

Abstract
Gastric adenocarcinoma is strongly associated with Helicobacter pylori infection; however, most infected persons never develop this malignancy. H. pylori strains harboring the cag pathogenicity island (cag+), which encodes CagA and a type IV secretion system (T4SS), induce more severe disease outcomes. H. pylori infection is also associated with iron deficiency, which similarly augments gastric cancer risk. To define the influence of iron deficiency on microbial virulence in gastric carcinogenesis, Mongolian gerbils were maintained on iron-depleted diets and infected with an oncogenic H. pylori cag+ strain. Iron depletion accelerated the development of H. pylori–induced premalignant and malignant lesions in a cagA-dependent manner. H. pylori strains harvested from iron-depleted gerbils or grown under iron-limiting conditions exhibited enhanced virulence and induction of inflammatory factors. Further, in a human population at high risk for gastric cancer, H. pylori strains isolated from patients with the lowest ferritin levels induced more robust proinflammatory responses compared with strains isolated from patients with the highest ferritin levels, irrespective of histologic status. These data demonstrate that iron deficiency enhances H. pylori virulence and represents a measurable biomarker to identify populations of infected persons at high risk for gastric cancer.
Introduction
Helicobacter pylori is the most common bacterial infection worldwide and represents the strongest known risk factor for the development of gastric adenocarcinoma.
H. pylori strains that harbor the cag pathogenicity island induce more severe gastric injury and further augment the risk for developing cancer of the stomach (1). The cag island encodes a bacterial type IV secretion system (T4SS), which translocates CagA, the product of the terminal gene within the island, into host cells. Intracellular CagA can become phosphorylated (2–4) or remain unphosphorylated; in either form, CagA affects multiple pathways that alter host cell morphology, signaling, and inflammatory responses (5–9). However, most persons infected by cag+ H. pylori strains never develop cancer. These observations underscore the importance of defining factors that may alone, or in tandem with known virulence determinants, increase the risk for gastric cancer, thereby providing a means to identify H. pylori–infected subpopulations at highest risk for disease.
Environmental conditions can also modify the risk for carcinogenesis. Iron deficiency is associated with an increased risk for neoplasms that arise within the gastrointestinal tract (10), including the stomach (11–13). H. pylori infection contributes to iron deficiency (14), and bacterial eradication results in reversal of this disorder (15). We recently demonstrated that CagA facilitates H. pylori colonization in polarized epithelial cells via iron acquisition (16), suggesting that iron deficiency influences virulence of this pathogen.
Gastric cancer in humans occurs only after decades of H. pylori infection, precluding large-scale studies that can comprehensively examine the combinatorial effects of disease modifiers. H. pylori infection of Mongolian gerbils can lead to the development of gastric adenocarcinoma in the distal stomach, as in humans (17, 18). Carcinoma develops in this model following infection with H. pylori alone, without the coadministration of carcinogens, and occurs within a time frame suitable for rigorous evaluation of endpoints. Thus, gerbils represent a robust model of H. pylori–induced human gastric cancer. The aim of this study was to define the role of bacterial virulence determinants in H. pylori–induced gastric carcinogenesis within the context of iron deficiency, using an animal model of H. pylori infection and cancer that closely resembles human disease. These results were then extended into a human population at increased risk for gastric cancer.
Results
Dietary iron depletion significantly reduces systemic and gastric iron levels.
To assess the effectiveness of dietary iron depletion in a rodent model of gastric cancer, noninvasive MRI was utilized to determine relative levels of hepatic iron, as a measure of systemic iron stores, in Mongolian gerbils maintained on iron-replete or iron-depleted diets. High-resolution, multi-slice T2-weighted images were collected in both coronal (Figure 1A) and axial (Figure 1B) planes for liver visualization, and a pixel-by-pixel representation (heat map, Figure 1C) showed spatial variations in the T2 signal. Gerbils maintained on iron-depleted diets demonstrated significantly (P < 0.05) slower rates of signal decay, reflecting lower levels of hepatic iron, compared with gerbils maintained on iron-replete diets (Figure 1, D–F). To confirm these findings, inductively coupled plasma-dynamic reaction cell-mass spectrometry (ICP-DRC-MS) was used to directly measure hepatic iron concentration (Figure 1G). Consistent with the MRI findings, ICP-DRC-MS demonstrated that gerbils maintained on iron-depleted diets had significantly (P < 0.001) less hepatic iron compared with gerbils maintained on iron-replete diets. To extend these findings, serum ferritin (Figure 1H), hemoglobin (Figure 1I), and hematocrit levels (Figure 1J) were assessed in a subset of gerbils. These results demonstrated that gerbils maintained on iron-depleted diets have significantly lower levels of serum ferritin (P < 0.0001), hemoglobin (P < 0.05), and hematocrit (P < 0.005) compared with gerbils maintained on iron-replete diets. To directly assess iron depletion in the target organ of interest, ICP-MS was used to measure iron concentrations in gastric mucosa in a subset of gerbils (Figure 1K); the results demonstrated that gerbils maintained on iron-depleted diets had significantly (P < 0.0001) lower gastric iron concentrations than gerbils maintained on iron-replete diets. Collectively, these data indicate that use of an iron-depleted diet results in significantly reduced systemic and gastric iron stores in this model of H. pylori infection and cancer.
Figure 1. Dietary iron depletion results in decreased hepatic iron, serum ferritin, hemoglobin, hematocrit, and gastric iron.

MRI was utilized to assess relative levels of hepatic iron in gerbils maintained on iron-replete or iron-depleted diets for 12 weeks. A representative (A) coronal scan (white box outlines liver tissue), (B) axial scan, (C) heat map, and (D) echo series (blue lines outline liver margins) are shown from an iron-replete gerbil. (E) Signal decay over time demonstrated that iron-depleted gerbils have a slower rate of signal decay, which is inversely proportional to the level of hepatic iron. (F) The cumulative rate of signal decay demonstrated that gerbils maintained on iron-depleted diets have significantly slower rates of signal decay, representing lower levels of hepatic iron. (G) ICP-DRC-MS indicated that hepatic iron concentration is significantly lower in gerbils maintained on iron-depleted diets (n = 65) versus iron-replete diets (n = 65) at 6 and 12 weeks. (H) ELISAs demonstrated that serum ferritin concentrations are significantly lower in gerbils maintained on iron-depleted (n = 40) versus iron-replete (n = 40) diets at 6 and 12 weeks. (I) Hemoglobin and (J) hematocrit values were assessed from blood isolated from gerbils maintained on iron-replete (n = 8) versus iron-depleted (n = 8) diets at 6 weeks. (K) ICP-MS demonstrated that gerbils maintained on iron-depleted diets (n = 8) have significantly lower levels of gastric iron versus gerbils maintained on iron-replete diets (n = 7) at 6 weeks. Data points represent values from individual gerbil samples. Mean values are shown, and error bars indicate standard error. Student’s t tests were used to determine statistical significance.
Iron depletion augments and accelerates H. pylori–induced gastric inflammation and carcinogenesis.
We next determined the effects of iron depletion on H. pylori–induced gastric inflammation and cancer. Uninfected gastric mucosa contained few inflammatory cells (Figure 2A and Figure 3, A and B). Infection with wild-type H. pylori strain 7.13 induced gastritis, which was characterized by a robust inflammatory infiltrate and formation of lymphoid follicles, and was more severe in H. pylori–infected iron-depleted gerbils compared with infected iron-replete gerbils (P < 0.01; Figure 2A and Figure 3, C and D). Gastritis also developed earlier in H. pylori–infected iron-depleted gerbils compared with infected iron-replete gerbils at 2 weeks (33% vs. 13%) and 6 weeks (79% vs. 55%) after challenge (Table 1). Based upon the bimodal distribution in inflammatory scores (Figure 2A), we examined the relationship between inflammation and H. pylori colonization density in infected iron-replete and iron-depleted gerbils (Figure 2B). A significant inverse relationship (P < 0.0001; r = –0.63) was observed between the severity of gastric inflammation and H. pylori colonization density under both conditions, such that gerbils with the lowest degree of gastric inflammation had the highest bacterial burden.
Figure 2. Iron depletion augments and accelerates H. pylori–induced gastric inflammation and carcinogenesis.

(A) Gastric inflammation at 6–12 weeks after challenge was assessed and scored (0 to 12) in uninfected (UI) and H. pylori–infected gerbils. Each data point represents the inflammation score from a single gerbil. Mean values are shown, and Mann-Whitney U tests were used to determine statistical significance between groups. (B) Correlation plot demonstrates an inverse relationship between the severity of gastric inflammation and H. pylori colonization density at 6–12 weeks after challenge. Each data point represents the inflammation score and colonization density from a single gerbil maintained on iron-replete (black symbols) or iron-depleted (gray symbols) diets. Spearman nonparametric correlations were used to determine the Spearman r correlation coefficient and statistical significance. (C and D) The percentage of gerbils that remained disease-free, as defined by absence of (C) dysplasia or (D) adenocarcinoma, at 2, 6, and 12 weeks after challenge is represented as Kaplan-Meier curves. Statistical differences were determined by log-rank tests.
Figure 3. Wild-type H. pylori strain 7.13 induces gastric inflammation, dysplasia, and adenocarcinoma in Mongolian gerbils.
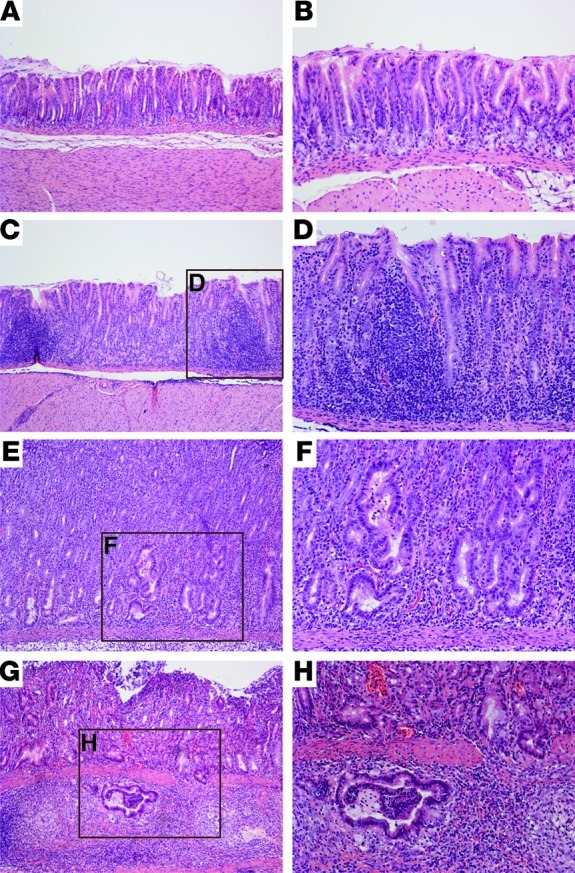
Hematoxylin and eosin–stained tissues (6 weeks after challenge) were evaluated for parameters of inflammation and injury by histopathology. Representative images of (A and B) uninfected gastric mucosa; (C and D) H. pylori–induced gastritis; (E and F) H. pylori–induced dysplasia; and (G and H) H. pylori–induced adenocarcinoma. A, C, E, and G (left panels): original magnification, ×100; B, D, F, and H (right panels): original magnification, ×200.
Table 1.
Iron depletion increases the frequency and severity of gastric inflammation, dysplasia, and adenocarcinoma induced by wild-type H. pylori strain 7.13
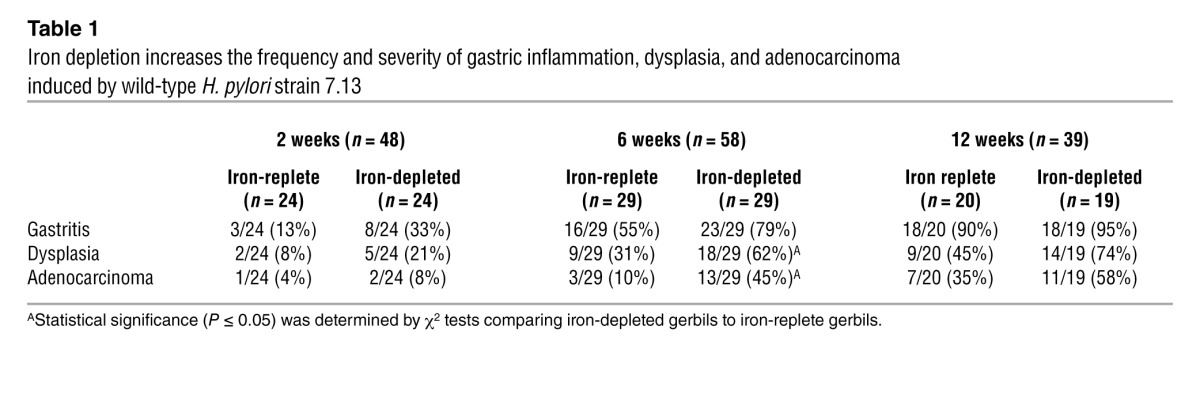
Consistent with the increased severity of gastric inflammation, dysplasia, as characterized by irregular glandular structures with pseudostratified nuclei (Figure 3, E and F), was more prevalent in H. pylori–infected iron-depleted gerbils compared with iron-replete gerbils at 2 weeks (21% vs. 8%), 6 weeks (62% vs. 31%), and 12 weeks (74% vs. 45%) after challenge (Table 1). Wild-type H. pylori also induced gastric adenocarcinoma, which was characterized by irregular glandular structures with enlarged nuclei penetrating through the muscularis mucosa (Figure 3, G and H), and was again observed more frequently in H. pylori–infected iron-depleted gerbils compared with iron-replete gerbils at 2 weeks (8% vs. 4%), 6 weeks (45% vs. 10%), and 12 weeks (58% vs. 35%) after infection (Table 1). The incidence of dysplasia and adenocarcinoma within the entire cohort of uninfected gerbils or gerbils infected with wild-type H. pylori strain 7.13 was also evaluated by Kaplan-Meier curves, which demonstrated that significantly more (P < 0.0001) H. pylori–infected iron-depleted gerbils developed dysplasia (Figure 2C) and adenocarcinoma (Figure 2D) compared with infected iron-replete gerbils. Irrespective of iron status, loss of cagA significantly attenuated the development of inflammation (P < 0.001; Figure 2A), dysplasia (0 of 64), and adenocarcinoma (0 of 64), indicating that CagA plays a critical role in H. pylori–induced gastric inflammation and carcinogenesis. These data demonstrate that dietary iron depletion significantly increases the severity and accelerates the development of H. pylori–induced premalignant and malignant lesions.
To determine whether iron depletion altered the topography of H. pylori colonization, immunofluorescence and confocal microscopy were performed on gastric sections from infected gerbils (Figure 4). H. pylori were present throughout the stomach, including the corpus (Figure 4A) and antrum (Figure 4B) under both iron-replete (Supplemental Video 1; supplemental material available online with this article; doi: 10.1172/JCI64373DS1) and iron-depleted (Supplemental Video 2) conditions. Within each anatomic region of the stomach, H. pylori localized to multiple foci within gastric glands (Figure 4, C–E) and formed microcolonies (Figure 4F). Although there were no differences in H. pylori colonization density between iron-replete and iron-depleted animals as determined by quantitative culture, colonization density within individual glands of iron-depleted animals, as determined by quantitative microscopy, was significantly (P < 0.05) higher than within glands from iron-replete animals (Figure 4, G–I). These data indicate that iron depletion alters the distribution of H. pylori within the stomach, which may contribute to accelerated oncogenesis.
Figure 4. Wild-type H. pylori strain 7.13 colonizes multiple sites within gerbil gastric tissue and colonizes gastric glands to higher levels under iron-depleted conditions.

Immunofluorescence and confocal microscopy were performed on gastric tissue from gerbils infected with H. pylori for 6 weeks. For all images, tissue sections were stained for H. pylori (green), actin (red), and cellular nuclei (blue). H. pylori was distributed throughout the entire glandular stomach, including the (A) corpus and (B) antrum. (C–E) Boxes designate representative areas shown at higher magnification. H. pylori localized to multiple sites within the gastric gland, including the (C) apical region, (D) glandular pit, and (E) glandular neck, and (F) formed microcolonies. Scale bars: 40 μm (A and B) and 10 μm (C–F). Representative examples of infected gastric glands from (G) iron-replete and (H) iron-depleted gerbils used for quantification of H. pylori. Scale bar: 10 μm (G and H). (I) The number of H. pylori in a subset of iron-replete (n = 31) and iron-depleted (n = 39) gastric glands were quantified and expressed as log CFU/gland.
To ensure that increased disease severity under conditions of iron depletion was specific for H. pylori, a model of ethanol-induced gastric inflammation (19) was utilized (Supplemental Figure 1A). Ethanol treatment induced significantly (P < 0.01) higher levels of gastric inflammation compared with vehicle control treatment; however, there were no differences in inflammation between gerbils maintained on an iron-replete diet and those on an iron-depleted diet (Supplemental Figure 1B). These data suggest that increased severity of gastric inflammation and injury under conditions of iron depletion among H. pylori–infected gerbils is not solely dependent upon the effects of iron deficiency in the uninfected host.
To further assess the effects of iron depletion on host inflammatory responses, quantitative real-time RT-PCR was utilized to measure proinflammatory cytokine levels within gastric tissue. Mongolian gerbils are outbred, which limits the number of reagents available for detailed analyses; therefore, we focused on cytokines that have previously been shown to be increased in H. pylori–infected gerbils (20). Infection with H. pylori strain 7.13 significantly increased levels of IL1B (P < 0.005), IFNG (P < 0.01), and TNFA (P < 0.01), compared with uninfected controls; however, there were no differences in expression levels of these cytokines between H. pylori–infected gerbils under iron-replete versus iron-depleted conditions (Supplemental Figure 2).
The ferric uptake regulator (Fur) is a master H. pylori regulator that controls expression of a large repertoire of genes by acting as a repressor under iron-replete conditions (21). To define the role of fur in H. pylori–induced injury in this model, wild-type H. pylori strain 7.13 and a 7.13 fur– isogenic mutant were used to infect Mongolian gerbils under iron-replete conditions (Supplemental Figure 3A). No differences in colonization were observed between wild-type strain 7.13 and the 7.13 fur– isogenic mutant (Supplemental Figure 3B). Consistent with our previous findings (Figure 2A), wild-type H. pylori strain 7.13 induced significantly (P < 0.01) higher levels of gastric inflammation in gerbils maintained on iron-depleted diets than in gerbils maintained on iron-replete diets. Loss of fur had no effect on the level of gastric inflammation compared with the wild-type strain under iron-replete conditions (Supplemental Figure 3C), suggesting that other regulators may be involved in mediating increased virulence of H. pylori in this model.
Iron depletion alters H. pylori proteomic profiles and enhances assembly and function of the cag T4SS.
To define mechanisms that mediate increased H. pylori virulence under iron-depleted conditions, minimally passaged H. pylori isolates were derived from single bacterial colonies harvested from iron-replete (n = 5) or iron-depleted (n = 5) gerbils and subjected to two-dimensional differential gel electrophoresis coupled with mass spectrometry (2D-DIGE/MS; Supplemental Figure 4), as previously described (22). Proteomic profiles of in vivo–adapted H. pylori strains isolated from iron-depleted gerbils differed significantly (P < 0.05) from profiles of strains harvested from iron-replete gerbils (Supplemental Figure 4C).
To identify candidates that reproducibly changed in iron-depleted strains, differentially abundant proteins were subjected to mass spectrometry. Based upon an increased abundance of proteins that mediate bacterial-host cell interactions (HopQ, enolase) and function of the cag T4SS (CagA) (Table 2), scanning electron microscopy was used to visualize H. pylori strains isolated from iron-replete (Figure 5, A–E) and iron-depleted (Figure 5, G–K) gerbils during coculture with human gastric epithelial cells to assess assembly and function of pilus components of the cag T4SS. Approximately 9 T4SS pili per bacterial cell were visualized on the surfaces of H. pylori strains isolated from iron-replete gerbils (Figure 5, A–E, and M). In contrast, the number of cag T4SS pili was significantly (P < 0.0001) increased to approximately 17 T4SS pili per bacterial cell for strains isolated from iron-depleted gerbils (Figure 5, G–K and M), suggesting that increased assembly of the T4SS is attributable to adaptation under conditions of iron depletion. Therefore, to directly assess the effects of iron depletion on this phenotype, the preinoculation parental H. pylori strain 7.13 was grown in vitro under iron-replete or iron-restricted conditions and then cocultured with gastric epithelial cells. Approximately 2 T4SS pili per bacterial cell were visualized on the surface of strain 7.13 grown under iron-replete conditions in vitro (Figure 5, F and N). In contrast, the number of T4SS pili significantly (P < 0.0001) increased to approximately 11 T4SS pili per bacterial cell for H. pylori grown under iron-restricted conditions (Figure 5, L and N), a phenotype that was abrogated following the addition of exogenous iron (Figure 5N).
Table 2.
Proteomic profiles of in vivo–adapted H. pylori isolated from iron-depleted gerbils differ significantly from profiles of strains harvested from iron-replete gerbils
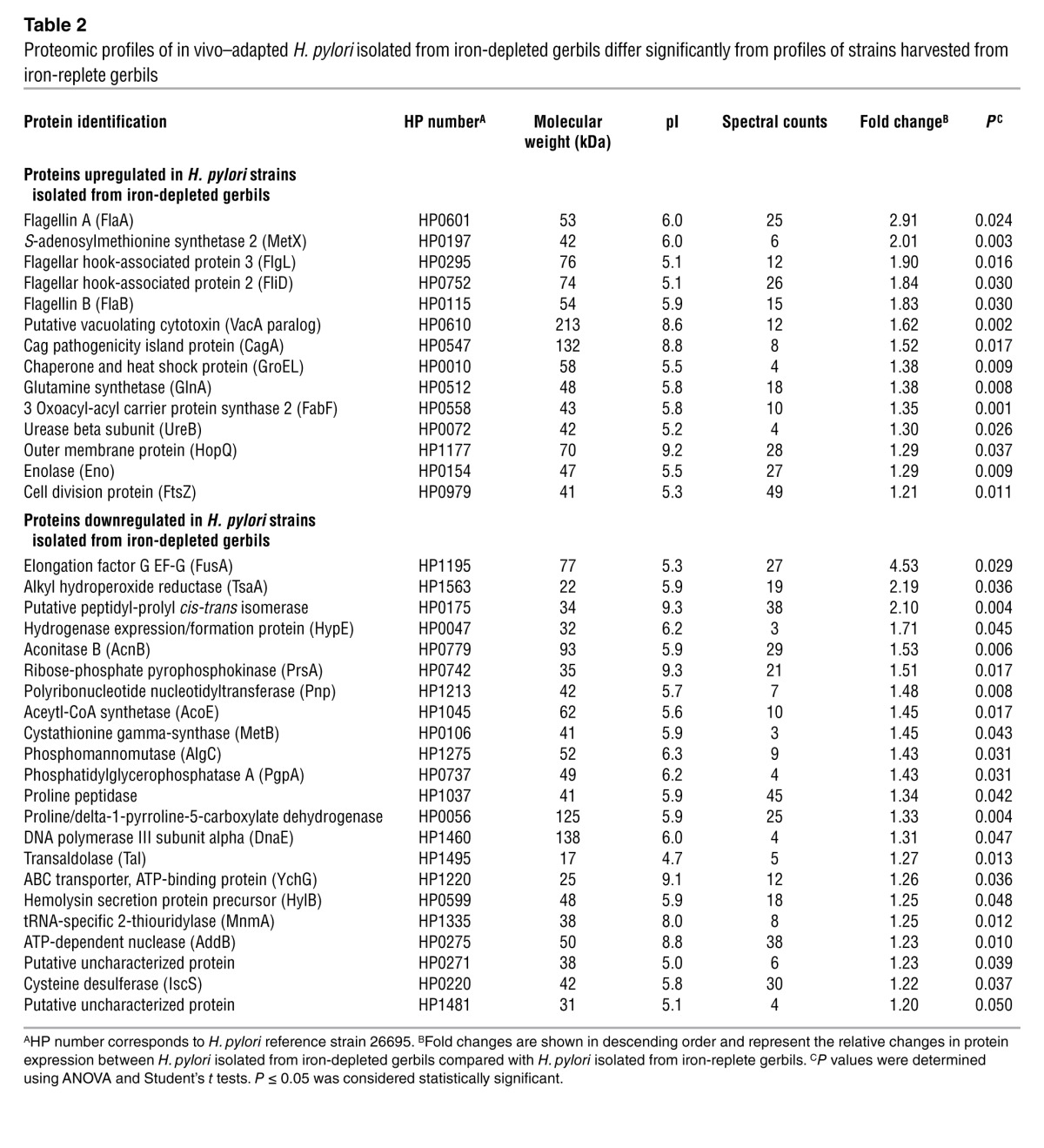
Figure 5. Exposure of H. pylori to iron-depleted conditions in vivo and in vitro augments the capacity to assembly pilus components of the cag T4SS.

In vivo-adapted H. pylori strains isolated from iron-replete (n = 5) and iron-depleted (n = 5) gerbils 12 weeks after infection as well as the parental preinoculation strain 7.13 grown in vitro under iron-replete, iron-restricted, or iron-restricted conditions with iron supplementation (Iron-restricted+) were cocultured with AGS human gastric epithelial cells for 4 hours, and cag T4SS pili were visualized by scanning electron microscopy. Images of 10 independent H. pylori strains isolated from (A–E) iron-replete or (G–K) iron-depleted gerbils and a representative image of strain 7.13 grown in vitro under (F) iron-replete and (L) iron-restricted conditions during coculture with AGS cells are shown. Arrows designate representative cag T4SS pili. Scale bars: 1 μm (A–L). (M and N) The number of cag T4SS pili per bacterial cell was enumerated based on analysis of at least 20 high-powered fields. Each data point represents the number of T4SS pili per bacterial cell (M, analysis of output strains from iron-replete, n = 57 and iron-depleted gerbils, n = 99; N, analysis of the preinoculation strain 7.13 in vitro under varying iron conditions; replicates: iron-replete, n = 20; iron-restricted, n = 20; iron-restricted+, n = 20). Mean values are shown, and Student’s t tests were used to determine statistical significance between groups.
To determine whether increased assembly of the cag T4SS corresponded to enhanced cag-mediated virulence responses, we cocultured in vivo–adapted strains with human gastric epithelial cells and quantified CagA expression (Figure 6, A and B) and translocation (Figure 6, A and C) as assessed by phosphorylated CagA (pCagA). In comparison to strains isolated from iron-replete gerbils, H. pylori isolated from iron-depleted gerbils expressed significantly higher levels of CagA, confirming results from the proteomics analysis (Table 2). Consistent with increased assembly of the cag T4SS (Figure 5), strains isolated from iron-depleted gerbils also translocated more CagA into gastric epithelial cells. These phenotypes were recapitulated when the preinoculation parental strain 7.13 was grown under iron-replete or iron-restricted conditions and then cocultured with gastric epithelial cells (Figure 6, D–F).
Figure 6. H. pylori harvested in iron-depleted conditions in vivo and in vitro increase expression and translocation of CagA into gastric epithelial cells and augment the host proinflammatory response.

In vivo-adapted H. pylori strains harvested from iron-replete (n = 5) or iron-depleted (n = 5) gerbils 12 weeks after infection, as well as strain 7.13 grown in vitro under iron-replete, iron-restricted, or iron-restricted conditions with iron supplementation (Iron-restricted+), were cocultured with AGS human gastric epithelial cells for 6 hours. In vivo-adapted H. pylori isolated from iron-depleted gerbils (A and B) exhibited increased CagA expression (A and C) and translocation (pCagA) into gastric epithelial cells. (D–F) In vitro, CagA expression and translocation were increased under iron-restricted conditions, a phenotype that was abrogated following the addition of exogenous iron. (G and H) In vivo-adapted H. pylori isolated from iron-depleted gerbils induced increased IL-8 expression in gastric epithelial cells compared with strains isolated from iron-replete gerbils, a phenotype that was recapitulated under iron-restricted conditions in vitro (I). Data represent fold change over parental preinoculation strain 7.13 (B and C) or over uninfected control cells (G–I). Error bars indicate standard error of the mean from experiments performed on at least 3 independent occasions, and Student’s t tests and ANOVAs were used to determine statistical significance between groups.
Expression of the proinflammatory chemokine IL-8 by gastric epithelial cells is mediated by cag-dependent signaling (23). Similar to increased CagA translocation into host cells, in vivo–adapted H. pylori strains isolated from iron-depleted gerbils induced significantly (P < 0.0001) higher levels of IL-8 compared with strains harvested from iron-replete gerbils (Figure 6, G and H), a phenotype that was recapitulated in vitro in response to low-iron conditions (Figure 6I). Collectively, these findings indicate that iron deficiency enhances the intensity of H. pylori interactions with its host via increased assembly of the cag T4SS, increased translocation of CagA into host cells, and enhanced induction of IL-8, all of which likely contribute to the heightened severity and acceleration of H. pylori–induced injury observed in vivo.
We next assessed whether an in vivo–adapted H. pylori strain harvested from an iron-depleted gerbil that developed cancer induced more severe gastric pathology than wild-type parental strain 7.13 when rechallenged into iron-replete gerbils. Colonization densities were similar in gerbils infected with parental strain 7.13 and the in vivo–adapted strain (Figure 7A). Infection with either strain induced significantly higher levels of inflammation when compared with uninfected gerbils; however, the severity of gastritis was similar in gerbils infected with parental strain 7.13 versus gerbils infected with the in vivo–adapted strain (Figure 7B). Consistent with similar levels of gastric inflammation, the frequency of gastritis and carcinoma did not differ in gerbils infected with strain 7.13 and the in vivo–adapted strain (Figure 7C). When readapted output strains were evaluated for their ability to induce chemokine expression in gastric epithelial cells, no differences were observed in the ability to induce IL-8 compared with output strains from parental 7.13-infected gerbils (Figure 7D). Collectively, these data support our in vitro results indicating that increased virulence of H. pylori strains under conditions of iron depletion can be abrogated by iron repletion.
Figure 7. Increased virulence of H. pylori isolated from iron-depleted gerbils is abrogated under conditions of iron repletion in vivo.

(A) Colonization density (log CFU/g of gastric tissue) was similar among gerbils infected with wild-type parental strain 7.13 or the in vivo-adapted strain isolated from an iron-depleted gerbil that developed cancer. (B) Gastric inflammation induced by parental strain 7.13 and the in vivo-adapted strain 6 weeks after challenge was assessed and scored (0 to12). Each data point represents the inflammation score from a single gerbil. Mean values are shown and Mann-Whitney U tests were used to determine statistical significance between groups. (C) The percentage of gerbils with gastritis or carcinoma is shown. Statistical differences were assessed by χ2 tests. (D) Readapted H. pylori strains induce similar levels of IL-8 expression in gastric epithelial cells compared with output strains from parental strain 7.13-infected gerbils. Data represent fold change over uninfected control cells. Error bars indicate standard error of the mean from experiments performed on at least 3 independent occasions, and Mann-Whitney U tests were used to determine statistical significance between groups.
Iron levels are inversely related to the severity of H. pylori–induced premalignant lesions and H. pylori virulence in a human population.
To extend findings from our rodent model of cancer into the natural niche of H. pylori, we quantified serum ferritin levels, as an indicator of systemic iron status, in a cohort of H. pylori–infected individuals at high risk for gastric cancer residing in the Andean mountain Nariño region of Colombia (Table 3). Serum ferritin concentrations decreased in a gradient fashion that was inversely related to the severity of premalignant lesions, whereby patients harboring the most advanced lesion, intestinal metaplasia, had significantly (P < 0.05) lower levels of ferritin compared with patients with only nonatrophic gastritis (Figure 8A). These results prompted us to investigate whether H. pylori strains isolated from patients with differing ferritin levels, but harboring similar histologic lesions, differed in virulence phenotype. When cocultured with human gastric epithelial cells, H. pylori strains isolated from patients with the lowest serum ferritin levels induced significantly (P < 0.05) higher levels of IL-8 compared with strains harvested from persons with the highest ferritin levels, in patients with gastritis only (Figure 8B) or with intestinal metaplasia (Figure 8C). This indicates that the ability of H. pylori strains to induce more severe pathogenic responses is more likely related to iron levels than the severity of histologic lesions in the stomach.
Table 3.
Frequency and severity of gastric lesions in H. pylori–infected patients residing in the Andean mountain Nariño region of Colombia

Figure 8. Iron levels are inversely related to the severity of H. pylori–induced premalignant lesions and H. pylori virulence in a human population.

(A) Serum samples, harvested from H. pylori–infected Colombian patients at high risk for gastric cancer, were segregated based on severity of gastric injury (non-atrophic gastritis, n = 55; atrophic gastritis, n = 40; intestinal metaplasia, n = 99) and analyzed for ferritin concentration by ELISA. Each data point represents the serum ferritin concentration from a single patient. (B and C) Strains harvested from patients with the lowest (n = 10) or highest (n = 10) serum ferritin concentrations from patients with gastritis alone (B) or intestinal metaplasia (C) were cocultured with AGS human gastric epithelial cells for 6 hours. Each strain was tested on 3 independent occasions and data points represent each independent replicate. Strains isolated from patients with the lowest ferritin concentrations (Low) induced significantly higher levels of IL-8 compared with strains isolated from patients with the highest ferritin concentrations (High) from both the gastritis alone and intestinal metaplasia groups. Mean values are shown and Mann-Whitney U tests were used to determine statistical significance between groups.
Discussion
Variable outcomes of H. pylori infection depend on host genetic diversity, strain-specific microbial factors, and/or environmental variables that ultimately influence the intensity of interactions between pathogen and host. We have now shown that, in the setting of iron depletion, H. pylori increases the severity and accelerates the development of gastric injury and carcinoma in an animal model of infection and cancer that closely resembles human disease. Increased pathogenesis is mediated at least in part by enhanced deployment and function of the cag T4SS, and mirrors the relationship between iron status and H. pylori virulence in a human population at increased risk for gastric cancer. Our results are consistent with previous reports linking iron deficiency and gastrointestinal cancer risk in both rodent and human studies. Iron deficiency in rats accelerates carcinogen-induced gastrointestinal cancer and metastasis (24), and case-control studies in humans have demonstrated an inverse relationship between dietary iron intake and gastric adenocarcinoma (11–13, 25); similar associations between low iron stores and colorectal cancer risk have also been reported (26, 27). The current study represents the first to not only investigate the combinatorial effect of iron deficiency and H. pylori virulence determinants, but also to demonstrate a synergistic influence of these elements on the development of gastric cancer. Thus, iron status, in conjunction with the presence of H. pylori cag+ strains, should be considered a risk factor for progression to gastric cancer.
Although the current data indicate that iron deficiency and H. pylori can increase gastric cancer risk, iron overload states have also been shown to contribute to cancer at other sites within the gastrointestinal tract. Some epidemiological studies have reported that increased iron exposure is linked to an elevated risk of colorectal carcinogenesis (28–30). Mutations in HFE result in hemochromatosis and iron overload, which is linked to hepatocellular carcinoma (31), possibly due to the ability of iron to induce oxidative stress and DNA damage; however, there is no evidence that iron overload syndromes increase the risk of gastric cancer. Of interest, iron deficiency can also increase oxidative stress and DNA damage. Heme iron deficiency and consequent imbalances in iron-containing mitochondrial enzymes can induce DNA damage, oxidative stress, and mitochondrial decay (32). Iron deficiency promotes oxidative damage due to abnormal assembly of mitochondrial electron transport chains, leading to increased levels of DNA damage and reduced antioxidant defenses in polymorphonuclear leukocytes (32–34). H. pylori CagA has also been shown to directly induce oxidative stress and DNA damage in gastric epithelial cells (35). Thus, a common pathway leading to oncogenic transformation within the gastrointestinal tract may be oxidative stress–induced DNA damage, whether mediated by iron overload or by enhanced CagA translocation within the context of iron deficiency. Another disease that can lead to iron deficiency is celiac disease. Malignancies, frequently lymphomas of the small intestinal tract, can develop in patients with this disorder. However, the risk of gastric malignancy conferred by celiac disease is not uniformly increased (36), which may reflect varying prevalence rates of H. pylori in different clinical populations (37). Based on the current data, the effects of iron on cancer risk appear to vary in a niche-specific manner, which likely reflects complex interactions between different microbial, host, and environmental constituents.
Although iron deficiency can clearly affect host cellular responses, the current data indicate that more severe pathologic outcomes are also likely due to the effects of iron depletion on H. pylori per se and that this occurs through amplification of H. pylori virulence. Iron depletion significantly altered the proteomic profiles of in vivo–adapted H. pylori, and proteins involved in survival and persistence were significantly upregulated. Motility, mediated by flagella, is required for H. pylori survival and colonization of the gastric niche (38, 39), and FlaA and FlaB, the major flagellin subunits, were significantly upregulated under conditions of iron deficiency. Iron depletion was associated with increased expression of H. pylori enolase (Eno), which mediates adherence, invasion, and immune suppression for many bacterial pathogens, including Staphylococcus aureus (40), Bacillus anthracis (40), and Borrelia burgdorferi (41). Additional H. pylori survival factors upregulated by iron deficiency included urease, the heat shock protein GroEL, and glutamine synthetase. Urease is required for colonization of the acidic gastric niche (42), while GroEL has been shown to be expressed in conjunction with other known virulence constituents, including FlaA, UreB, VacA, and CagA (43). Further, heat shock proteins, such as GroEL, are associated with the presence of gastric cancer (44). Glutamine synthetase mediates virulence in many bacterial pathogens, including Salmonella typhimurium (45), Mycobacterium tuberculosis (46), and Streptococcus pneumonia (47). Of interest, FlaA was also differentially abundant in a previous proteomics analysis that compared the noncarcinogenic progenitor H. pylori strain B128 with the carcinogenic strain 7.13 (22). However, this was the only protein altered in both studies. A novel finding from the current work is that iron deficiency promotes increased assembly of the cag T4SS and increased expression and translocation of CagA into host cells. The cag pathogenicity island is strongly associated with an increased risk of gastric cancer in humans (1), and transgenic mice overexpressing CagA develop gastric adenocarcinoma (48). Thus, one consequence of in vivo adaptation of H. pylori to low iron conditions is upregulation of a bacterial oncoprotein.
Iron is an essential micronutrient for virtually all microorganisms and is critical for the survival of H. pylori. Due to its limited availability in the host, H. pylori has evolved exquisite regulatory networks for iron acquisition and sequestration, including those mediated by Fur, which functions as a repressor under iron-replete conditions. In contrast to studies focused on other iron-regulated effectors, our results have shown that the ability of H. pylori strain 7.13 to induce more severe gastric injury under iron-depleted conditions is mediated by a fur-independent mechanism. Further, our proteomics data also support the premise that increased virulence of H. pylori under conditions of iron deficiency in this model is not Fur dependent, as we did not detect many Fur-regulated proteins when comparing H. pylori strains isolated from iron-replete and iron-depleted gerbils. Similar to H. pylori, however, other bacterial pathogens can alter virulence potential in response to iron concentrations. Shiga toxin from Shigella dysenteriae and the Shiga-like toxin from Escherichia coli are iron regulated, and toxin expression is enhanced under conditions of iron deficiency (49, 50). Diphtheria toxin from Clostridium diphtheriae (51) and exotoxin A from Pseudomonas aeruginosa (52) are secreted bacterial toxins that are also induced by iron depletion. Data from the current study have extended these findings by demonstrating that iron depletion can also accelerate microbe-induced cancer.
Our findings in a human population at high risk for gastric cancer support these experimental data by revealing a link between host iron status and the severity of H. pylori–induced premalignant lesions. The annual incidence of gastric cancer has been estimated to be 0.1% for patients with atrophy and 0.25% for patients with intestinal metaplasia (53). Persons with more severe gastric premalignant lesions typically harbor reduced acid levels, which can, in turn, lower iron levels via decreased absorption. However, our current results in patients with only gastritis suggest that lower iron levels in H. pylori–infected persons likely accelerate disease early in the carcinogenic process, leading to an augmentation of cancer risk.
In conclusion, this study has identified iron deficiency as an environmental factor that augments and accelerates H. pylori–induced gastric carcinogenesis by capitalizing on an animal model of H. pylori infection and cancer that recapitulates human disease. Further, we have identified a mechanism of increased H. pylori virulence that involves enhanced assembly and function of the cag T4SS under conditions of iron depletion, emphasizing the importance of strain-specific virulence constituents acting in concert with environmental factors to influence pathogenic outcomes. Global test-and-treat strategies for H. pylori have not been fully embraced primarily due to the relatively low incidence of cancer that develops among infected individuals. Carriage of H. pylori is also inversely related to esophageal and atopic diseases (54, 55), emphasizing the importance of identifying colonized persons that are predisposed to pathologic outcomes. Thus, on a broader scale, our studies may enable diagnostic testing and eradication strategies to be more sharply focused on high-risk populations to effectively prevent the development of gastric cancer.
Methods
Mongolian gerbil model.
Male Mongolian gerbils were purchased from Charles River Laboratories and housed in the Vanderbilt University Animal Care Facilities. Rodent diets were modified from TestDiet AIN-93M (Purina Mills) to contain 0 ppm iron (iron-depleted, TestDiet 5TWD) or 250 ppm iron (iron-replete, TestDiet 5STQ) (Supplemental Table 1). Gerbils were maintained on diets throughout the duration of each experiment. Wild-type carcinogenic H. pylori strain 7.13, a 7.13 cagA– isogenic mutant, a 7.13 fur– isogenic mutant, gerbil in vivo–adapted strains, and human clinical H. pylori isolates were minimally passaged on trypticase soy agar plates with 5% sheep blood (TSA/SB, BD Biosciences) and in Brucella broth (BD Biosciences) supplemented with 10% fetal bovine serum (Atlanta Biologicals) for 16 hours at 37°C with 5% CO2. Gerbils were orogastrically challenged with sterile Brucella broth, wild-type H. pylori strain 7.13, the cagA– isogenic mutant (Supplemental Figure 5), or the fur– isogenic mutant (Supplemental Figure 3A). Gerbils were euthanized 2, 6, or 12 weeks after challenge. A model of ethanol-induced inflammation (19) was also used, whereby gerbils were treated biweekly with water as a vehicle control or 50% ethanol for 6 weeks and euthanized following 6 weeks of treatment (Supplemental Figure 1A). A separate cohort of gerbils was maintained on standard rodent chow (Purina Mills) and then orogastrically challenged with sterile Brucella broth, wild-type carcinogenic H. pylori strain 7.13, or an in vivo–adapted strain harvested from an iron-depleted gerbil. Gerbils were euthanized 6 weeks after challenge. The Vanderbilt University Institutional Animal Care and Use Committee approved all experiments and procedures.
MRI.
Gerbils were anesthetized and placed in a Varian 4.7T horizontal bore imaging system (Varian Inc.) for data collection. High-resolution images were collected in both the coronal and axial planes for liver visualization. The signal from each voxel within the region of interest was fit to a monoexponential signal decay model to determine T2*: S = S0e–t/T2*, where T2* is inversely proportional to the amount of hepatic iron.
ICP-DRC-MS.
Livers were harvested, processed, and subjected to trace iron analysis by ICP-DRC-MS (Supplemental Table 2).
Rodent ferritin ELISA.
Serum samples from gerbils were stored at –80°C. Levels of ferritin were determined by ELISA (Kamiya Biomedical Co.), according to the manufacturer’s instructions, and analyzed with Gen5 software (Synergy 4, BioTek).
Hemoglobin and hematocrit tests.
Blood was collected from gerbils maintained on either iron-replete or iron-depleted diets for 6 weeks in tubes containing EDTA (Fisher Scientific) and complete blood counts (CBCs) with and without blood smears were performed.
Quantitative H. pylori culture.
Gastric tissue was harvested and homogenized in sterile PBS. Following serial dilution, samples were plated on selective trypticase soy agar plates with 5% sheep blood (Hemostat Laboratories) containing vancomycin (20 μg/ml; Sigma-Aldrich), nalidixic acid (10 μg/ml; Sigma-Aldrich), bacitracin (30 μg/ml; Calbiochem), and amphotericin B (2 μg/ml; Sigma-Aldrich) for isolation of H. pylori. Plates were incubated for 3–5 days at 37°C with 5% CO2. Colonies were identified as H. pylori based on characteristic morphology, Gram stain (BD), urease, and oxidase (BD) activities. Colony counts were expressed as log CFU per gram of gastric tissue.
Histopathology.
Linear strips of gastric tissue, extending from the squamocolumnar junction through the proximal duodenum, were fixed in 10% neutral-buffered formalin (Azer Scientific Inc.), embedded in paraffin, and stained with hematoxylin and eosin. A pathologist, blinded to treatment groups, assessed indices of inflammation and the presence of dysplasia and adenocarcinoma. Severity of acute and chronic inflammation was graded 0–3 in both the gastric antrum and corpus, as previously described (56), for a cumulative score of 0–12.
Immunofluorescence and confocal microscopy.
Gastric samples were processed for immunofluorescence and confocal microscopy, as previously described (57). Briefly, gastric tissue samples were fixed in 2% paraformaldehyde and embedded in agar, and 100 μm sections were cut using a vibratome (Leica). Rabbit anti–H. pylori antibodies (Dako), Alexa Fluor 594 phalloidin (Molecular Probes; Invitrogen), and DAPI (Molecular Probes; Invitrogen) were used for visualization of H. pylori, actin cytoskeleton, and cellular nuclei, respectively. Samples were imaged with a Zeiss LSM 700 confocal microscope, and z-stacks were reconstructed into 3D using Volocity software (Improvision). Quantification of H. pylori within individual gastric glands was performed using a Volocity Image Analysis Measurement Protocol (Improvision). This protocol was applied across all analyzed samples. Briefly, glands were selected, and the total volume (μm3) of H. pylori per gland was measured. The average volume of an individual bacterium was used to calculate the total number of bacteria per gland.
2D-DIGE/MS.
In vivo–adapted H. pylori strains harvested from iron-replete (n = 5) or iron-depleted (n = 5) gerbils were minimally passaged and subjected to French press–mediated lysis for 2D-DIGE, as previously described (22). Briefly, each protein sample was individually labeled with either Cy3 or Cy5 (Supplemental Figure 4A). Each sample was combined and labeled en masse with Cy2 to generate the pooled internal standard. First-dimensional pH 4-7/pH 7-11 separations and second-dimensional SDS-PAGE were performed according to the manufacturer’s instructions. DeCyder v6.5 (GE Healthcare) was used for 2D-DIGE analyses. The normalized ratio of each individual protein feature from a Cy3- or Cy5-labeled sample was standardized to the Cy2 signal from the internal standard. Principal component analysis (PCA) was performed using the extended data analysis module. Resulting peptides were subjected to C18 reverse-phase liquid chromatography coupled in-line with tandem mass spectrometry (LC-MS/MS) using an LTQ linear ion trap tandem mass spectrometer equipped with a MicroAS autosampler and Surveyor HPLC pump, nanospray source, and Xcalibur 2.0 instrument control (Thermo Fisher Scientific Inc.). To identify candidate proteins that formed individual spots of interest in the 2D gels, the tandem MS/MS data were searched against the UniProtKB database ( www.uniprot.org) with the taxonomy tag “Helicobacter pylori B128 (taxon identifier 544406),” which was concatenated with the database sequences in reverse to enable false-discovery rate calculations, and also contained common laboratory contaminants. Searches were performed using both the Sequest and X! Tandem algorithms, allowing for cysteine carbamidomethylation and partial methionine oxidation. Results were validated and assembled into protein identifications using Scaffold (version Scaffold_3.1.2; Proteome Software Inc.). Peptide identifications were accepted if they could be established at greater than 95.0% probability as specified by the Peptide Prophet algorithm. Protein identifications were accepted if they could be established at greater than 99.0% probability and contained at least 2 identified peptides (enumerated as “spectral counts” listed in Table 2). Protein probabilities were assigned by the Protein Prophet algorithm. Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony.
Gastric epithelial cell culture.
AGS human gastric epithelial cells were grown in RPMI 1640 (Life Technologies) with 10% fetal bovine serum at 37°C with 5% CO2. H. pylori coculture studies with AGS cells were conducted at an MOI of 100:1 for 4 hours.
Scanning electron microscopy.
In vivo–adapted H. pylori strains or strain 7.13 grown under iron-replete (100 μM FeCl3; Sigma-Aldrich), iron-restricted (100 μM dipyridyl; Sigma-Aldrich), or iron-restricted conditions with iron supplementation (100 μM dipyridyl with 100 μM FeCl3; Sigma-Aldrich) were cocultured with AGS cells for 4 hours. Cells were then processed and imaged to assess assembly of cag T4SS pili, as previously described (58).
Western blot analysis.
In vivo–adapted H. pylori strains or strain 7.13 grown under iron-replete (100 μM FeCl3, Sigma-Aldrich), iron-restricted (100 μM dipyridyl; Sigma-Aldrich), or iron-restricted conditions with iron supplementation (100 μM dipyridyl with 100 μM FeCl3; Sigma-Aldrich) were cocultured with AGS cells for 6 hours. Protein lysates were harvested, separated by SDS-PAGE, and transferred to PVDF membranes. Levels of total CagA (anti-CagA antibody; Austral Biologicals), phosphorylated CagA (anti-pY99 antibody; Santa Cruz Biotechnology Inc.) as a measure of translocated CagA, and total H. pylori protein (anti-Hp) (59) were determined. Protein intensities were quantified using the ChemiGenius Gel Bio Imaging System (Syngene).
Real-time RT-PCR.
In vivo–adapted H. pylori strains were cocultured with AGS cells for 6 hours and RNA was extracted (QIAGEN) according to the manufacturer’s instructions. RT-PCR and quantitative PCR were performed according to the manufacturer’s instructions to determine relative differences in IL8 expression normalized to levels of human GAPDH (TaqMan; Applied Biosystems). Gastric tissue was harvested from uninfected and infected gerbils maintained on either iron-replete or iron-depleted diets. Gastric tissue was homogenized in lysis buffer and RNA was extracted (QIAGEN) according to the manufacturer’s instructions. Contaminating DNA was removed by digestion with RNase-free DNase (Promega). Primers were generated based on IL1B, IFNG, and TNFA sequences using the following GenBank accession numbers: AB177840.1, L37782.1, AB177841.1, respectively. RT-PCR and quantitative PCR were performed according to the manufacturer’s instructions to determine relative differences in levels of cytokine expression (IL1B, IFNG, and TNGA) normalized to levels of gerbil 18S (Applied Biosystems). Expression levels were determined by the ΔΔCt method and are represented as 2Δ.
IL-8 ELISA.
In vivo–adapted H. pylori strains; strain 7.13 grown under iron-replete (100 μM FeCl3; Sigma-Aldrich), iron-restricted (100 μM dipyridyl; Sigma-Aldrich), or iron-restricted conditions with iron supplementation (100 μM dipyridyl with 100 μM FeCl3; Sigma-Aldrich); or human clinical H. pylori isolates were cocultured with AGS cells for 6 hours, and cell supernatants were collected. Levels of IL-8 were determined by Quantikine IL-8 ELISA (R&D Systems Inc.), according to the manufacturer’s instructions, and analyzed with Gen5 software (Synergy 4; BioTek).
Human ferritin ELISA.
Serum samples from human subjects from the Andean mountain Nariño region of Colombia (60) were previously obtained under the Vanderbilt University Institutional Review Board-approved protocols and stored at –80°C. Levels of ferritin were determined by ELISA (Calbiotech Inc.), according to the manufacturer’s instructions, and analyzed with Gen5 software (Synergy 4; BioTek).
Statistics.
Mean values with standard error are shown from experiments performed on at least 3 independent occasions. Mann-Whitney U, Spearman nonparametric correlation, log-rank, χ2, 2-tailed Student’s t, and ANOVA tests were used for comparisons. A P value less than or equal to 0.05 was considered significant.
Study approval.
All animal and human studies were conducted in accordance with the Declaration of Helsinki principles and were approved by the Vanderbilt University Institutional Animal Care and Use Committee and the Vanderbilt University Institutional Review Board, respectively. All human subjects provided written informed consent.
Supplementary Material
Acknowledgments
We acknowledge Russ Gerads of Applied Speciation and Consulting LLC for performing the ICP-DRC-MS studies on hepatic tissue. We also thank Scott Merrell at Uniformed Services University for providing the 7.13 fur– isogenic mutant strain. We acknowledge the following core laboratories and personnel at Vanderbilt University for their contributions to these studies: Mass Spectrometry Research Center and Proteomics Laboratory (Sarah Stuart and Yaofang Zhang); Institute for Small Animal Imaging (Fuxue Xin); Translational Pathology Shared Resource (Stephanie Doss and Anne Pate); Tissue Acquisition and Pathology Core; Division of Animal Care; Institute for Clinical and Translational Research (VICTR); and the Digestive Disease Research Center. We acknowledge the following funding sources for support of this work: F32CA153539 (to J.M. Noto); R01CA077955, R01DK058587, P01CA116087, and P30DK058404 (to R.M. Peek Jr.); P01CA028842 (to P. Correa); R01AI068009, and the Department of Veterans Affairs (to T.L. Cover); and R01DK053620 (to K.T. Wilson).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(1):479–492. doi:10.1172/JCI64373.
See the related Commentary beginning on page 113.
References
- 1.Polk DB, Peek RM., Jr Helicobacter pylori: gastric cancer and beyond. . Nat Rev Cancer. 2010;10(6):403–414. doi: 10.1038/nrc2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stein M, Bagnoli F, Halenbeck R, Rappuoli R, Fantl WJ, Covacci A. c-Src/Lyn kinases activate Helicobacter pylori CagA through tyrosine phosphorylation of the EPIYA motifs. . Mol Microbiol. 2002;43(4):971–980. doi: 10.1046/j.1365-2958.2002.02781.x. [DOI] [PubMed] [Google Scholar]
- 3.Tammer I, Brandt S, Hartig R, Konig W, Backert S. Activation of Abl by Helicobacter pylori: a novel kinase for CagA and crucial mediator of host cell scattering. . Gastroenterology. 2007;132(4):1309–1319. doi: 10.1053/j.gastro.2007.01.050. [DOI] [PubMed] [Google Scholar]
- 4.Mueller D, et al. c-Src and c-Abl kinases control hierarchic phosphorylation and function of the CagA effector protein in Western and East Asian Helicobacter pylori strains. . J Clin Invest. 2012;122(4):1553–1566. doi: 10.1172/JCI61143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Segal ED, Cha J, Lo J, Falkow S, Tompkins LS. Altered states: involvement of phosphorylated CagA in the induction of host cellular growth changes by Helicobacter pylori. . Proc Natl Acad Sci U S A. 1999;96(25):14559–14564. doi: 10.1073/pnas.96.25.14559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Odenbreit S, Puls J, Sedlmaier B, Gerland E, Fischer W, Haas R. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. . Science. 2000;287(5457):1497–1500. doi: 10.1126/science.287.5457.1497. [DOI] [PubMed] [Google Scholar]
- 7.Mimuro H, Suzuki T, Tanaka J, Asahi M, Haas R, Sasakawa C. Grb2 is a key mediator of Helicobacter pylori CagA protein activities. . Mol Cell. 2002;10(4):745–755. doi: 10.1016/S1097-2765(02)00681-0. [DOI] [PubMed] [Google Scholar]
- 8.Amieva MR, Vogelmann R, Covacci A, Tompkins LS, Nelson WJ, Falkow S. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. . Science. 2003;300(5624):1430–1434. doi: 10.1126/science.1081919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saadat I, et al. Helicobacter pylori CagA targets PAR1/MARK kinase to disrupt epithelial cell polarity. . Nature. 2007;447(7142):330–333. doi: 10.1038/nature05765. [DOI] [PubMed] [Google Scholar]
- 10.Pra D, Rech Franke SI, Pegas Henriques JA, Fenech M. A possible link between iron deficiency and gastrointestinal carcinogenesis. Nutr Cancer. 2009;61(4):415–426. doi: 10.1080/01635580902803701. [DOI] [PubMed] [Google Scholar]
- 11.Akiba S, et al. Serum ferritin and stomach cancer risk among a Japanese population. Cancer. 1991;67(6):1707–1712. doi: 10.1002/1097-0142(19910315)67:6<1707::AID-CNCR2820670638>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 12.Nomura A, Chyou PH, Stemmermann GN. Association of serum ferritin levels with the risk of stomach cancer. Cancer Epidemiol Biomarkers Prev. 1992;1(7):547–550. [PubMed] [Google Scholar]
- 13.Harrison LE, Zhang ZF, Karpeh MS, Sun M, Kurtz RC. The role of dietary factors in the intestinal and diffuse histologic subtypes of gastric adenocarcinoma: a case-control study in the U.S. Cancer. 1997;80(6):1021–1028. doi: 10.1002/(SICI)1097-0142(19970915)80:6<1021::AID-CNCR3>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 14.Muhsen K, Cohen D. Helicobacter pylori infection and iron stores: a systematic review and meta-analysis. . Helicobacter. 2008;13(5):323–340. doi: 10.1111/j.1523-5378.2008.00617.x. [DOI] [PubMed] [Google Scholar]
- 15.Yuan W, et al. Iron deficiency anemia in Helicobacter pylori infection: meta-analysis of randomized controlled trials. . Scand J Gastroenterol. 2010;45(6):665–676. doi: 10.3109/00365521003663670. [DOI] [PubMed] [Google Scholar]
- 16.Tan S, Noto JM, Romero-Gallo J, Peek RM, Jr, Amieva MR. Helicobacter pylori perturbs iron trafficking in the epithelium to grow on the cell surface. . PLoS Pathog. 2011;7(5):e1002050. doi: 10.1371/journal.ppat.1002050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Honda S, Fujioka T, Tokieda M, Satoh R, Nishizono A, Nasu M. Development of Helicobacter pylori-induced gastric carcinoma in Mongolian gerbils. . Cancer Res. 1998;58(19):4255–4259. [PubMed] [Google Scholar]
- 18.Watanabe T, Tada M, Nagai H, Sasaki S, Nakao M. Helicobacter pylori infection induces gastric cancer in Mongolian gerbils. . Gastroenterology. 1998;115(3):642–648. doi: 10.1016/S0016-5085(98)70143-X. [DOI] [PubMed] [Google Scholar]
- 19.Hur K, et al. Insufficient role of cell proliferation in aberrant DNA methylation induction and involvement of specific types of inflammation. Carcinogenesis. 2011;32(1):35–41. doi: 10.1093/carcin/bgq219. [DOI] [PubMed] [Google Scholar]
- 20.Toyoda T, et al. Anti-inflammatory effects of caffeic acid phenethyl ester (CAPE), a nuclear factor-kappaB inhibitor, on Helicobacter pylori-induced gastritis in Mongolian gerbils. . Int J Cancer. 2009;125(8):1786–1795. doi: 10.1002/ijc.24586. [DOI] [PubMed] [Google Scholar]
- 21.Danielli A, Roncarati D, Delany I, Chiarini V, Rappuoli R, Scarlato V. In vivo dissection of the Helicobacter pylori Fur regulatory circuit by genome-wide location analysis. . J Bacteriol. 2006;188(13):4654–4662. doi: 10.1128/JB.00120-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Franco AT, et al. Delineation of a carcinogenic Helicobacter pylori proteome. . Mol Cell Proteomics. 2009;8(8):1947–1958. doi: 10.1074/mcp.M900139-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crabtree JE, Farmery SM, Lindley IJ, Figura N, Peichl P, Tompkins DS. CagA/cytotoxic strains of Helicobacter pylori and interleukin-8 in gastric epithelial cell lines. . J Clin Pathol. 1994;47(10):945–950. doi: 10.1136/jcp.47.10.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jagadeesan V, Rao NJ, Sesikeran B. Effect of iron deficiency on DMH-induced gastrointestinal tract tumors and occurrence of hepatocyte abnormalities in Fischer rats. Nutr Cancer. 1994;22(3):285–291. doi: 10.1080/01635589409514354. [DOI] [PubMed] [Google Scholar]
- 25.Knekt P, Reunanen A, Takkunen H, Aromaa A, Heliovaara M, Hakulinen T. Body iron stores and risk of cancer. Int J Cancer. 1994;56(3):379–382. doi: 10.1002/ijc.2910560315. [DOI] [PubMed] [Google Scholar]
- 26.van Lee L, Heyworth J, McNaughton S, Iacopetta B, Clayforth C, Fritschi L. Selected dietary micronutrients and the risk of right- and left-sided colorectal cancers: a case-control study in Western Australia. Ann Epidemiol. 2011;21(3):170–177. doi: 10.1016/j.annepidem.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 27.Ioannou GN, Rockey DC, Bryson CL, Weiss NS. Iron deficiency and gastrointestinal malignancy: a population-based cohort study. Am J Med. 2002;113(4):276–280. doi: 10.1016/S0002-9343(02)01214-7. [DOI] [PubMed] [Google Scholar]
- 28.Selby JV, Friedman GD. Epidemiologic evidence of an association between body iron stores and risk of cancer. Int J Cancer. 1988;41(5):677–682. doi: 10.1002/ijc.2910410507. [DOI] [PubMed] [Google Scholar]
- 29.Stevens RG, Jones DY, Micozzi MS, Taylor PR. Body iron stores and the risk of cancer. N Engl J Med. 1988;319(16):1047–1052. doi: 10.1056/NEJM198810203191603. [DOI] [PubMed] [Google Scholar]
- 30.Chua AC, Klopcic B, Lawrance IC, Olynyk JK, Trinder D. Iron: an emerging factor in colorectal carcinogenesis. World J Gastroenterol. 2010;16(6):663–672. doi: 10.3748/wjg.v16.i6.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jin F, Xiong WJ, Jing JC, Feng Z, Qu LS, Shen XZ. Evaluation of the association studies of single nucleotide polymorphisms and hepatocellular carcinoma: a systematic review. J Cancer Res Clin Oncol. 2011;137(7):1095–1104. doi: 10.1007/s00432-010-0970-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Atamna H, Killilea DW, Killilea AN, Ames BN. Heme deficiency may be a factor in the mitochondrial and neuronal decay of aging. Proc Natl Acad Sci U S A. 2002;99(23):14807–14812. doi: 10.1073/pnas.192585799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walter PB, et al. Iron deficiency and iron excess damage mitochondria and mitochondrial DNA in rats. Proc Natl Acad Sci U S A. 2002;99(4):2264–2269. doi: 10.1073/pnas.261708798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Aslan M, et al. Lymphocyte DNA damage and oxidative stress in patients with iron deficiency anemia. Mutat Res. 2006. 601 (1-2): 144 149 [DOI] [PubMed] [Google Scholar]
- 35.Chaturvedi R, et al. Spermine oxidase mediates the gastric cancer risk associated with Helicobacter pylori CagA. . Gastroenterology. 2011;141(5):1696–1708. doi: 10.1053/j.gastro.2011.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Elfstrom P, Granath F, Ye W, Ludvigsson JF. Low risk of gastrointestinal cancer among patients with celiac disease, inflammation, or latent celiac disease. Clin Gastroenterol Hepatol. 2012;10(1):30–36. doi: 10.1016/j.cgh.2011.06.029. [DOI] [PubMed] [Google Scholar]
- 37.Villanacci V, Bassotti G, Liserre B, Lanzini A, Lanzarotto F, Genta RM. Helicobacter pylori infection in patients with celiac disease. . Am J Gastroenterol. 2006;101(8):1880–1885. doi: 10.1111/j.1572-0241.2006.00621.x. [DOI] [PubMed] [Google Scholar]
- 38.Josenhans C, Labigne A, Suerbaum S. Comparative ultrastructural and functional studies of Helicobacter pylori and Helicobacter mustelae flagellin mutants: both flagellin subunits, FlaA and FlaB, are necessary for full motility in Helicobacter species. . J Bacteriol. 1995;177(11):3010–3020. doi: 10.1128/jb.177.11.3010-3020.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eaton KA, Suerbaum S, Josenhans C, Krakowka S. Colonization of gnotobiotic piglets by Helicobacter pylori deficient in two flagellin genes. . Infect Immun. 1996;64(7):2445–2448. doi: 10.1128/iai.64.7.2445-2448.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carneiro CR, Postol E, Nomizo R, Reis LF, Brentani RR. Identification of enolase as a laminin-binding protein on the surface of Staphylococcus aureus. . Microbes Infect. 2004;6(6):604–608. doi: 10.1016/j.micinf.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 41.Nogueira SV, Smith AA, Qin JH, Pal U. A surface enolase participates in Borrelia burgdorferi-plasminogen interaction and contributes to pathogen survival within feeding ticks. . Infect Immun. 2012;80(1):82–90. doi: 10.1128/IAI.05671-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scott DR, Marcus EA, Wen Y, Oh J, Sachs G. Gene expression in vivo shows that Helicobacter pylori colonizes an acidic niche on the gastric surface. . Proc Natl Acad Sci U S A. 2007;104(17):7235–7240. doi: 10.1073/pnas.0702300104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Momynaliev KT, Rogov SI, Selezneva OV, Chelysheva VV, Akopian TA, Govorun VM. Comparative analysis of transcription profiles of Helicobacter pylori clinical isolates. . Biochemistry. 2005;70(4):383–390. doi: 10.1007/s10541-005-0129-9. [DOI] [PubMed] [Google Scholar]
- 44.Lin YF, et al. Comparative immunoproteomics of identification and characterization of virulence factors from Helicobacter pylori related to gastric cancer. . Mol Cell Proteomics. 2006;5(8):1484–1496. doi: 10.1074/mcp.M600111-MCP200. [DOI] [PubMed] [Google Scholar]
- 45.Klose KE, Mekalanos JJ. Simultaneous prevention of glutamine synthesis and high-affinity transport attenuates Salmonella typhimurium virulence. . Infect Immun. 1997;65(2):587–596. doi: 10.1128/iai.65.2.587-596.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tullius MV, Harth G, Horwitz MA. Glutamine synthetase GlnA1 is essential for growth of Mycobacterium tuberculosis in human THP-1 macrophages and guinea pigs. . Infect Immun. 2003;71(7):3927–3936. doi: 10.1128/IAI.71.7.3927-3936.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kloosterman TG, et al. Regulation of glutamine and glutamate metabolism by GlnR and GlnA in Streptococcus pneumoniae. . J Biol Chem. 2006;281(35):25097–25109. doi: 10.1074/jbc.M601661200. [DOI] [PubMed] [Google Scholar]
- 48.Ohnishi N, et al. Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. . Proc Natl Acad Sci U S A. 2008;105(3):1003–1008. doi: 10.1073/pnas.0711183105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Van Heyningen WE, Gladstone GP. The neurotoxin of Shigella shigae. III. The effect of iron on production of the toxin. . Br J Exp Pathol. 1953;34(2):221–229. [PMC free article] [PubMed] [Google Scholar]
- 50.Calderwood SB, Mekalanos JJ. Iron regulation of Shiga-like toxin expression in Escherichia coli is mediated by the fur locus. . J Bacteriol. 1987;169(10):4759–4764. doi: 10.1128/jb.169.10.4759-4764.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tai SP, Krafft AE, Nootheti P, Holmes RK. Coordinate regulation of siderophore and diphtheria toxin production by iron in Corynebacterium diphtheriae. . Microb Pathog. 1990;9(4):267–273. doi: 10.1016/0882-4010(90)90015-I. [DOI] [PubMed] [Google Scholar]
- 52.Bjorn MJ, Iglewski BH, Ives SK, Sadoff JC, Vasil ML. Effect of iron on yields of exotoxin A in cultures of Pseudomonas aeruginosa PA-103. . Infect Immun. 1978;19(3):785–791. doi: 10.1128/iai.19.3.785-791.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.de Vries AC, et al. Gastric cancer risk in patients with premalignant gastric lesions: a nationwide cohort study in the Netherlands. Gastroenterology. 2008;134(4):945–952. doi: 10.1053/j.gastro.2008.01.071. [DOI] [PubMed] [Google Scholar]
- 54.Vicari JJ, et al. The seroprevalence of CagA-positive Helicobacter pylori strains in the spectrum of gastroesophageal reflux disease. . Gastroenterology. 1998;115(1):50–57. doi: 10.1016/S0016-5085(98)70364-6. [DOI] [PubMed] [Google Scholar]
- 55.Chen Y, Blaser MJ. Helicobacter pylori colonization is inversely associated with childhood asthma. . J Infect Dis. 2008;198(4):553–560. doi: 10.1086/590158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Franco AT, et al. Regulation of gastric carcinogenesis by Helicobacter pylori virulence factors. . Cancer Res. 2008;68(2):379–387. doi: 10.1158/0008-5472.CAN-07-0824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pentecost M, Otto G, Theriot JA, Amieva MR. Listeria monocytogenes invades the epithelial junctions at sites of cell extrusion. . PLoS Pathog. 2006;2(1):e3. doi: 10.1371/journal.ppat.0020003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shaffer CL, et al. Helicobacter pylori exploits a unique repertoire of type IV secretion system components for pilus assembly at the bacteria-host cell interface. . PLoS Pathog. 2011;7(9):e1002237. doi: 10.1371/journal.ppat.1002237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cao P, McClain MS, Forsyth MH, Cover TL. Extracellular release of antigenic proteins by Helicobacter pylori. . Infect Immun. 1998;66(6):2984–2986. doi: 10.1128/iai.66.6.2984-2986.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mera R, et al. Long term follow up of patients treated for Helicobacter pylori infection. . Gut. 2005;54(11):1536–1540. doi: 10.1136/gut.2005.072009. [DOI] [PMC free article] [PubMed] [Google Scholar]