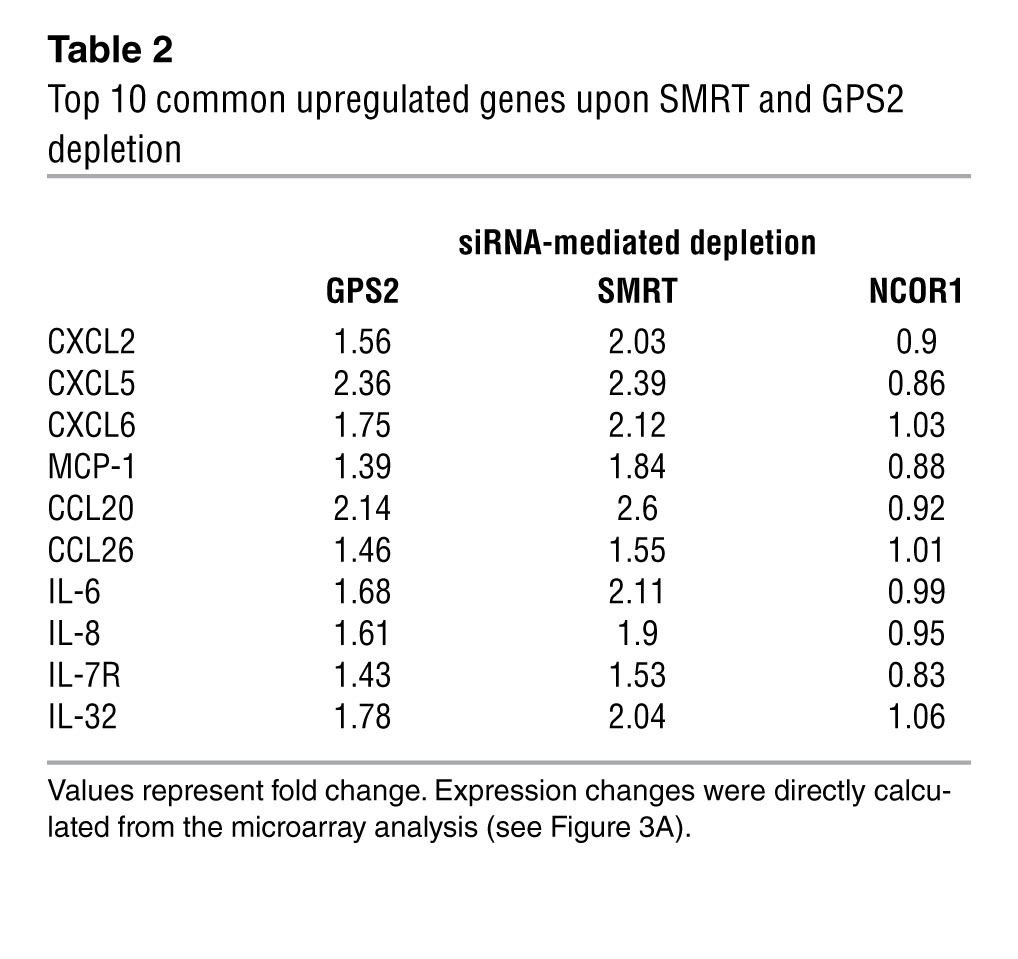
The critical involvement of SMRT and GPS2, but not of NCOR1, in regulating inflammatory gene expression in adipocytes was surprising, given the established role of NCOR1-containing complexes in inflammatory pathways in other tissues, such as macrophages and liver (29, 34). Our results suggest that, at least in part, distinct complexes could control transcription of different gene subsets in adipocytes. To identify which subset is controlled by GPS2, SMRT, and NCOR1 in human adipocytes, we depleted these 3 components individually by RNAi in preadipocytes and in isolated mature adipocytes (Figure 3A). We first validated the effectiveness and specificity of the individual siRNAs targeting GPS2, SMRT, or NCOR1 at the mRNA and protein levels (Supplemental Figure 4, A–F). We then analyzed and compared the genome-wide transcriptome signatures of preadipocytes and adipocytes in control and knockdown conditions. Depletion of GPS2, SMRT, and NCOR1 in preadipocytes caused upregulation of 159, 97, and 244 genes, respectively (Figure 3B). Unexpectedly, the number of upregulated (i.e., derepressed) genes was higher in differentiated adipocytes for GPS2 and SMRT (1,006 and 511, respectively), whereas only 14 genes were derepressed by NCOR1 (Figure 3C). Functional GO classification revealed that genes upregulated upon SMRT or GPS2 depletion that overlapped were linked to the NF-κB cascade, inflammatory response pathways, and immune cell chemotaxis (Supplemental Figure 5, A and B, Supplemental Figure 6, A–C, and Supplemental Table 7). In agreement with our correlation data, IL-6 expression was significantly upregulated in adipocytes depleted for GPS2 and SMRT, but not for NCOR1. Notably, GPS2 and SMRT appeared to control the expression of MCP-1 and CXCL2 (Table 2), which, being chemoattractants, play main roles in recruiting immune cells to proinflammatory adipocytes, thereby establishing the inflammatory status of obese adipose tissue.
Figure 3. Depletion of GPS2 or SMRT, but not of NCOR1, derepresses a subset of inflammatory genes in primary human adipocyte cultures.

(A) Genome-wide mRNA expression analysis using microarrays was performed in undifferentiated preadipocytes and adipocytes upon siRNA-mediated depletion of GPS2, SMRT, and NCOR1 (n = 6). (B and C) Repressed genes that were upregulated upon siRNA-mediated depletion as in A. Numbers in parentheses denote the number of upregulated/downregulated genes.
Table 2.
Top 10 common upregulated genes upon SMRT and GPS2 depletion
Microarray results were essentially confirmed by qPCR analysis of selected genes (Figure 4A and Supplemental Figure 7). Upregulation of key inflammatory genes, such as IL-6, IL-8, and MCP-1, upon depletion of GPS2 and SMRT was confirmed using different sets of individual siRNAs (Supplemental Figure 4 and data not shown). Moreover, the expression results based on mRNA quantification were further confirmed at the protein level by measuring the secretion of a large panel of cytokines (secretome) into the culture media (Figure 4B). Confirming the microarray results, GPS2 and SMRT depletion in adipocytes provoked secretion of chemoattractant cytokines, such as IL-8, CCL5, MCP-1, CXCL1, and CXCL10, that was higher in isolated adipocytes. Interestingly, inflammatory mediators, such as TNF-α and IL-11, were significantly less affected by depletion of GPS2 or SMRT. Notably, depletion of NCOR1 seemed to be associated with reduced expression of some inflammatory genes (such as MCP-1, CXCL10, CCL5, or MIF), fully consistent with recent findings on adipocyte-specific NCOR1 knockout mice (31).
Figure 4. Depletion of GPS2 and SMRT induces gene expression and secretion of inflammatory cytokines in primary human adipocyte cultures.

(A) Verification of the microarray data using qPCR and color map representation of inflammatory gene expression (transcriptome) under the experimental conditions outlined in Figure 3A. (B) Secretion of cytokines (secretome) was monitored by a membrane array (n = 3). (C) ChIP assays were performed to measure recruitment of GPS2, SMRT, POL2, and histone markers at the IL-6 promoter upon siRNA-mediated depletion of GPS2 and SMRT. CTL, control. IgG served as nonspecific control antibody. *P < 0.05.
We then analyzed the consequences of siRNA depletion on promoter occupancy and chromatin marks at the IL-6 promoter using ChIP assays, analogous to the experiments described in Figure 2J. SMRT and GPS2 were specifically recruited onto the IL-6 promoter under control conditions, whereas depletion of SMRT or GPS2 derepressed transcription, as shown by POL2 recruitment, reduced me2H3K9, and increased me2H3K4 and acH3 (Figure 4C). The combined results of expression analysis at the gene and protein level, chromatin recruitment, and siRNA depletion further supported the concept that regulation of proinflammatory genes in human mature adipocytes involves a corepressor complex containing SMRT and GPS2, but not NCOR1.
Adenovirus-mediated expression of GPS2 reverses the inflammatory phenotype of obese adipocytes and rescues siRNA-mediated depletion.
To further confirm the involvement of the SMRT-GPS2 complex in the transcriptional repression of inflammatory gene expression, we ectopically expressed HA-tagged human GPS2 using recombinant adenovirus particles in human isolated mature adipocytes from lean and obese subjects (Figure 5, A and B, and Supplemental Figure 9). We found that virus-mediated GPS2 expression significantly inhibited IL-6 and IL-8 expression in obese adipocytes (Figure 5A), consistent with recruitment of both SMRT and GPS2 onto the IL-6 promoter and decreased POL2 recruitment (Figure 5B).
Figure 5. Adenoviral expression of GPS2 represses cytokine expression in isolated human adipocytes and reverses siRNA- or cytokine-mediated derepression of IL-6 expression.

(A and B) Adenoviral expression of HA-GPS2 or GFP (as control) in human isolated mature adipocytes from subcutaneous adipose tissue of lean and obese subjects. (A) Gene expression of IL-6 and IL-8 was measured by qPCR. (B) Recruitment of GPS2, SMRT, and POL2 onto the IL-6 promoter was quantified by ChIP assays. (C–E) Gain-of-function experiments. Adenoviral expression of HA-GPS2 or GFP (as control) in isolated mature adipocytes from subcutaneous adipose tissue of lean subjects upon treatment with siRNAs and/or isolated cytokines, including ATCM. IL-6 mRNA expression was measured by qPCR. All data are representative of 5 different experiments. *P < 0.05.
A series of gain-of-function experiments was performed to study IL-6 regulation in human adipocytes under endogenous conditions (Figure 5, C–E) and using an ectopically expressed IL-6 reporter vector (Supplemental Figure 10). Adenoviral expression of GPS2 reversed the derepression of IL-6 expression mediated by RNAi-induced GPS2 depletion, but not SMRT depletion (Figure 5C), which confirmed that SMRT is required to repress IL-6 expression by GPS2. In addition, using the IL-6 reporter assay, we observed that plasmid-mediated overexpression of SMRT or GPS2 was capable of partially reversing siRNA-mediated derepression of IL-6 promoter activity (Supplemental Figure 10, B and C).
Cytokine stimulation by individual cytokines or by adipose tissue–conditioned medium from obese subjects (ATCM) in the above experiments revealed that virus-mediated GPS2 expression and rescue of siRNA depletion counteracted cytokine induction of IL-6 and MCP-1 expression (Figure 5, D and E, Supplemental Figure 10, E and F, and Supplemental Figure 11). Intriguingly, GPS2 depletion and cytokine stimulation had additive effects on the endogenous expression of IL-6 (Figure 5, D and E) and MCP-1 (Supplemental Figure 11C) and on IL-6 reporter activation (Supplemental Figure 10, E and F). This suggests a close mechanistic relationship of (a) derepression by corepressor removal and (b) activation by inflammatory transcription factor signaling in order to induce key inflammatory genes in adipocytes.
Identification of GPS2-regulated inflammatory transcription factors in adipocytes, and their role in controlling IL-6 expression.
Inflammatory transcription factors, such as AP-1, C/EBP, STATs, and NF-κB, mediate inflammatory responses in immune cells and hepatocytes; however, with the exception of NF-κB, little is known about their role in regulating inflammatory gene expression in human adipocytes. To identify such transcription factors and to determine whether their activity is controlled by GPS2, we analyzed the activation of 47 transcription factors in nuclear extracts from human adipocytes that were siRNA-depleted for GPS2 and SMRT, in conjunction with adenovirus expression of GPS2 and/or cytokine stimulation (Figure 6A and Supplemental Table 1).
Figure 6. The SMRT-GPS2 complex represses IL-6 expression in human adipocytes through inhibition of the proinflammatory transcription factors NF-κB, AP-1, and C/EBP.

(A and B) Identification of GPS2 and SMRT-dependent inflammatory transcription factors (TF) in adipocytes using a transcription factor profiling assay. Transcription factor activation was analyzed in nuclear extracts of human adipocytes upon treatment with GPS2 siRNA or ATCM coupled with adenoviral expression of GPS2 (or GFP as control). Reporter activation was measured as relative luminescence activity (RLU). Data are derived from 4 different experiments. (C) Human IL-6 promoter with known binding sites for inflammatory transcription factors. (D and E) ChIP analysis of transcription factor binding at R1 and R2. See Supplemental Figure 12 for schematic and ChIP analysis of the nonresponsive control region R3. (F and G) Re-ChIP. Human adipocytes were infected with adenovirus HA-GPS2 or GFP (as control). ChIP (first IP) using anti-HA was followed by re-ChIP (second IP) using the indicated antibodies. Data are representative of 5 different experiments. *P < 0.05.
Depletion of GPS2 and SMRT induced activation of major inflammatory transcription factors, including AP-1, C/EBPs, Ets, NF-κB, and STAT3, whereas other candidate factors, such as IRFs and STAT1, were not affected (Figure 6A and Supplemental Table 1). Notably, we observed that transcription factor activation by GPS2 depletion was comparable to the level of cytokine-induced (i.e., ATCM) activation (Figure 6B), in agreement with the results described above. Rescue experiments using adenovirus-expressed GPS2 under inflammatory conditions (siRNA-mediated GPS2 depletion or ATCM treatment) provoked repression of inflammatory transcription factor activation and abolished transcription factor II D (TFIID) binding (Figure 6B and Supplemental Table 2), consistent with genomic antagonism of a GPS-containing corepressor complex with the basal transcription machinery.
ChIP assays were performed to study the possible involvement of these factors in regulating IL-6 expression in adipocytes and to test whether GPS2 and SMRT are capable of forming genomic corepressor complexes at the suspected transcription factor binding sites at the human IL-6 promoter. Binding site search and previous data (35, 36) suggested that 2 core regulatory regions at the IL-6 promoter can be distinguished within the human IL-6 promoter (Figure 6C): (a) the region composed of NF-κB and C/EBPβ binding sites (referred to herein as R1), and (b) the region including the AP-1 binding site (referred to herein as R2). Using specific primers for R1 versus R2, we found that GPS2 depletion or ATCM treatment promoted the recruitment of C/EBPβ and NF-κB p65 onto R1, whereas AP-1 was specifically recruited onto R2 (Figure 6, D and E). Importantly, ectopic expression of GPS2 induced corecruitment of GPS2 with SMRT and antagonized the recruitment of inflammatory transcription factors onto IL-6 promoter regions (Figure 6, D and E), fully consistent with our transcription factor profiling results (Figure 6, A and B). Additionally, ChIP and re-ChIP experiments in adipocytes upon adenovirus-mediated GPS2 expression revealed that GPS2 and SMRT were part of the same transcriptional complex with C/EBPβ, NF-κB p65, and AP-1 at the specific IL-6 promoter regions (Figure 6, F and G). Collectively, these results lent further support to a major genomic function of GPS2, in conjunction with the SMRT corepressor complex, in antagonizing the activation of inflammatory transcription factors in human adipocytes.
TWIST1, a PPARγ-inducible gene, controls GPS2 and SMRT expression in human adipocytes.
To identify potential regulators of SMRT and GPS2 expression in adipocytes, we analyzed the corresponding promoter sequences for putative binding sites for transcription factors that were differentially regulated in obese adipose tissue based on our transcriptomic data (Supplemental Figure 1 and data not shown). This analysis revealed TWIST1, an E-box–binding transcription factor recently described as being differentially expressed in lean versus obese adipocytes (37, 38), to be a strong candidate. Indeed, we found in ChIP assays that TWIST1, along with POL2, was recruited onto the SMRT and GPS2 promoter regions containing E-boxes in lean adipocytes, whereas recruitment was significantly reduced in obese adipocytes (Figure 7, A and B). In agreement with the previous studies (37, 38), we found that TWIST1 expression was reduced by more than 50% in isolated adipocytes from obese subjects (Figure 7C). Correlation studies highlighted the strong positive association of GPS2 and SMRT expression with that of TWIST1 (Figure 7, D and E). TWIST1 siRNA knockdown in adipocytes caused downregulation of GPS2 and SMRT expression and upregulated expression of inflammatory genes, such as IL-6 and IL-8 (Figure 7F). In addition, siRNA-mediated depletion of TWIST1 reduced the recruitment of GPS2 and SMRT onto the IL-6 promoter (Figure 7G), whereas no direct recruitment of TWIST1 to this region was observed (data not shown). Finally, adenovirus-mediated GPS2 expression in adipocytes depleted for TWIST1 partially reversed IL-6 and IL-8 expression (Figure 7H), which is probably the consequence of SMRT downregulation induced by TWIST1 depletion. In line with these results, induction of an IL-6 luciferase reporter by TWIST1 depletion was antagonized by plasmid-mediated overexpression of GPS2 and SMRT (Supplemental Figure 10D). Overall, these data support the concept that TWIST1 could be one of the key factors controlling inflammatory gene expression in human adipocytes, here via positive regulation of GPS2 and SMRT expression.
Figure 7. TWIST1 controls transcription of GPS2 and SMRT in human adipocytes.

(A and B) Recruitment of TWIST1 and POL2 was analyzed on promoter and enhancer regions of GPS2 and SMRT in isolated adipocytes from subcutaneous adipose tissue of lean (n = 6) and obese (n = 5) subjects by ChIP assays. (C) TWIST1 mRNA expression was measured in isolated adipocytes of lean (n = 8) and obese (n = 12) subjects. (D and E) Correlation of TWIST1 expression with GPS2 and SMRT was analyzed using Spearman statistical test. (F) Adipocytes were transfected with siRNA targeting TWIST1. mRNA expression of TWIST1, GPS2, SMRT, NCOR1, IL-6, and IL-8 was measured by qPCR. (G) POL2, SMRT, and GPS2 recruitment onto the IL-6 promoter upon siRNA depletion of TWIST1 was analyzed by ChIP assay. IgG served as nonspecific control antibody. (H) Adenoviral overexpression of HA-GPS2 or GFP (as control) upon siRNA depletion of TWIST1. Gene expression of IL-6 and IL-8 were measured by qPCR. *P < 0.05.
Improvement of adipose tissue inflammation induced by weight loss is associated with upregulation of TWIST1 and the SMRT-GPS2 complex pathway.
Weight loss is known to improve the metabolic status of morbidly obese subjects, which is corroborated by reports of reduced systemic and improved adipose tissue inflammation (39). Therefore, we examined the expression of corepressor complex components, in relation to TWIST1 and inflammatory markers, in obese subjects before and 6 month after gastric bypass surgery–induced weight loss (Table 3). Expression of TWIST1, SMRT, and GPS2 increased after weight loss, in contrast to decreased expression of inflammatory cytokines, such as IL-6, IL-8, and MCP-1 (Figure 8, A–D, and data not shown). Expression of NCOR1, TBL1, and TBLR1 did not change (data not shown). Spearman association analysis indicated that the expression of SMRT and GPS2 positively correlated with that of TWIST1 and negatively correlated with that of IL-6 in mature adipocytes (Figure 8, E–G). Collectively, these data further support the relevance of the identified TWIST1-SMRT-GPS2 pathway in relation to obesity and adipose tissue inflammation, in particular highlighting its reversibility upon clinical intervention.
Table 3.
Clinical parameters of the female obese population before and 6 months after gastric bypass surgery
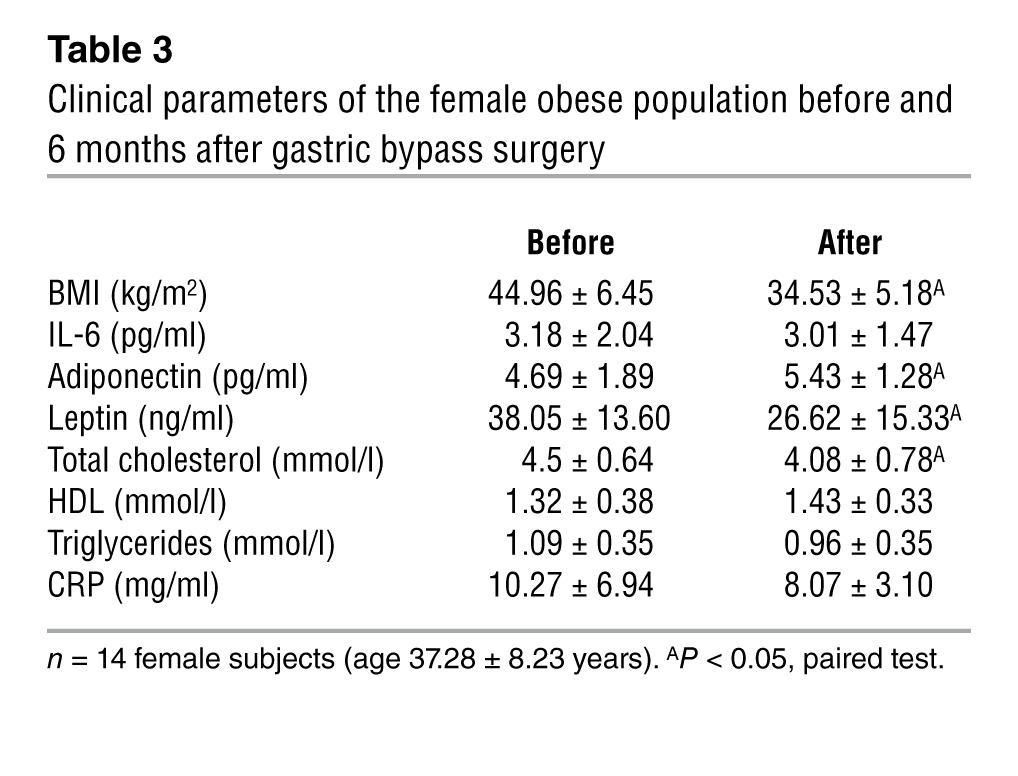
Figure 8. Increased expression of GPS2 and SMRT in obese adipose tissue after gastric bypass surgery–induced weight loss.

(A–D) qPCR analysis of SMRT, GPS2, IL-6, and TWIST1 expression in subcutaneous adipose tissue of obese subjects before and 6 months after surgery-induced weight loss (n = 14). (E–G) Correlations were determined with Spearman statistical test. (H) qPCR analysis of PPARγ expression in subcutaneous adipose tissue of obese subjects before and 6 months after surgery-induced weight loss (n = 14). (I and J) Correlations were determined with Spearman statistical test. *P < 0.05.
Thiazolidinedione drug treatment improves adipose tissue inflammation by regulation of SMRT and GPS2 expression.
PPARγ is known to be a master regulator of fat cell biology and adipocyte differentiation and the target for antidiabetic thiazolidinedione drugs (TZDs) such as rosiglitazone (40, 41). Notably, TWIST1 has been previously reported to be regulated by PPARs (42). To investigate the possibility that PPARγ acts upstream of the identified TWIST1-SMRT-GPS2 pathway, we analyzed the expression of PPARγ in subcutaneous adipose tissue of obese subjects before and 6 months after gastric bypass surgery–induced weight loss. PPARγ expression was significantly increased 6 months after surgery and positively correlated with expression levels of GPS2 and TWIST1 (Figure 8, H–J). To confirm these observations in vitro, we treated human adipocytes with rosiglitazone for 24 hours and then analyzed the gene expression profile. Rosiglitazone induced expression of FABP4, a known PPARγ target gene, as well as that of TWIST1, GPS2, and SMRT, but not that of NCOR1 (Figure 9A). ChIP assays revealed that at the TWIST1 promoter, rosiglitazone treatment increased PPARγ recruitment, in association with CBP/P300 and POL2 (Figure 9B), reflective of transcription activation. At the promoters of SMRT and GPS2, rosiglitazone treatment increased recruitment of TWIST1 (Figure 9, C and D).
Figure 9. PPARγ controls a TWIST1-SMRT-GPS2 cascade in human adipocytes.

(A–D) Adipocytes were differentiated for 10 days and then treated with the PPARγ agonist rosiglitazone (1 μM) or vehicle (DMSO) for 24 hours. (A) Gene expression of TWIST1, GPS2, NCOR1, SMRT, and FABP4 was analyzed by qPCR. (B) PPARγ, CBP/300, and POL2 recruitment onto the TWIST1 promoter was measured by ChIP assays. (C and D) TWIST1 and POL2 recruitment onto the SMRT and GPS2 promoters. (E) Expression analysis. (F–H) ChIP assays were performed on TWIST1, GPS2, and IL-6 promoters. Adipocytes were differentiated for 10 days, then cotreated for 24 hours with ATCM or control media and with vehicle (DMSO) or rosiglitazone (1 μM). *P < 0.05.
To investigate the link to inflammatory status, we treated adipocytes with ATCM in combination with rosiglitazone (1 μM) or DMSO vehicle. As expected, ATCM markedly induced expression of inflammatory genes such as IL-6, IL-8, MCP-1, IL-1, and TNF-α (Figure 9E). Under these conditions, rosiglitazone treatment increased the recruitment of PPARγ and TWIST1 onto the TWIST1 and GPS2 promoters, respectively (Figure 9, F and G). Similarly, recruitment of GPS2 and SMRT onto the IL-6 promoter was also increased (Figure 9H).
Our in vitro observations corroborated the putative link among the antiinflammatory actions of PPARγ. We then analyzed adipose tissue transcripts of previously reported diabetic subjects treated with the TZD pioglitazone for 6 months (43). Although all treated patients had improvement of their diabetic status (Table 4), we observed a difference regarding the adipose tissue gene signatures. Half of the patients responded to pioglitazone treatment, with reduced adipose tissue inflammation marked by decreased expression levels of IL-6, MCP-1, CD68, and CRP (Figure 10A). Using these parameters, we distinguished 2 patient profiles: those with and without improvement of adipose tissue inflammation. Interestingly, patients with improvement of adipose tissue inflammation demonstrated significantly increased expression of GPS2, SMRT, and TWIST1, while these were not regulated in the group without improvement (Figure 10B). In agreement with our observations in adipose tissue of obese subjects, GPS2 and SMRT expression negatively correlated with IL-6 systemic concentration (Figure 10, C and D) and with IL-6 mRNA expression (Figure 10E) in all patients. Moreover, expression of SMRT correlated positively with GPS2 and PPARγ (Figure 10F and Supplemental Figure 13). Collectively, these results indicated that adipose tissue inflammation seemed to be associated with expression levels of GPS2 and SMRT, which could be modulated by clinical responses to TZD treatment in patients.
Table 4.
Clinical parameters of diabetic patients before and after 6 months of pioglitazone treatment
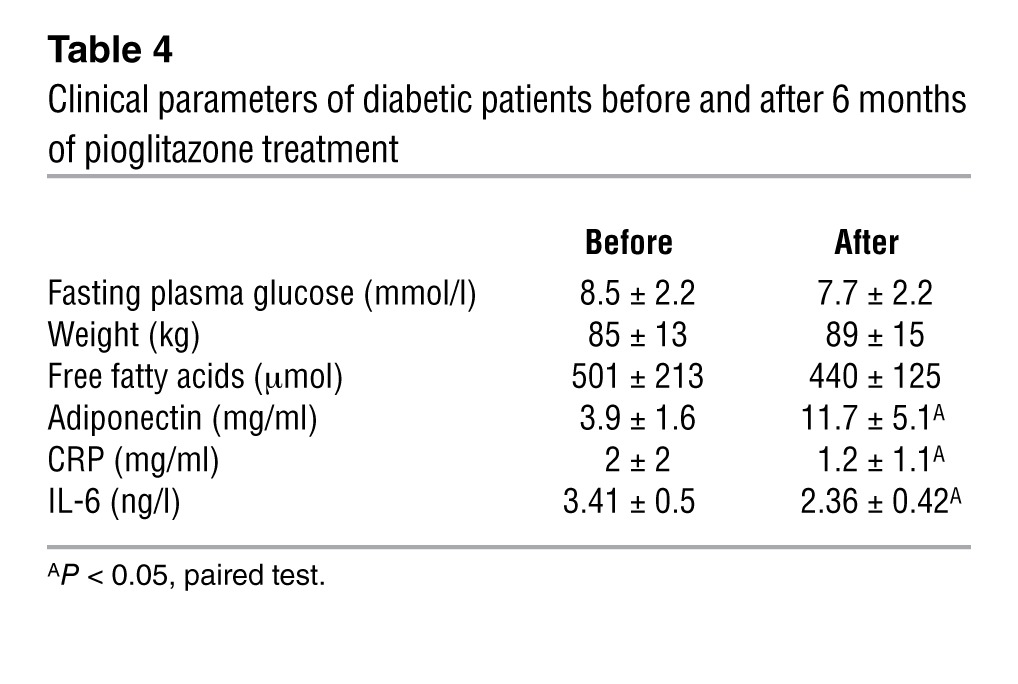
Figure 10. Expression of GPS2 and SMRT in adipocytes negatively correlates with IL-6 levels in diabetic subjects treated for 6 months with pioglitazone.

(A and B) Gene expression profiling of subcutaneous adipose tissue from 12 diabetic subjects before and after pioglitazone treatment. *P < 0.05, paired test. (C–F) Correlation analysis was performed with Spearman statistical test.
Discussion
The SMRT-GPS2 corepressor complex as a genomic checkpoint for adipocyte-specific reprogramming toward metaflammation.
Cell type–specific epigenetic and transcriptional reprogramming events, and their underlying regulatory components, are fundamentally linked to human diseases and are attracting increasing attention in the areas of obesity, metaflammation, and adipose tissue biology (21). The molecular characterization of such events not only promises to reveal insights into the causality of disease-associated physiological alterations, but also promises to reveal targets and strategies for therapeutic intervention and, optimally, disease prevention.
In this study, we sought to identify novel regulatory pathways and components that participate in adipocyte-specific transcriptional reprogramming events toward obesity-associated inflammation. We report that 2 specific transcriptional corepressor complex subunits, SMRT and GPS2, were differentially expressed and regulated in adipocytes of obese subjects. We provide evidence that this dysregulation of corepressor complex function contributed to adipose tissue inflammation by accounting for the derepression of a subset of inflammatory genes in adipocytes (Figure 11). We further identified a molecular pathway implicating TWIST1 and PPARγ in the transcriptional regulation of SMRT and GPS2 expression in adipocytes. Finally, we demonstrated that resolution of adipose tissue inflammation by gastric bypass surgery–induced weight loss in obese subjects or by treatment with a PPARγ agonist in diabetic subjects was associated with restored expression of SMRT and GPS2 (Figure 11).
Figure 11. Sequence of events leading to adipocyte inflammation in obesity.

Based on integration of the present data into previous models, we propose the following. Metabolic excess/overnutrition triggers adipocyte hypertrophy, which — via lipid toxicity and/or ER stress — initiates inflammatory gene expression in adipocytes. Initiation may occur via direct activation of inflammatory signaling cascades and/or via epigenetic mechanisms linked to derepression. Our present work demonstrated that a subset of key inflammatory genes was induced by clearance of the SMRT-GPS2 complex (IL-6, IL-8, and MCP-1), promoting chromatin remodeling and recruitment of the transcriptional machinery (POL2 and TFIID). Subsequently, adipocyte-derived chemoattractants and cytokines stimulate immune cell activation that finally results in the chronic inflammatory status of obese adipose tissue. Both obesity-associated epigenetic alterations and inflammatory signaling are likely to cause downregulation of GPS2 and SMRT expression, in part via TWIST1. Improvement (resolution) of adipose tissue inflammation by TZD treatment or by gastric bypass–induced weight loss stimulates expression and activity of PPARγ, an upstream regulator of the TWIST1-SMRT-GPS2 cascade. Consequently, these interventions restore the expression and function of the SMRT-GPS2 corepressor complex at inflammatory genes in adipocytes, thereby acting antiinflammatory.
These data, along with our findings in gain-of-function studies using human adipocytes, suggest that the corepressor pathway toward inflammation is reversible and thus may present an attractive target for future intervention. Our study provides the first evidence that a specific transcriptional corepressor complex is dysregulated in human obesity and describes a transcriptional and potentially epigenetic and reprogramming pathway that could be causally linked to metaflammation: the chronic inflammatory status of obese adipose tissue.
The present findings further refined the recent hypothesis that obesity complications are initiated by alterations in adipocytes that cause transcriptional reprogramming, leading to the secretion of adipose tissue–derived inflammatory mediators. Our newly identified pathway, controlled by the SMRT-GPS2 corepressor complex in human adipocytes, is likely to contribute to the well-documented attraction of immune cells via regulating MCP-1 (44, 45) as well as to the expression of adipocytokines involved in inflammatory responses, macrophage activation, and polarization via regulating IL-6 (16, 20, 46). These alterations, if not resolved by clinical or pharmacological intervention, are likely to establish the chronic low-grade inflammation status of obese adipose tissue.
Our results also indicate that the transcriptional regulation of inflammatory responses in adipocytes and macrophages, at least in part, requires different components and thus can be mechanistically distinguished. For example, MCP-1 expression in adipocytes is controlled by a SMRT-GPS2 corepressor complex, while in macrophages, MCP-1 is controlled by a related yet distinct complex containing NCOR1 and coronin 2A, but not GPS2 or SMRT (28, 34). Although we currently lack any data on human adipose tissue–associated macrophages, it is likely that the obesity-associated alterations that reduce GPS2 and SMRT expression in obese adipose tissue define a pathway that occurs specifically in adipocytes.
Dysregulation of corepressor expression: cause or consequence of adipose tissue inflammation?
Our observations demonstrated a correlation of corepressor expression with adipose tissue inflammation in vivo and provided direct evidence that experimental reduction of SMRT-GPS2 expression was sufficient to induce proinflammatory gene expression in cultured human adipocytes. However, whether reduced corepressor expression is causal to the development of adipose tissue inflammation in obesity remains to be addressed. It will be especially challenging to fully describe and separate molecular events, and the obesity-associated alterations thereof, that occur in adipocytes versus adipose tissue–associated immune cells, such has macrophages and T cells.
Regardless, it is likely that metabolic and inflammatory signalling in obese adipocytes, as well as in adipose tissue, converge in complex regulatory circuits with multiple levels of interdependence. For example, alterations of corepressor expression in noninflamed adipocytes (healthy obese) could be initiated by nutritional signaling linked to enigmatic epigenetic changes, whereas with disease progression (inflamed obese), inflammatory signaling cascades could have a more direct negative effect on the expression of GPS2 and SMRT, along with a variety of other genes and miRNAs, as reported previously (47).
The involvement of dynamic epigenetic processes is supported by our demonstration that the SMRT-GPS2 pathway toward inflammation had degrees of reversibility, both upon experimental manipulation (i.e., siRNA-mediated depletion and adenovirus expression) and upon clinical intervention (i.e., TZD treatment and gastric bypass surgery). The correlations also suggested that individual genetic or epigenetic differences regarding the expression of SMRT and/or GPS2 could be distinctive in causing variations concerning the inflammatory status of obese people. To further explore such a possibility, studies involving healthy obese or early-onset obese individuals would be of great benefit. Additionally, it would be interesting to determine the expression levels of GPS2 and SMRT, and their relation to inflammation, in childhood obesity.
Derepression is crucial to induce inflammatory gene expression in human adipocytes.
Our data should also be useful in developing and refining models of how inflammatory gene expression is regulated in specific nonimmune cell types, such as adipocytes. In the absence of any functional studies in human adipocytes, we can currently assume that most inflammatory genes will be regulated by a variety of inflammatory transcription factors, some of which are targets for repression by SMRT-GPS2.
Numerous findings of the present study suggested that the classic activation model does not fully describe the mechanisms that account for induced gene transcription at inflammatory sites in adipocytes. First, our transcription factor profiling assay indicated that depletion of GPS2, similar to the effects of SMRT depletion, increases the activity of many, but not all, inflammatory factors, along with increased TFIID activity, in human adipocytes. Second, RNAi-mediated depletion of corepressor subunits, and the reversal thereof by adenovirus expression, clearly induced transcription of inflammatory genes, such as IL-6 and MCP-1, in human adipocytes, even in the apparent absence of any stimulating inflammatory signal (e.g., cytokines and ATCM). Third, re-ChIP experiments at the IL-6 promoter detected promoter-bound complexes of SMRT and GPS2 as well as inflammatory factors, including NF-κB p65, AP-1, and C/EBP. Intriguingly, the latter 2 factors are known to be already promoter-bound under basal conditions, and thus represent good candidates for recruiting SMRT-GPS2.
Together, these data suggest that appropriate genomic corepressor action is required for maintaining a transcriptional silent state of inflammatory gene expression in adipocytes. We speculate that any stimulus that reduces promoter occupancy of corepressors can potentially initiate the inflammatory program in adipocytes and contribute to initiation and maintenance of subsequent chronic inflammatory states. Such stimulus could be either the activation of inflammatory factors, such as NF-κB p65 (classic activation mechanism), or the reduced expression — and thereby occupancy — of corepressor subunits (alternative activation by derepression). In reality, it will be difficult to separate these 2 key events from each other, and they likely will act in a synergistic manner. Indeed, we showed that corepressor removal (using siRNA depletion) and cytokine stimulation synergistically affected the expression of IL-6 and other genes, although each stimulus alone was sufficient to partially induce transcription.
Related corepressors NCOR1 and SMRT have different functions in adipocytes.
According to available data, the highly related corepressors SMRT and NCOR1 share the same domain architecture, interact with the same set of transcription factors, and assemble into similar corepressor complexes, which suggests that they have redundant functions. Our results provide evidence that this may not be the case with respect to control of inflammatory responses, in which NCOR1 and SMRT complexes obviously have separate functions.
Glass and coworkers have demonstrated that in murine macrophages, NCOR1 and SMRT seem to function independently and in a mutually dependent manner to regulate some specific subsets of inflammatory genes (29, 34). A large set of inflammatory genes, including MCP-1 and IL-1, seems to be controlled by NCOR1 and SMRT, indicative of an unexpected requirement of both corepressors. In contrast, we previously reported that in human and murine hepatocytes, key genes associated with the acute phase response, such as haptoglobin or SAA, are controlled specifically by a NCOR1-GPS2 complex, but not by SMRT (29).
Here, we report that in human adipocytes, depletion of NCOR1 and SMRT induced different gene expression profiles. As far as we can determine from the gene expression, SMRT acts as a key repression checkpoint of inflammatory gene expression, whereas NCOR1 seems to be more involved in metabolic processes in adipocytes. However, our microarray data also revealed that 43 genes were commonly regulated by SMRT and NCOR1 in preadipocytes, which suggests that SMRT and NCOR1 function in a mutually dependent manner for these genes, as described in macrophages. Unexpectedly, NCOR1 did not seem to regulate many genes in mature human adipocytes compared with preadipocytes. One possible explanation for this could be that NCOR1 acts as major corepressor for (unliganded) PPARγ in preadipocytes, while the addition of rosiglitazone during differentiation causes dissociation of NCOR1 from PPARγ. This could indicate that SMRT and NCOR1 act at different stages of adipocyte differentiation by repressing distinct gene programs. Indeed, GPS2, SMRT, and NCOR1 did not appear to regulate the same gene expression profiles during human adipogenesis (Supplemental Figure 8, Supplemental Table 8, and our unpublished observations), in support of this hypothesis.
Our data are consistent with a recent study suggesting that NCOR1 is the main corepressor of PPARγ actions in adipocytes, as specific knockout of NCOR1 in mature adipocytes derepressed PPARγ activity, at least in mice (31). Our human data are also supported by a previous study in mice, showing that SMRT mutants that disrupted interactions with nuclear receptors presented widespread adipose tissue defects, including adipocyte hypertrophy and increased expression of inflammatory genes (30, 48). Although our study suggests that SMRT function is uncoupled from regulating PPARγ activity, a direct comparison of the respective NCOR1 versus SMRT knockout phenotypes in mice would be needed to draw further conclusions.
Genomic and antiinflammatory roles of the corepressor complex subunit GPS2 in human adipocytes.
Recent biochemical studies have highlighted the assembly of the subunit GPS2 into both SMRT and NCOR1 complexes and suggest a structural core function of GPS2 in both (49). However, ChIP assays combined with RNAi also indicated the existence of SMRT and NCOR1 complexes, along with the HDAC3, TBL1, and TBLR1 subunits, that occupied different genes — in macrophages or hepatocytes — despite the lack of GPS2 (29, 34). In our adipose studies, we demonstrated that GPS2 depletion provoked the same gene expression alterations as did SMRT depletion, which suggests that GPS2 is mainly part of a SMRT-containing corepressor complex in adipocytes. These data indicate that GPS2 and SMRT function in a mutually dependent manner, independent of NCOR1. Our observations further support the notion that GPS2 seems to be linked to a number of antiinflammatory pathways, making it an interesting regulatory molecule. While we describe here a genomic pathway wherein GPS2 and SMRT, along with the corepressor complex, converge at the inflammatory promoters in adipocytes, we note that GPS2, unlike SMRT, may exert additional nongenomic functions by inhibiting the cytoplasmic TNF-α–JNK signaling cascade (50). To what extent this pathway contributes to the inflammatory phenotype of human adipose tissue, and whether it occurs in human adipocytes and/or in adipose tissue–associated macrophages, remain to be addressed. Whether the general antiinflammatory role of GPS2 is a suitable target for future therapeutic strategies also remains to be explored. Interestingly, we observed that GPS2 expression negatively correlated with systemic IL-6 concentration in patients that responded to clinical treatment with the antidiabetic PPARγ agonist pioglitazone. GPS2 was also found to be strongly associated with adiponectin expression, linking GPS2 expression with insulin sensitivity status. This is consistent with a related observation in transgenic mice (50). Of note in this respect is that GPS2, in addition to being upregulated by PPARγ agonists, also directly interacts with SUMOylated PPARγ, thus providing a realistic route for antiinflammatory drug design.
Upstream regulators of GPS2 and SMRT expression in human adipocytes.
In our search for upstream regulators of GPS2 and SMRT expression in adipocytes, we herein identified TWIST1, a PPARγ-inducible gene, as a strong candidate (42). TWIST1, which belongs to the family of basic helix-loop-helix transcription factors, acts as a key player in embryogenesis, particularly in gastrulation and subsequent mesoderm formation, and knockout mice were embryonic lethal due to neuronal abnormalities (51). Interestingly, TWIST1 has been characterized as a critical regulator of energy homeostasis and inflammation in metabolic tissues, including adipose tissue (37, 38, 42).
Studies using our molecular model suggested that TWIST1 regulates the expression of GPS2 and SMRT, but not that of NCOR1. Thus, reduced expression of TWIST1 in obese adipocytes is likely to contribute to the downregulation of GPS2 and SMRT. Our data additionally indicated that treatment of inflamed adipocytes with a PPARγ agonist improved their inflammatory phenotype by increasing the binding of SMRT and GPS2 onto inflammatory promoters, such as IL-6. This binding of GPS2 and SMRT was directly related to increased recruitment of TWIST1 onto GPS2 and SMRT promoters, consistent with their increased expression levels. In agreement with previous observations (38), we found that reduced levels of TWIST1 were associated with an inflammatory phenotype of adipose tissue. However, the precise role of TWIST1 in the regulation of inflammatory gene expression in adipocytes remains to be clarified, in particular with regard to the possible direct positive or negative regulation of IL-6 as a promoter-bound factor (37).
Available data indicate that several mechanisms could be involved in the transcription regulation of inflammatory genes by TWIST1. While our present findings demonstrated positive regulation of GPS2 and SMRT, TWIST family members have been previously reported to repress transcription in other promoter contexts (51). Mechanisms include the promotion of histone deacetylation — via HAT inhibition or HDAC recruitment — or direct inhibition of promoter-bound transcriptional activators. This second mechanism may account for the possible antiinflammatory action of TWIST1 on NF-κB activation of TNF-α expression and would not require the recruitment of corepressor complexes to inhibit inflammatory genes. In support of this model, our results indicated that GPS2 and SMRT have a minor role in the regulation of TNF-α expression, compared with IL-6 and MCP-1.
Emerging antiinflammatory PPARγ pathways in human adipocytes.
Our findings suggest PPARγ to be an upstream regulator of the TWIST1-SMRT-GPS2 cascade. That TWIST1 could be a direct PPAR target gene was evident from previous work describing the related PPARδ subtype as a regulator of TWIST1 expression in brown adipose tissue. Whereas PPARδ-induced TWIST1 in brown adipose tissue blocked transactivation of the coregulator PGC1-α (42), our study identified that the PPARγ-TWIST1-SMRT-GPS2 corepressor cascade in white adipose tissue seems to be mechanistically distinct. Additionally, our finding that PPARγ acted as a regulator of GPS2 expression in human adipocytes is opposed by the prior finding that in mouse adipocytes, GPS2 acts as a regulator of PPARγ (50), further highlighting notable species differences in regulatory adipocyte cascades.
Our identified pathway may help to further dissect the role of PPARγ, and its ligands, in human adipose tissue. While the key role of PPARγ in promoting adipogenesis is well established (52, 53), a currently debated issue is whether the antiinflammatory effect of PPARγ agonists in adipose tissue is caused by PPARγ action in macrophages, adipocytes, or both. PPARγ is induced during differentiation of monocytes into macrophages, is highly expressed in activated macrophages, acts as a main regulator of M2 macrophage polarization (54), and functions as a potent antiinflammatory regulator in murine macrophages (55). Other studies have revealed that specific activation of PPARγ in adipocytes is sufficient to improve insulin sensitivity and to repress adipose tissue inflammation, at least in mouse models (56).
We have shown here that activation of PPARγ in human adipocytes is associated with repression of inflammatory genes, mainly due to the activation of a TWIST1-SMRT-GPS2 cascade. In addition, mRNA levels of GPS2 and SMRT in adipose tissue were inversely correlated with systemic levels of IL-6 in patients who responded to treatment with the TZD pioglitazone, which suggests that the antiinflammatory action of PPARγ in adipocytes involves the regulation of SMRT and GPS2 expression levels.
An intriguing open issue concerns the possibility that PPARγ in human adipocytes also directly inhibits inflammatory gene expression via a transrepression pathway related to those previously identified in macrophages or hepatocytes (29, 34, 56). Moreover, because an inflammatory environment promotes phosphorylation of PPARγ by CDK5, which affects its transrepressive activity, the interplay of agonist action with both modifications in the contexts of human adipocytes awaits further clarification (57). Hypothetically, the adipocyte pathway could involve ligand-modulated docking of SUMOylated PPARγ to the SMRT-GPS2 corepressor complex at inflammatory genes, thereby adding yet another regulatory level to the transcriptional cascade identified herein.
Methods
Isolation of human preadipocytes and adipocytes from subcutaneous adipose tissue.
Isolation and culture of human preadipocytes and adipocytes from biopsies of subcutaneous adipose tissue of lean subjects (after plastic surgery) and obese subjects (after bariatric surgery) was essentially performed as previously described (58). See Supplemental Methods for details.
Culture of human adipocytes.
Preadipocytes were cultured for 24 hours in 1 ml DMEM with 10% FBS in 12-well plates at a cell density of 105 cells per well. Cells were then incubated with 0.75 ml DMEM/F12 differentiation medium (50 nM insulin, 100 nM dexamethasone, 0.25 mM inhibitor-1-methyl-3-isobutylxanthine, and 100 nM rosiglitazone) for 4 days. This medium was then replaced by 0.75 ml DMEM/F12 culture medium (50 nM insulin, 100 nM dexamethasone, and 100 nM rosiglitazone), and the culture medium was changed every 2 days until day 10, at which time most of the preadipocytes contained large lipid droplets. The preadipocytes were then incubated with DMEM/F12 without dexamethasone and rosiglitazone treatment for 2 days to avoid the antiinflammatory effects of these compounds.
Recombinant virus construction and adipocyte infection.
Adenoviral expression vectors were constructed using the AdEasy system (Agilent Technologies). Briefly, HA-tagged human GPS2 was subcloned into the pAdTrack shuttle vector containing a CMV promoter in front of the insert and GFP under the control of a second CMV promoter. Empty pAdTrack vector, only expressing GFP, was used as a negative control. The shuttle vectors were recombined in BJ5183 bacteria. Virus particles were then packaged, amplified, and harvested in 293A cells. The titer was determined by TCID50 assay. For infections, cultured adipocytes were incubated with adenovirus particles for 2 days at MOI 5 and MOI 50. Transduction was efficient after 2 days of culture, as determined by Western blot analysis and GFP fluorescence.
ELISA and cytokine membrane array.
The secretion medium (cell culture medium) of preadipocytes and adipocytes was collected 48 hours after siRNA cell transfection. See Supplemental Table 3 for details of individual siRNAs. The secretion profiles of inflammatory cytokines were measured using Human Cytokines array Panel A Array (R&D Systems), according to the manufacturer’s instructions. Cell culture supernatants were mixed with a cocktail of biotinylated detection antibodies. Nitrocellulose membranes (spotted with different cytokine antibodies) were then incubated the sample/antibody mixture. After several washes, streptavidin-HRP and chemiluminescent detection reagents were added, which produced light at each spot proportional to the amount of cytokine bound. To quantify cytokine secretion in cultured cells, ELISA was performed according to the manufacturer’s instructions (R&D).
DNA microarray.
An Illumina RNA amplification kit (Ambion) was used according to the manufacturer’s instructions to obtain biotin-labeled cDNA from 250 ng total RNA. cDNA was hybridized to Illumina Human HT-12 v3.0 Expression BeadChips (Illumina Inc.) containing probes for 48,000 transcripts. 12 samples were hybridized on the same chip. Hybridized probes were detected with cyanin-3–streptavidin (1 mg/ml; Amersham Biosciences, GE Healthcare) using Illumina BeadArray Reader (Illumina Inc.) and BeadStudio v3 software (Illumina Inc.). Raw data (i.e., average probe signals) were extracted using the numerical results with Illumina Bead Studio software using default settings without any additional normalization. Differential gene expression was determined by standard analysis of microarray (SAM) analysis using a false discovery rate (FDR) of approximately 0.5%. We performed a functional analysis of these gene lists using the FunNet tool (59). KEGG pathways and Gene Ontology (GO) Cellular Component annotations were examined in order to identify those functional annotations that were significantly enriched in the different gene lists.
Transcription factor profiling array.
The activity of 47 human transcription factors in human adipocytes was assayed using a plate array according to the manufacturer’s instructions (Signosis Inc.). Briefly, nuclear proteins were isolated from adipocytes, and 12 g nuclear extract was added to a mixture of DNA sequences that encoded 47 different human transcription factor–binding sites. This mixture was incubated at 16°C for 30 minutes to allow for the formation of transcription factor–DNA complexes, then passed through an isolation column to separate the transcription factor–DNA complexes from free DNA probes. 100 μl elution buffer was then added to the column to elute the transcription factor–DNA complexes, which were subsequently denatured by incubation at 98°C. 95 μl of each sample was then added to each well of a 96-well plate containing an immobilized complementary sequence to 1 of the 47 transcription factors. The 96-well plate was sealed and incubated overnight at 42°C to hybridize the complementary strands of DNA. After hybridization, the plate was washed 3 times, streptavidin-HRP conjugate and substrate was added, and resulting chemiluminescence was detected using a multidetection microplate reader.
ChIP assays.
ChIP assays were performed as described previously (29). Briefly, adipocytes isolated from adipose tissue were crosslinked for 10 minutes with 1% formaldehyde, and the reaction was stopped with 125 mM glycine for 5 minutes. Cells were rinsed by PBS and incubated with Farnham lysis buffer (5 mM PIPES [pH 8.0], 85 mM KCl, and 0.5% NP-40 plus protease inhibitor) at 4°C to isolate the nuclei. Nuclei were then centrifuged at 380 g for 5 minutes at 4°C. The nuclei were resuspended in 1 ml RIPA buffer (1× PBS, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% SDS plus protease inhibitor cocktail) at 4°C and sonicated using Diogenode Bioruptor Sonicator at high setting for 15 minutes (30 seconds on, 30 seconds off) to generate DNA fragments 0.2–0.8 kb in length. Sonicates were centrifuged at 16,060 g at 4°C, and supernatant was collected. 200 μl resuspended magnetic bead slurry was added in the microfuge tube containing the collected supernatant. 2–5 μg primary antibody was then added. The microfuge was gently mixed on a rotor platform overnight. The next day, 100 μl antibody-coupled beads was added to the chromatin preparation and incubated at 4°C overnight on a rotor. After several washes, immune-bound chromatin was eluted from the beads and centrifuged, and the supernatant containing the ChIPed DNA was incubated at 65°C overnight to complete the reversal of the formaldehyde crosslinks. Qiagen Kit was then used to elute the DNA. Quantification of the precipitated DNA regions was performed by PCR. See Supplemental Tables 4–6 for primer sequences and antibodies.
Clinical study populations.
Morbidly obese patients, candidates for bariatric surgery, were recruited in the nutrition division of Pitié-Salpêtrière Hospital, Institute of Cardiometabolism and Nutrition (ICAN). As part of this study, we used biopsies of subcutaneous adipose tissue located in the periumbilical area obtained during gastric bypass surgery (obese subjects) or during plastic surgery (lean subjects) (Table 1). 14 obese patients were followed for 6 months after gastric bypass. Subcutaneous adipose tissue pieces were taken from these patients before and 6 months after gastric bypass (Table 3) in the same area. Clinical investigation was performed as described previously (43); we used adipose tissue biopsies from this prior study (Table 4). Tissue samples were stored at –80°C until RNA extraction.
Statistics.
Data are expressed as mean ± SEM. Experiments were performed at least 3 times, each using preadipocytes and adipocytes from different human subjects. Statistical analysis was performed using 2-tailed Student’s t test. Comparisons between more than 2 groups were carried out using 1-way ANOVA. Correlations analyses were made using Spearman statistical test. A P value less than 0.05 was considered statistically significant.
Study approval.
The ethics committee of Hotel-Dieu Hospital approved the clinical investigations. All subjects gave written informed consent after individual explanation of the research program.
Supplementary Material
Acknowledgments
We thank Marta Palmieri (University of Verona, Verona, Italy) for providing the IL-6 reporter plasmid, Ronald Evans (Salk Institute, UCLA, Los Angeles, California, USA) for SMRT plasmids and antibodies, and Christopher Glass (UCSD, San Diego, California, USA) and Thorsten Heinzel (University of Jena, Jena, Germany) for NCOR1 plasmids and antibodies. We are grateful to colleagues at U872, NutriOmic for sharing reagents and biological samples and to Judith Aron-Wisnewsky, Michele Guerre-Millo, and Elise Dalmas for helpful discussions. We thank Rohia Alili, Edi Prifti, and Jean-Philippe Bastard for their technical support. We thank Assistance Publique des Hôpitaux de Paris (APHP; Programs of Clinical Investigation [PHRC] no. 0702) and TAKEDA pharma for providing pioglitazone used in the clinical study. Nutriomic team support was provided by the European Community seventh framework program, ADAPT: Adipokines as Drug to combat Adverse Effects of Excess Adipose tissue project (grant agreement no. 201100), French National Agency of Research (ANR; Adipofib to K. Clément, CONRAD to N. Venteclef), and Region Ile de France (CORDDIM to N. Venteclef). This work was supported by a French government grant managed by the ANR (program “Investments for the Future”; grant no. ANR-10-IAHU). E. Treuter was supported by grants from the Center for Biosciences, the Swedish Research Council, the Novo Nordisk Foundation, and the Swedish Cancer Society.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(1):362–379. doi:10.1172/JCI64052.
References
- 1.Calder PC, et al. Inflammatory disease processes and interactions with nutrition. Br J Nutr. 2009;101(suppl 1):S1–S45. doi: 10.1017/S0007114509377867. [DOI] [PubMed] [Google Scholar]
- 2.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444(7121):860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 3.Kolb H, Mandrup-Poulsen T. The global diabetes epidemic as a consequence of lifestyle-induced low-grade inflammation. Diabetologia. 2010;53(1):10–20. doi: 10.1007/s00125-009-1573-7. [DOI] [PubMed] [Google Scholar]
- 4.Fernandez-Real JM, Pickup JC. Innate immunity, insulin resistance and type 2 diabetes. Trends Endocrinol Metab. 2008;19(1):10–16. doi: 10.1016/j.tem.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 5.Glass CK, Olefsky JM. Inflammation and lipid signaling in the etiology of insulin resistance. Cell Metab. 2012;15(5):635–645. doi: 10.1016/j.cmet.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Michaud A, Drolet R, Noël S, Paris G, Tchernof A. Visceral fat accumulation is an indicator of adipose tissue macrophage infiltration in women. Metabolism. 2012;61(5):689–698. doi: 10.1016/j.metabol.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 7.Cancello R, et al. Reduction of macrophage infiltration and chemoattractant gene expression changes in white adipose tissue of morbidly obese subjects after surgery-induced weight loss. Diabetes. 2005;54(8):2277–2286. doi: 10.2337/diabetes.54.8.2277. [DOI] [PubMed] [Google Scholar]
- 8.Tilg H, Moschen AR. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol. 2006;6(10):772–783. doi: 10.1038/nri1937. [DOI] [PubMed] [Google Scholar]
- 9.Trayhurn P, Wood IS. Adipokines: inflammation and the pleiotropic role of white adipose tissue. Br J Nutr. 2004;92(3):347–355. doi: 10.1079/BJN20041213. [DOI] [PubMed] [Google Scholar]
- 10.Wellen KE, Hotamisligil GS. Obesity-induced inflammatory changes in adipose tissue. J Clin Invest. 2003;112(12):1785–1788. doi: 10.1172/JCI20514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dalmas E, et al. Variations in circulating inflammatory factors are related to changes in calorie and carbohydrate intakes early in the course of surgery-induced weight reduction. Am J Clin Nutr. 2011;94(2):450–458. doi: 10.3945/ajcn.111.013771. [DOI] [PubMed] [Google Scholar]
- 12.Skurk T, Alberti-Huber C, Herder C, Hauner H. Relationship between adipocyte size and adipokine expression and secretion. J Clin Endocrinol Metab. 2007;92(3):1023–1033. doi: 10.1210/jc.2006-1055. [DOI] [PubMed] [Google Scholar]
- 13.Tordjman J, Guerre-Millo M, Clement K. Adipose tissue inflammation and liver pathology in human obesity. Diabetes Metab. 2008;34(6 pt 2):658–663. doi: 10.1016/S1262-3636(08)74601-9. [DOI] [PubMed] [Google Scholar]
- 14.Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. 2011;11(2):98–107. doi: 10.1038/nri2925. [DOI] [PubMed] [Google Scholar]
- 15.Arner P, et al. Dynamics of human adipose lipid turnover in health and metabolic disease. Nature. 2011;478(7367):110–113. doi: 10.1038/nature10426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dalmas E, Clement K, Guerre-Millo M. Defining macrophage phenotype and function in adipose tissue. Trends Immunol. 2011;32(7):307–314. doi: 10.1016/j.it.2011.04.008. [DOI] [PubMed] [Google Scholar]
- 17.Kumar A, Takada Y, Boriek AM, Aggarwal BB. Nuclear factor-kappaB: its role in health and disease. J Mol Med (Berl). 2004;82(7):434–448. doi: 10.1007/s00109-004-0555-y. [DOI] [PubMed] [Google Scholar]
- 18.Sigal LH. Basic science for the clinician 39: NF-kappaB-function, activation, control, and consequences. J Clin Rheumatol. 2006;12(4):207–211. doi: 10.1097/01.rhu.0000231385.94784.e4. [DOI] [PubMed] [Google Scholar]
- 19.Suganami T, et al. Role of the Toll-like receptor 4/NF-kappaB pathway in saturated fatty acid-induced inflammatory changes in the interaction between adipocytes and macrophages. Arterioscler Thromb Vasc Biol. 2007;27(1):84–91. doi: 10.1161/01.ATV.0000251608.09329.9a. [DOI] [PubMed] [Google Scholar]
- 20.Osborn O, Olefsky JM. The cellular and signaling networks linking the immune system and metabolism in disease. Nat Med. 2012;18(3):363–374. doi: 10.1038/nm.2627. [DOI] [PubMed] [Google Scholar]
- 21.Rosenfeld MG, Lunyak VV, Glass CK. Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev. 2006;20(11):1405–1428. doi: 10.1101/gad.1424806. [DOI] [PubMed] [Google Scholar]
- 22.Medzhitov R, Horng T. Transcriptional control of the inflammatory response. Nat Rev Immunol. 2009;9(10):692–703. doi: 10.1038/nri2634. [DOI] [PubMed] [Google Scholar]
- 23.Rosell M, Jones MC, Parker MG. Role of nuclear receptor corepressor RIP140 in metabolic syndrome. Biochim Biophys Acta. 2011;1812(8):919–928. doi: 10.1016/j.bbadis.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Villanueva CJ, et al. TLE3 is a dual-function transcriptional coregulator of adipogenesis. Cell Metab. 2011;13(4):413–427. doi: 10.1016/j.cmet.2011.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Siersbæk R, Mandrup S. Transcriptional networks controlling adipocyte differentiation. Cold Spring Harb Symp Quant Biol. 2011;76:247–255. doi: 10.1101/sqb.2011.76.010512. [DOI] [PubMed] [Google Scholar]
- 26.Saijo K, Glass CK. Microglial cell origin and phenotypes in health and disease. Nat Rev Immunol. 2011;11(11):775–787. doi: 10.1038/nri3086. [DOI] [PubMed] [Google Scholar]
- 27.Venteclef N, Jakobsson T, Steffensen KR, Treuter E. Metabolic nuclear receptor signaling and the inflammatory acute phase response. Trends Endocrinol Metab. 2011;22:333–343. doi: 10.1016/j.tem.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 28.Huang W, et al. Coronin 2A mediates actin-dependent de-repression of inflammatory response genes. Nature. 2011;470(7334):414–418. doi: 10.1038/nature09703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Venteclef N, et al. GPS2-dependent corepressor/SUMO pathways govern anti-inflammatory actions of LRH-1 and LXRbeta in the hepatic acute phase response. Genes Dev. 2010;24(4):381–395. doi: 10.1101/gad.545110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nofsinger RR, et al. SMRT repression of nuclear receptors controls the adipogenic set point and metabolic homeostasis. Proc Natl Acad Sci U S A. 2008;105(50):20021–20026. doi: 10.1073/pnas.0811012105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li P, et al. Adipocyte NCoR knockout decreases PPARgamma phosphorylation and enhances PPARgamma activity and insulin sensitivity. Cell. 2011;147(4):815–826. doi: 10.1016/j.cell.2011.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Henegar C, et al. Adipose tissue transcriptomic signature highlights the pathological relevance of extracellular matrix in human obesity. Genome Biol. 2008;9(1):R14. doi: 10.1186/gb-2008-9-1-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maury E, Brichard SM. Adipokine dysregulation, adipose tissue inflammation and metabolic syndrome. Mol Cell Endocrinol. 2010;314(1):1–16. doi: 10.1016/j.mce.2009.07.031. [DOI] [PubMed] [Google Scholar]
- 34.Ghisletti S, et al. Cooperative NCoR/SMRT interactions establish a corepressor-based strategy for integration of inflammatory and anti-inflammatory signaling pathways. Genes Dev. 2009;23(6):681–693. doi: 10.1101/gad.1773109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen FH, Crist SA, Zhang GJ, Iwamoto Y, See WA. Interleukin-6 production by human bladder tumor cell lines is upregulated by bacillus Calmette-Guerin through nuclear factor-kappaB and Ap-1 via an immediate early pathway. J Urol. 2002;168(2):786–797. doi: 10.1016/S0022-5347(05)64745-3. [DOI] [PubMed] [Google Scholar]
- 36.Dandrea M, Donadelli M, Costanzo C, Scarpa A, Palmieri M. MeCP2/H3meK9 are involved in IL-6 gene silencing in pancreatic adenocarcinoma cell lines. Nucleic Acids Res. 2009;37(20):6681–6690. doi: 10.1093/nar/gkp723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pettersson AT, et al. A possible inflammatory role of twist1 in human white adipocytes. Diabetes. 2010;59(3):564–571. doi: 10.2337/db09-0997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pettersson AT, et al. Twist1 in human white adipose tissue and obesity. J Clin Endocrinol Metab. 2011;96(1):133–141. doi: 10.1210/jc.2010-0929. [DOI] [PubMed] [Google Scholar]
- 39.Clement K, et al. Weight loss regulates inflammation-related genes in white adipose tissue of obese subjects. FASEB J. 2004;18(14):1657–1669. doi: 10.1096/fj.04-2204com. [DOI] [PubMed] [Google Scholar]
- 40.Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell. 1994;79(7):1147–1156. doi: 10.1016/0092-8674(94)90006-X. [DOI] [PubMed] [Google Scholar]
- 41.Willson TM, Lambert MH, Kliewer SA. Peroxisome proliferator-activated receptor gamma and metabolic disease. Annu Rev Biochem. 2001;70:341–367. doi: 10.1146/annurev.biochem.70.1.341. [DOI] [PubMed] [Google Scholar]
- 42.Pan D, Fujimoto M, Lopes A, Wang YX. Twist-1 is a PPARdelta-inducible, negative-feedback regulator of PGC-1alpha in brown fat metabolism. Cell. 2009;137(1):73–86. doi: 10.1016/j.cell.2009.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hartemann-Heurtier A, et al. Effects of bed-time insulin versus pioglitazone on abdominal fat accumulation, inflammation and gene expression in adipose tissue in patients with type 2 diabetes. Diabetes Res Clin Pract. 2009;86(1):37–43. doi: 10.1016/j.diabres.2009.06.028. [DOI] [PubMed] [Google Scholar]
- 44.Kanda H, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest. 2006;116(6):1494–1505. doi: 10.1172/JCI26498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsou CL, et al. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest. 2007;117(4):902–909. doi: 10.1172/JCI29919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117(1):175–184. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Arner E, et al. Adipose tissue microRNAs as regulators of CCL2 production in human obesity. Diabetes. 2012;61(8):1986–1993. doi: 10.2337/db11-1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fang S, et al. Corepressor SMRT promotes oxidative phosphorylation in adipose tissue and protects against diet-induced obesity and insulin resistance. Proc Natl Acad Sci U S A. 2011;108(8):3412–3417. doi: 10.1073/pnas.1017707108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oberoi J, et al. Structural basis for the assembly of the SMRT/NCoR core transcriptional repression machinery. Nat Struct Mol Biol. 2011;18(2):177–184. doi: 10.1038/nsmb.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cardamone MD, et al. A protective strategy against hyperinflammatory responses requiring the nontranscriptional actions of GPS2. Mol Cell. 2012;46(1):1–14. doi: 10.1016/j.molcel.2012.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Franco HL, Casasnovas J, Rodríguez-Medina JR, Cadilla CL. Redundant or separate entities? Roles of Twist1 and Twist2 as molecular switches during gene transcription. Nucleic Acids Res. 2011;39(4):1177–1186. doi: 10.1093/nar/gkq890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.He W, et al. Adipose-specific peroxisome proliferator-activated receptor gamma knockout causes insulin resistance in fat and liver but not in muscle. Proc Natl Acad Sci U S A. 2003;100(26):15712–15717. doi: 10.1073/pnas.2536828100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Imai T, et al. Peroxisome proliferator-activated receptor gamma is required in mature white and brown adipocytes for their survival in the mouse. Proc Natl Acad Sci U S A. 2004;101(13):4543–4547. doi: 10.1073/pnas.0400356101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Odegaard JI, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007;447(7148):1116–1120. doi: 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hevener AL, et al. Macrophage PPAR gamma is required for normal skeletal muscle and hepatic insulin sensitivity and full antidiabetic effects of thiazolidinediones. J Clin Invest. 2007;117(6):1658–1669. doi: 10.1172/JCI31561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pascual G, et al. Anti-inflammatory and antidiabetic roles of PPARgamma. Novartis Found Symp. 2007;286:183–196. doi: 10.1002/9780470985571.ch16. [DOI] [PubMed] [Google Scholar]
- 57.Choi JH, et al. Antidiabetic actions of a non-agonist PPARgamma ligand blocking Cdk5-mediated phosphorylation. Nature. 2011;477(7365):477–481. doi: 10.1038/nature10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Keophiphath M, Rouault C, Divoux A, Clement K, Lacasa D. CCL5 promotes macrophage recruitment and survival in human adipose tissue. Arterioscler Thromb Vasc Biol. 2010;30(1):39–45. doi: 10.1161/ATVBAHA.109.197442. [DOI] [PubMed] [Google Scholar]
- 59.Prifti E, Zucker JD, Clement K, Henegar C. FunNet: an integrative tool for exploring transcriptional interactions. Bioinformatics. 2008;24(22):2636–2638. doi: 10.1093/bioinformatics/btn492. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.