

Abstract
Cyclin D1b is a splice variant of the cell cycle regulator cyclin D1 and is known to harbor divergent and highly oncogenic functions in human cancer. While cyclin D1b is induced during disease progression in many cancer types, the mechanisms underlying cyclin D1b function remain poorly understood. Herein, cell and human tumor xenograft models of prostate cancer were utilized to resolve the downstream pathways that are required for the protumorigenic functions of cyclin D1b. Specifically, cyclin D1b was found to modulate the expression of a large transcriptional network that cooperates with androgen receptor (AR) signaling to enhance tumor cell growth and invasive potential. Notably, cyclin D1b promoted AR-dependent activation of genes associated with metastatic phenotypes. Further exploration determined that transcriptional induction of SNAI2 (Slug) was essential for cyclin D1b–mediated proliferative and invasive properties, implicating Slug as a critical driver of disease progression. Importantly, cyclin D1b expression highly correlated with that of Slug in clinical samples of advanced disease. In vivo analyses provided strong evidence that Slug enhances both tumor growth and metastatic phenotypes. Collectively, these findings reveal the underpinning mechanisms behind the protumorigenic functions of cyclin D1b and demonstrate that the convergence of the cyclin D1b/AR and Slug pathways results in the activation of processes critical for the promotion of lethal tumor phenotypes.
Introduction
Cyclin D1b is a highly oncogenic variant of the cell cycle regulator cyclin D1 (cyclin D1a) (1, 2). In marked contrast to cyclin D1a, cyclin D1b expression alone is sufficient to promote cellular transformation, both in vitro and in vivo (1, 3). Moreover, cyclin D1b has been suggested, in a limited number of model systems, to promote aggressive tumor phenotypes (4). Accordingly, emerging evidence demonstrates that preferential upregulation of the cyclin D1b isoform is tightly associated with tumor development, tumor progression, and poor outcome (5–7). Although evidence to date strongly suggests that cyclin D1b represents a gain-of-function D-cyclin variant of clinical relevance, the underlying mechanism or mechanisms by which cyclin D1b promotes protumorigenic phenotypes remain unknown.
Whereas cyclin D1a functions to promote tumor formation through advancing cell cycle progression, this biochemical function does not appear to be conserved in cyclin D1b. While cyclin D1b retains the ability to bind cyclin-dependent kinase 4 (CDK4), a potent effector of the G1-S phase transition, cyclin D1b is a poor catalyst of CDK4 activity (1, 8). Consistent with this observation, cyclin D1b expression does not independently correlate with increased proliferative capacity in clinical specimens (5). Little insight into the underpinning mechanisms of cyclin D1b–mediated oncogenic capacity was gained from motif analyses of the transcript encoding cyclin D1b (transcript b). Transcript b harbors a 3′ end that is highly divergent from that of full-length cyclin D1 transcript, which arises as a failure to splice at the exon 4/intron 4 boundary of CCND1 pre-mRNA, resulting in read-through into intron 4 and incorporation of an early stop codon (1, 8, 9). Loss of the exon 5–encoded sequences eliminates a phosphorylation site in cyclin D1a that was suggested to promote nuclear export, and indeed, cyclin D1b appears to be constitutively nuclear, but surprisingly maintains a half-life identical to that of cyclin D1a (1, 3, 8).
Interestingly, both oncogenic and nononcogenic events that impinge upon cyclin D1b production have been identified. The oncogenic splicing factor SRSF1 (also known as SF2/ASF1) was recently shown to enhance production of transcript b and positively correlates with cyclin D1b status in prostate tumors (10). Further investigation revealed that, through direct binding at the critical exon 4/intron 4 boundary of the CCND1 transcript, SRSF1 functions to limit efficient splicing at this site, resulting in a surge of transcript b and ultimately cyclin D1b production (10). Moreover, a polymorphism at the critical exon 4 splice junction (G/A870) has been associated with enhanced cyclin D1b production in normal prostatic tissue (the A allele), while SRSF1 was found to bind with higher affinity to transcripts containing the G allele (6, 10). Notably, while the A allele appears to be required for transcript b production in normal tissue, this requirement is lost as a function of disease progression, potentially due to the induction of SRSF1 expression. Additional splicing factors, including SAM68, also enhance production of cyclin D1b in hormone-dependent cancers, although the mechanisms by which this is accomplished remain unclear (11). Thus, while multiple oncogenic and nononcogenic events that impinge upon cyclin D1b production in human disease have been identified, studies to date have yet to rigorously delineate the mechanism or mechanisms by which cyclin D1b affects the processes of pathological importance in cancer.
Recent in vivo genomic/proteomic screens from multiple organs found that an overwhelming majority of the cyclin D1a interactome belongs to the super-family of transcriptional regulators, including a large number of steroid nuclear receptors (12). These unbiased analyses are consistent with previous findings that demonstrated that cyclin D1a associates with and modulates activity of both estrogen receptor α (ERα) (2, 13, 14) and the androgen receptor (AR) (2, 15–18) in breast and prostate cancer (PCa) cells, respectively. Further investigation of selected ER- and AR–dependent target genes revealed that cyclin D1b is selectively altered in the ability to modulate nuclear receptor function (19, 20). For example, cyclin D1b induction proved sufficient to promote resistance to ERα-directed therapeutics (20), and analyses of clinical specimens demonstrated that cyclin D1b expression significantly correlated with poor prognosis (7). Conversely, while cyclin D1b retains the ability to bind AR, the ability to modulate AR activity is divergent from that of cyclin D1a (19). Specifically, it was shown that unlike cyclin D1a, cyclin D1b lacks the ability to attenuate AR induced expression of prostate specific antigen (PSA), an AR target gene used to monitor PCa development and progression (15, 17, 19, 21, 22). These initial observations were of potentially strong clinical relevance, as examination of a large cohort of clinical specimens revealed that cyclin D1b (but not cyclin D1a) was dramatically upregulated in a subset of AR-positive human prostatic adenocarcinomas (6). Combined, these findings implicate cyclin D1b as a major effector of disease progression in hormone-dependent cancers.
Given the established clinical relevance of cyclin D1b expression in hormone-dependent cancers, a myriad of in vitro and in vivo prostate adenocarcinoma model systems were used to identify the underlying means through which cyclin D1b elicits protumorigenic phenotypes. It was discovered that a major function of cyclin D1b is to induce nuclear receptor–dependent transcriptional events that result in markedly enhanced invasion and prometastatic potential activities that required Slug, a prooncogenic transcription factor. Clinical analyses established the translational relevance of the findings, wherein it was demonstrated that cyclin D1b and Slug were coexpressed in a large number of human tumors. The present study establishes a critical and previously unknown link between 2 major oncogenic pathways of clinical relevance and identifies a key mechanism by which cyclin D1b promotes aggressive tumor phenotypes.
Results
Cyclin D1b induces a unique gene expression program associated with prometastatic phenotypes.
Given the observation that cyclin D1b harbors heightened oncogenic activity distinct from full-length cyclin D1a, it is imperative to assess the mechanism or mechanisms underlying cyclin D1b–mediated protumorigenic activity. While cyclin D1b expression has previously been associated with tumor progression in a large number of tissue types (4, 20, 23), there is substantial evidence of cyclin D1b upregulation in PCa. Indeed, cyclin D1b is selectively induced as a function of tumorigenesis (6) and has been demonstrated to alter AR function (19) in this tumor type. Therefore, cells of prostatic origin that express low levels of endogenous cyclin D1b were utilized to assess the molecular and cellular consequence of increased cyclin D1b expression. As shown, LNCaP cells express little endogenous cyclin D1b. Therefore, multiple clones were generated which resulted in low or high expression (Figure 1A), so as to mimic the spectrum of expression observed in human tumors. Consistent with previous studies (6), androgen status had no effect on transcript b stability/levels (Supplemental Figure 1A; supplemental material available online with this article; doi: 10.1172/JCI64750DS1), and cyclin D1b expression was maintained in androgen-depleted conditions (Figure 1A). Analyses of cyclin D1b function in the presence and absence of androgen are critical, as first-line therapeutic intervention for disseminated PCa is androgen deprivation therapy (ADT) and AR signaling in ADT-sensitive cells is required for translation of both cyclin D1 isoforms (6, 24). Thus, the models generated allow for analyses of cyclin D1b function under conditions mimicking both the early stage of disease (prior to ADT challenge) and that after ADT administration.
Figure 1. Cyclin D1b induces protumorigenic phenotypes and a unique gene expression program associated with metastasis.

(A) Control (LN-vec) and cyclin D1b clones expressing low (LN-D1b [L]) and high (LN-D1b [H]) levels of cyclin D1b were hormone deprived for 72 hours. Expression of cyclin D1b protein was analyzed in the presence or absence of DHT 24 hours after treatment. (B) Control or cyclin D1b–expressing LNCaPs were plated in soft agar in androgen-proficient (FBS) or androgen-depleted (CDT) conditions and cultured for a period of 4 weeks, after which colonies greater than 75 μm in size were counted. (C) Control or cyclin D1b–expressing cells were seeded in the upper chamber of a Boyden invasion chamber and allowed to invade through the matrix toward androgen-proficient (FBS) or androgen-deprived (CDT) chemoattractants for 24 hours. Cells were fixed and DAPI stained; the total number of invading cells was reported. (D) Heat map of the differential gene expression profile regulated by cyclin D1a and cyclin D1b (cluster 1), cyclin D1a only (cluster 2), or cyclin D1b only (cluster 3). Genes shown demonstrated a false discovery rate of 1% or less and an absolute fold change of 2 or more. (E) Venn diagram comparing all cyclin D1a vs. cyclin D1b genes using a 2.0-fold cut off. Error bars represent mean ± SEM. **P < 0.01; ***P < 0.001.
To assess the potential impact of cyclin D1b on cancer cell phenotypes, colony formation assays were initially performed. Parental cells are weakly tumorigenic and have a limited capacity for anchorage-independent growth (25). As shown, cyclin D1b expression significantly enhanced colony formation capacity in soft agar (Figure 1B). However, this was dependent on AR function, as androgen depletion ablated the ability of cyclin D1b to confer an anchorage-independent phenotype, suggesting that cooperation between cyclin D1b and androgen-signaling pathways is required for cyclin D1b function. Further investigation assessed the impact of cyclin D1b on invasive phenotypes utilizing invasion chamber assays. As shown, cyclin D1b–expressing cells demonstrated approximately a 20% increase in the total number of invading cells over control (Figure 1C). Of note, neither control nor cyclin D1b–expressing cells invaded through the matrix toward a gradient depleted of androgens, indicating that the invasive phenotypes attributable to cyclin D1b expression are androgen dependent. Collectively, these findings highlight the ability of cyclin D1b to illicit protumorigenic phenotypes and underscore the importance of androgen signaling to potentiate cyclin D1b function.
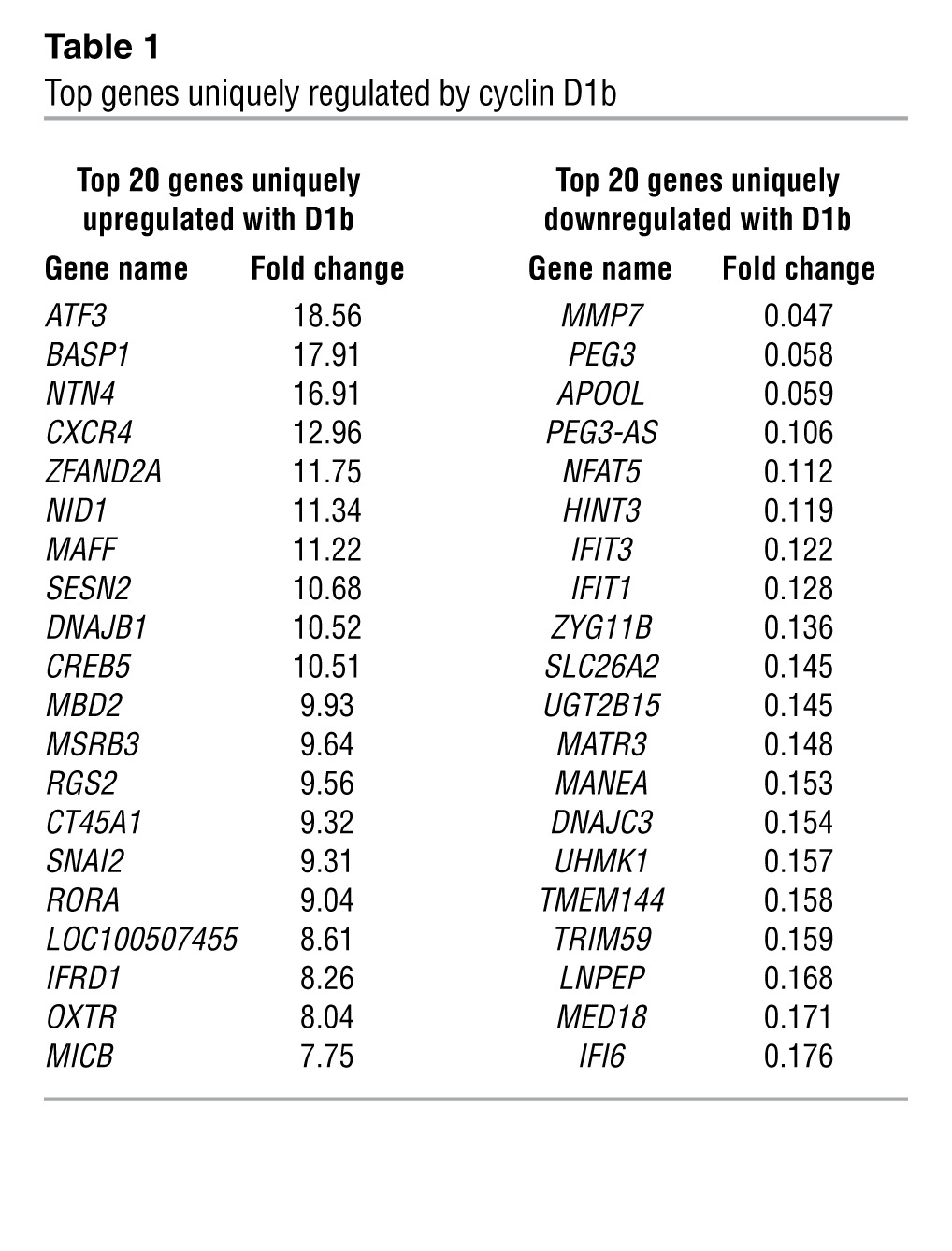
To determine the underlying mechanisms by which cyclin D1b promotes AR-dependent effects on cell growth and invasion, unbiased gene expression analyses were performed. For these studies, cells were cultured transiently under conditions of androgen ablation, transduced with either control (GFP), cyclin D1b, or full-length cyclin D1a, and subsequently stimulated with 1 nM dihydrotestosterone (DHT) prior to harvest, RNA collection, and microarray analyses (Supplemental Figure 1B). Relative protein levels are shown in Supplemental Figure 1B. Transcripts were filtered using a cut-off of P < 0.05 and a stringent false discovery filter and organized based on their patterns of up- and downregulation. Three distinct classes of transcripts were uncovered: those that are regulated by both cyclin D1a and cyclin D1b (cluster 1), those that are regulated only by cyclin D1a (cluster 2) and those that are uniquely regulated by cyclin D1b (cluster 3). Surprisingly, there was very little overlap between the transcriptomes of cyclin D1a and cyclin D1b, with only 117 transcripts common between the 2 clusters. Consistent with previous findings (16) the AR-regulated gene KLK3 (PSA) was present only in the cyclin D1a–regulated cluster, highlighting potential differences in the roles of D1a and D1b cyclins in AR action. Furthermore, cluster 3 is the largest of all the gene sets (790 genes upregulated and 685 downregulated), demonstrating cyclin D1b can serve as a unique regulator of transcription in the presence of androgen (Figures 1, D and E). Gene Ontology analysis of biological pathways uncovered a variety of known processes regulated by cyclin D1a, including cell cycle, mitosis, response to DNA damage, and DNA repair (Supplemental Figure 1C), with minimal enrichment for any one pathway in the cyclin D1b cluster (Supplemental Figure 1C). Strikingly, however, a number of the top 20 genes uniquely regulated by cyclin D1b are functionally associated with altered cell migration, invasion, or differentiation, including NTN4, CXCR4, and SNAI2 (Table 1). Of these genes, only CXCR4 and SNAI2 have been shown to be regulated by androgens (16, 26). Moreover, comparison of the cyclin D1b–specific gene set (cluster 3) with a recently identified metastasis-associated gene signature across multiple cancer types uncovered commonalities between the 2 gene sets (Supplemental Figure 1D), with SNAI2 as the top cyclin D1b–regulated gene present in this signature (whereas no overlap was seen with the cyclin D1a, cluster 2, gene set). Further investigation in multiple models of cyclin D1b upregulation (data not shown) identified the transcriptional regulator SNAI2 (Slug), a known inducer of both growth and invasion in different systems (27–29) as strongly associated with cyclin D1b. Since, SNAI2 expression promotes phenotypes similar to those induced by cyclin D1b, is regulated as a function of androgen action, and is clinically associated with metastatic progression, subsequent efforts were focused on defining the role of SNAI2 as a mediator of cellular phenotypes associated with cyclin D1b expression.
Table 1.
Top genes uniquely regulated by cyclin D1b
Cyclin D1b induces SNAI2 (Slug) expression through cooperation with the AR axis.
In light of the strong and specific link between cyclin D1b and SNAI2 expression and the established requirement of androgen for cyclin D1b–mediated phenotypes, the impact of androgen status on cyclin D1b–induced Slug upregulation was assessed in multiple model systems. Initially, the impact of suppressing AR signaling was determined. As shown in Figure 2A, SNAI2 mRNA was sensitive to AR activation, as the AR competitive antagonist Casodex suppressed SNAI2 mRNA levels in control cells. Consistent with the data herein, cyclin D1b enhanced SNAI2 expression in a dose-dependent manner (Figure 2A). Of note, while knockdown of D1 cyclins in cyclin D1b–expressing cells drastically reduced SNAI2 levels (Supplemental Figure 2A), upregulation of cyclin D1a alone had no effect on SNAI2 mRNA (Supplemental Figure 2B), indicating cyclin D1b (but not cyclin D1a) is sufficient to drive SNAI2 expression. Interestingly, this effect was also inhibited by Casodex, indicating that the ability of cyclin D1b to regulate SNAI2 expression is AR dependent. Consonantly, protein analyses revealed that cyclin D1b expression resulted in marked accumulation of Slug protein in the presence of androgen (Figure 2A), whereas Slug levels remained low under conditions of androgen ablation (Figure 2A) with no change observed in total AR protein levels (data not shown). These findings highlight the interdependency of cyclin D1b and AR as regulators of Slug expression.
Figure 2. Cyclin D1b enhances SNAI2 (Slug) expression through cooperation with the AR axis.

(A) Left: control and cyclin D1b–expressing cells were incubated with the AR inhibitor Casodex or ethanol (EtOH) control for 24 hours, RNA harvested, and relative SNAI2 levels determined. Right: control and cyclin D1b cells were cultured in androgen-proficient or androgen-depleted conditions for 72 hours and relative levels of Slug determined. Control cells cultured in androgen-proficient medium serve as a positive control. (B) Left: LNCaP and LAPC4 cells were cultured in androgen-proficient conditions and relative expression of cyclin D1b and SNAI2 levels determined. CDK4 (protein) and GAPDH (transcript) serve as controls. Right: LAPC4 cells were hormone deprived and stimulated with EtOH (0.1%), DHT (1 nM), and/or Casodex (10 μM) for 24 hours and SNAI2 expression analyzed (normalizing to GAPDH). (C) The AR-negative cell line PC3 was transfected with cyclin D1b constructs in biological duplicate, and relative levels of transcript b and SNAI2 transcript determined. ddH2O serves as a non-template control (NTC). (D) Schematic of Morpholino mechanism of action in CCND1 alternative splicing. (E) Increasing amounts of Morpholino were introduced to LNCaP cells and relative levels of transcript b determined by qPCR after 72 hours (normalized to GAPDH). (F) LNCaP cells were treated with 12 μM Morpholino for 72 hours and levels of cyclin D1b determined. (G) Cells were treated as in E, and levels of transcript b and SNAI2 are shown. GAPDH serves as a control. Error bars represent mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001.
Comparison of SNAI2 transcript levels between cells expressing various levels of cyclin D1b revealed a positive correlation between cyclin D1b and SNAI2 levels. As compared with LNCaP control, a 3-fold increase in SNAI2 transcripts was detected in LAPC4 (which endogenously expresses cyclin D1b; Figure 2B), whose levels were also dependent on AR activity (Figure 2B). Additionally, VCaP cells engineered to express low levels of cyclin D1b (comparable with endogenous levels in LAPC4) (Supplemental Figure 2C) showed a significant increase in SNAI2 levels as compared with vector control (Supplemental Figure 2C). Conversely, in AR-negative PCa cells (PC3), cyclin D1b induction had no impact on SNAI2 expression (Figure 2C). Combined, these data demonstrate that cyclin D1b acts in concert with AR to induce expression of Slug, thus demonstrating cooperation between AR and an established oncogene of PCa relevance.
Modulation of the endogenous cyclin D1 alternative splicing event results in SNAI2 upregulation.
The data presented above strongly suggest that cyclin D1b and AR cooperate to induce Slug expression. To directly address this hypothesis, the impact of directly altering splicing of the CCND1 transcript was assessed using Morpholino technology. As previously described, the cyclin D1b transcript arises due to a failure to splice at the exon 4/intron 4 boundary of the cyclin D1 pre-mRNA. Thus, Morpholinos were designed to specifically bind the cyclin D1 mRNA, which suppressed splicing at the exon 4/intron 4 boundary (Figure 2D). Accordingly, introduction of the Morpholino into cells that produce little endogenous transcript b effectively suppressed the splicing event, resulting in a dose-dependent upregulation of transcript b (Figure 2E and Supplemental Figure 2D) and cyclin D1b protein (Figure 2F). While treatment with the Morpholino had no effect on overall CCND1 levels (Supplemental Figure 2E), SNAI2 levels increased in a dose-dependent manner (Figure 2G). These data demonstrate that manipulation of the cyclin D1 splicing event within cells facilitates upregulation of the cyclin D1b transcript, and results in increased SNAI2 expression.
Cyclin D1b enhances AR occupancy at SNAI2 regulatory loci.
To elucidate the means by which cyclin D1b and AR cooperate to induce Slug expression, kinetic analyses of SNAI2 mRNA induction were performed in hormone therapy–sensitive cells after DHT stimulation. As expected, expression of the mRNA encoding the AR target gene KLK3/PSA was significantly induced (as early as 120 minutes after DHT stimulation), and its expression continued to increase over the time of monitoring (Figure 3A). Importantly, SNAI2 transcript accumulation closely mirrored that of KLK3/PSA, thus suggesting that the effect of AR on Slug expression could be direct.
Figure 3. Cyclin D1b interacts with AR on chromatin and enhances AR occupancy at SNAI2 regulatory loci.

(A) Androgen-depleted LNCaP cells were stimulated with 1 nM DHT for the indicated time points and relative expression of SNAI2 and KLK3 transcript levels determined. (B) Androgen proficient LNCaP lysates expressing a 3× flag cyclin D1b construct were fractionated into soluble and chromatin-tethered lysates and subjected to immunoprecipitation of AR or Flag. 10% input and IgG served as positive and negative controls, respectively, while GAPDH and histone H4 served as soluble and chromatin-tethered specific controls, respectfully. (C) LNCaP vector and cyclin D1b cells were androgen depleted for 72 hours and then stimulated with either DHT (10 nM) or EtOH (0.1%) for 3 hours. Samples were harvested for ChIP analysis, and percentage (input) occupancy of AR (top panel), cyclin D1b (middle), and acetylated histone H4 (bottom panel) are reported for the SNAI2 and the KLK3 loci (D). Error bars represent mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001
While previous biochemical analyses have demonstrated that cyclin D1b and AR maintain the ability to interact (19, 30), it remained unclear if such complexes exist on chromatin. Thus, chromatin-tethering–coupled immunoprecipitation assays, which have previously been shown to distinguish between nuclear and chromatin-associated complexes (31), were performed. As expected, AR is enriched in chromatin-tethered fractions in the presence of androgen in multiple model systems (Supplemental Figure 3A). Interestingly, the majority of cyclin D1b protein was also enriched in chromatin-tethered fractions, consistent with the identified role of D-type cyclins as transcriptional regulators (refs. 2, 12, and Supplemental Figure 3A). As cyclin D1b and AR were independently shown to be associated with chromatin, fractionated lysates were immunoprecipitated for AR or cyclin D1b to determine whether AR/cyclin D1b complexes reside on chromatin. As seen previously, minimal cyclin D1b was detected in the soluble fraction (Figure 3B, lane 1), and no cyclin D1b/AR complexes were detected by either cyclin D1b or AR IP (Figure 3B, lanes 2 and 3). Conversely, cyclin D1b/AR complexes were detected in chromatin-tethered fractions isolated from androgen-dependent or castration-resistant models (Figure 3B and Supplemental Figure 3B), indicating that cyclin D1b associates with AR complexes on chromatin.
Analyses of publically available genome-wide coupled deep sequencing (ChIP-Seq) or ChIP-chip studies that assessed AR binding to chromatin in multiple cell types (including prostate, breast, and muscle-derived tissues) (32–35) led to the identification of 2 potential AR occupied sites within the SNAI2 regulatory locus: 1 proximal to the TSS (∼600 b, AR occupied region 2, AROR2) and 1 distal to the TSS (∼3600 bp AROR1) (Supplemental Figure 3C), reminiscent of AR occupancy at the KLK3 regulatory enhancer and promoter elements (Supplemental Figure 3D). As shown, DHT stimulation in PCa cells resulted in significant enrichment of AR at both loci (Figure 3C). Parallel studies using the isogenic models described in Figure 1 (LN-D1b [L]) revealed that even modest upregulation of cyclin D1b expression significantly enhanced DHT-dependent occupancy of AR at AROR2 (P < 0.05). AR occupancy also trended higher at the distal site (AROR1) in the presence of cyclin D1b. Previous genome-wide analysis of cyclin D1 occupancy on chromatin found cyclin D1 enrichment within the murine Snai2 regulatory locus (12), suggestive of potential similar occupancy by cyclin D1b in prostate cells. Consistent with this hypothesis and data in Figure 3B, cyclin D1b was present at AR-occupied loci within the SNAI2 locus, whose occupancy was dependent upon androgen status. Thus, cyclin D1b resides at regulatory elements within the SNAI2 locus and promotes AR residency at sites likely to regulate SNAI2 expression. To further address this hypothesis, the impact on histone modifications associated with sites of active transcription was analyzed. As shown, DHT enhanced the presence of acetylated histone H4 at the promoter as well as the recruitment of RNA polymerase II proximal to the SNAI2 transcriptional start site, which was enhanced in the presence of cyclin D1b (Supplemental Figure 3E). Importantly, while cyclin D1b expression resulted in a significant enrichment of acetylated histone H4 at AROR1 and trended higher at AROR2 in response to androgen (Figure 3C), induction of cyclin D1a resulted in a lack of histone H4 enrichment and AR in response to DHT (Supplemental Figure 3F). Thus, it is likely that cyclin D1b specifically affects AR-dependent SNAI2 transcription through enrichment of acetylated histones. In contrast, while cyclin D1b was also present at the KLK3 enhancer region, AR occupancy was unchanged by cyclin D1b expression, and there was little effect on DHT-induced histone H4 acetylation (Figure 3D), consistent with the inability of cyclin D1b to promote KLK3 expression. Together, these findings reveal that cyclin D1b enhances site-specific AR occupancy and histone acetylation, events which track with DHT-mediated induction of SNAI2 expression. Based on these findings, it is proposed that the interaction of AR with specific androgen response elements within the regulatory regions of the SNAI2 gene is enhanced by cyclin D1b expression and that these cooperative activities lead to increased expression of SNAI2 (Slug).
Cyclin D1b promotes chromosomal conformations associated with active transcription.
Cyclin D1b was shown to promote SNAI2 transcription in response to DHT stimulation though enhanced AR occupancy and enrichment of acetylated histones. To further delineate the mechanisms that underlie cyclin D1b action at the SNAI2 locus, chromosome conformation capture (3C) experiments were performed to assess the relative positioning of chromosomal elements required for transcriptional activation. Previous reports demonstrated that upon transcriptional initiation, enhancer elements within the genome mobilize to “promoter-like” regions in close proximity to transcriptional start sites (36). Such action “bridges” distal chromosomal elements necessary for transcriptional induction, creating a “DNA loop.” 3C quantifies the relative distance between 2 DNA elements by restriction digestion of DNA and subsequent ligation of adjacent sequences. DNA regions bridged by a chromosomal loop ligate with higher frequencies than those that are not, and relative distance from an enhancer element can be quantified utilizing the highly sensitive TaqMan quantitative PCR (qPCR system). Thus, this system was used to determine the consequence of cyclin D1b expression on chromosomal architecture associated with active transcription. As such, a control region adjacent to the previously identified “enhancer-like” AROR1 of the SNAI2 locus was chosen as a constant region and 4 distal (test) sites spanning the SNAI2 gene and downstream sequences were tested for frequency of ligation (Figure 4A). As shown, there was little evidence of looping in the absence of DHT in control cells (LNCaP), indicative of large distances spanning the region between the constant and test sites (Figure 4A). However, after DHT stimulation, there was a large induction in the ligation frequency between the control (constant site) and all test regions (which diminishes as a function of distance), indicating that there is likely a looping event within the SNAI2 locus that occurs in response to DHT (Figure 4A). Quantification of this event confirmed a 2-fold increase in ligation frequency at proximal test sites in response to DHT, an effect that was abolished at sites distal to the enhancer element (Figure 4B). Importantly, cells expressing cyclin D1b demonstrated a heightened response to DHT at sites within, and distal to, the SNAI2 gene, indicating cyclin D1b promotes bridging of distal enhancer elements, likely required for transcriptional initiation (Figure 4B). Of note, recent unpublished AR ChIP-Seq analyses (C. McNair and K. Knudsen, unpublished observations) uncovered a potential third AR-occupied region within the SNAI2 gene itself (Figure 4C). As cyclin D1b has been shown to enhance AR occupancy at site-specific loci (Figure 3C) and 3C analyses determined that the most common looping event in cyclin D1b cells was seen at a site proximal to this region, cyclin D1b appears to enhance DNA looping from the enhancer region to this distal AR-occupied region through AR interactions. Taking these observations together, cyclin D1b promotes DNA looping at the SNAI2 locus in response to DHT, likely achieved through enhanced interaction with a distal AR-occupied site with the SNAI2 gene, culminating in DNA conformations known to promote transcription. These results confirm cyclin D1b as a potent AR cofactor and provide much needed insight into the mechanisms behind cyclin D1b–mediated transcriptional control.
Figure 4. Cyclin D1b promotes chromosomal confirmations associated with active transcription in response to androgen.

(A) LNCaP cells were starved of hormone for 72 hours and treated with 10 nM DHT for 3 hours. Cells were fixed, digested with the HindIII endonuclease, and ligated; total DNA was purified. Relative distance between the constant region (proximal to AROR1 of the SNAI2 gene) and 4 test regions spanning the SNAI2 gene and downstream sequences was determined using TaqMan qPCR. Top: 100 ng of purified ligated DNA was used to test individual primer sets for each test site to ensure formation of single bands specific to ligation products. Bottom: representative images of PCR products of each ligated site in the presence or absence of DHT. A control region lacking HindIII restriction sites serves as a genomic loading control. (B) LNCaP-Vec and LNCaP-D1b cells were treated as in A, and frequency of ligation is plotted as relative to ligation frequency of parental EtOH controls, after normalizing for total DNA content (control region). (C) ChIP sequencing data of AR occupancy across the cell cycle (C. McNair and K.E. Knudsen, unpublished observations) in LNCaP cells in response to 3 hours of (10 nM) DHT. AR occupancy within the SNAI2 gene is indicated by peaks, and proximal 3C test sites are designated by triangles. Error bars represent mean ± SEM. ***P < 0.001.
Slug is necessary and sufficient to induce cyclin D1b–mediated oncogenic and prometastatic phenotypes.
Slug has been previously identified as causal for a number of phenotypes associated with aggressive human malignancies (27, 37, 38). Given the observed association of cyclin D1b expression with prostate tumorigenesis (6) and aggressive tumor cell phenotypes (Figure 1), the requirement of Slug for cyclin D1b function was assessed. For these studies, a validated pool of siRNA constructs directed against the SNAI2 transcript was introduced into cells, resulting in a dose-dependent reduction in transcript levels and a greater than 80% reduction in Slug protein (as compared with siRNA control; Figure 5A). Parallel studies were performed with limiting cotransfection of a plasmid encoding GFP, so as to allow for tracking of cells with siRNA uptake. Notably, SNAI2 knockdown markedly suppressed invasive capacity in control cells (∼70% reduction in invading cells), but this effect was more pronounced in cells with cyclin D1b upregulation (reduction of ∼80%) (Figure 5B). Thus, Slug expression is not only required for the limited invasive potential exhibited by parental models, but is also critical for cyclin D1b–induced invasive phenotypes.
Figure 5. Slug is necessary and sufficient for cyclin D1b–mediated prometastatic and tumorigenic phenotypes.

(A) Control and cyclin D1b cells were treated with a pool of siRNAs directed against the SNAI2 transcript for 72 hours and harvested for RNA (left) and protein (right). Relative levels of SNAI2 and Slug are reported. (B) siRNA-treated cells were cotransfected with GFP and allowed to invade the Boyden chamber matrix toward an androgen-proficient gradient for 24 hours, after which they were fixed and GFP-positive cells counted. Left panel shows representative fields of GFP-positive cells. Original magnification, ×10. (C) LNCaP control, Slug-expressing, and Slug- and cyclin D1b–expressing cells (left) were plated in soft agar and cultured in androgen-proficient conditions for 4 weeks. Colonies greater than 75 μm were counted for colony formation (middle). Right panel shows representative colony growth for each cell line after 4 weeks. Original magnification, ×20 (inset). (D) A polyclonal population of cells expressing Slug was probed by immunofluorescence for DAPI (panel 1), Slug (panel 2) and E-cadherin (panel 3). Merged images are represented in panel 4. Original magnification, ×40. DAPI serves as a nuclear control. Error bars represent mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
To better define the role of Slug in anchorage-independent growth, cells with stable upregulation of Slug alone or Slug together with cyclin D1b were generated (Figure 5C) and assessed in soft agar assays. It was observed that Slug alone was sufficient to markedly increase colony formation (Figure 5C). Interestingly, coexpression with cyclin D1b enhanced colony formation (∼10%) over that of Slug alone, indicating that while Slug expression is sufficient to drive cyclin D1b–mediated phenotypes, cyclin D1b regulates additional networks that promote anchorage independent growth. Previous studies in other model systems suggested that the ability of Slug to directly suppress CDH1 (encoding E-cadherin) underlies the prometastatic phenotypes associated with Slug expression (39, 40). Surprisingly, induction of Slug was not sufficient to diminish extracellular levels of E-cadherin in this system (Figure 5D). Further investigation of relative CDH1 transcript levels in LN-vector (LN-vec), LN-SLUG polyclonal, and LN-SLUG (clonal) cell lines showed no significant changes in CDH1 expression (Supplemental Figure 4A), supporting the notion that Slug regulated phenotypes are independent of changes in E-cadherin levels or localization. Similarly, the expression of several other targets of Slug implicated in prometastatic phenotypes (derived from mouse models) (41–43) demonstrated an insensitivity to Slug expression (Supplemental Figure 4B). Collectively, these data implicate Slug as an obligate effector of cyclin D1b function and provide evidence of CDH1-independent Slug functions.
Slug expression enhances human tumor xenograft formation and proliferation in vivo.
Given the novel and unexpected effects of the cyclin D1b/Slug axis in vitro, the in vivo impact on tumor development and growth was assessed. Initially, luciferase-expressing isogenic cell models with and without Slug upregulation were injected subcutaneously into the hind limbs of nude mice and monitored for growth. Notably, Slug-expressing tumors developed more quickly than parental controls (Figure 6A), consistent with in vitro colony formation assays (Figure 5C). Tumor growth measurements revealed that Slug-expressing tumors began to exhibit a significant growth advantage 8 weeks after tumor formation (Figure 6B). Prior to sacrifice at 8 weeks (required due to tumor size), mice were injected with BrdU and proliferative indices quantified. Consistent with the observed growth advantage, tumors expressing Slug demonstrated a significantly enhanced proliferative rate as compared with isogenic controls (∼40% vs. ∼20% BrdU positive) (Figure 6C). No altered cellular morphology was observed by H&E analyses (Figure 6D). Importantly, Slug expression remained elevated in LN-SLUG tumors, whereas expression was variable and only weakly detected in control xenografts (Figure 6D). No change in AR levels was observed between the 2 xenografts, as monitored by both IHC and immunoblot (Figure 6D), further validating the maintenance of a luminal-like phenotype in tumors with high Slug expression. Similar to in vitro analyses, there was no detectable difference in the relative expression levels of cyclin D1a or E-cadherin in the 2 xenograft lines as determined by immunoblot (Supplemental Figure 5, A and B) and immunofluorescence (Supplemental Figure 5C), indicating that the growth advantage observed in LN-SLUG tumors occurs independently of changes in the expression of cyclin D1 or E-cadherin. These data reveal unanticipated functions of Slug in potentiating in vivo tumor development and tumor cell proliferation in AR-positive PCa cells.
Figure 6. Slug enhances growth of AR-positive PCa cells.

(A) Control or LN-SLUG cells were injected into the flanks of nude mice and the percentage of tumor-free mice is reported as a function of time. (B) Left: relative tumor growth of control or LN-SLUG cells is plotted as fold increase in volume relative to time of detectable tumor formation. Right: control and LN-SLUG tumors at 8 weeks after palpable tumor formation. (C) Left: Prior to sacrifice, animals were injected with BrdU for 24 hours. Tumor sections were stained for the presence of BrdU incorporation, and 3 random fields from each tumor were counted. BrdU incorporation is reported as percentage positive divided by total cell number. (D) Left: tumors were sectioned and stained for H&E, Slug, and AR expression. Right: total protein was isolated from tumors, and expression of AR and Slug is shown from 3 representative samples of each tumor type. Original magnification, ×20. Statistical analyses are representative of mean ± SEM. *P < 0.05; ***P < 0.001.
Slug enhances prometastatic phenotypes of PCa cells in vivo.
Previously, Slug has been implicated as a regulator of pathways known to be important for metastatic phenotypes, consistent with in vitro observations presented above. Recent findings implicating AR in the regulation of processes associated with metastasis heightened the importance of assessing Slug function in the context of AR-positive tumor cells (26). It is important to note that while the vast majority of metastatic human PCa retains AR, none of the AR-positive model systems reliably spontaneously metastasize in immunocompromised mice (44, 45). To test whether Slug expression was sufficient to promote metastatic phenotypes in vivo, cells were implanted in the upper chorioallantoic membrane (CAM) of a fertilized chick embryo and organs harvested and analyzed for the presence of human metastatic cells. Such assays have previously been demonstrated to accurately assess the metastatic potential of PCa cells in vivo (46). As shown, Slug-expressing cells metastasized to the lung with much higher frequency than control cells and also promoted liver metastasis (Figure 7A). Similar prometastatic phenotypes were recapitulated in murine models after tail-vein injection, where Slug enhanced the total number of cells localizing to both the lung and liver (Figure 7B), indicating Slug promotes prometastatic phenotypes in multiple models of metastatic progression. Since metastasis in the clinical setting is linked to the aggressive castration resistant stage (castration-resistant PCa [CRPC]) and is the major cause of patient mortality, the ability of Slug to promote aggressive prometastatic phenotypes in CRPC models was examined in vivo after cardiac injection (47). As such, fluorescently labeled isogenic CRPC models were generated with and without Slug upregulation (GFP-C4-2-SLUG or RFP-C4-2-vec) (Figure 7C), and injected into the left ventricles of 6-week-old male mice. While Slug expression did not affect arrival to the bone, examination of whole liver sections identified the presence of C4-2-SLUG but not C4-2-vec cells scattered throughout the liver (Figure 7C). Additionally, while few control cells were detected in the lungs, C4-2-SLUG cells homed to the lung with greater frequency, displaying an approximately 7-fold increase in cell number (Figure 7C), indicating that Slug affords CRPC cells an enhanced ability to home to distant tissues, likely attributed to increased adhesion to the endothelium of the capillary beds of soft-tissues (48, 49). Additional time points were also analyzed (0.5, 1.5, and 2 hours after injection) with comparable cell numbers seen at each time point for both cell lines in each organ (data not shown). Such data are of high clinical relevance, as liver metastases have been identified in patients with advanced metastatic PCa, which highly correlates with the extent of lung metastases (50). Additionally, while these liver tumors respond to chemotherapeutic intervention similarly to other metastatic PCa tumors, the overall survival of patients with liver metastases was significantly shorter than that of patients where such metastases were not apparent, indicating that metastatic liver tumors may represent a particularly aggressive and lethal subset of PCa (50). Collectively, these data suggest that Slug not only confers a proinvasive phenotype in vitro, but remarkably promotes prometastatic phenotypes in vivo and may represent a novel biomarker of aggressive soft tissue metastatic disease.
Figure 7. Slug enhances prometastatic phenotypes of PCa cells in vivo.

(A) Fertilized special pathogen–free eggs were incubated for 10 days at 38°C in a rotary humidified incubator. After 10 days of incubation, small holes were drilled over the air sac and near the allantoic vein. 2 × 106 cultured human prostate adenocarcinoma cells (stable LNCaP-vec and LNCaP-Slug cell lines) were implanted onto the membrane in each egg. After sealing the windows, the eggs were incubated in a stationary incubator for 7 days and the embryos were sacrificed after 17 total days of incubation. The embryonic livers and lungs were harvested and analyzed for the presence of tumor cells using quantitative human Alu-specific PCR. (B) LN-SLUG or control cells (150,000 cells/mouse) were injected via the tail vein, after which whole organs were harvested, sectioned completely through; total number of fluorescent cells are reported/organ (left) and representative images shown (right). (C) C42-SLUG-GFP or C42-vec-RFP cells were injected into the left ventricle of nude mice along with fluorescent beads (used as a marker of proper injection). One hour after injection, organs were harvested, whole tissues were sectioned through, and total number of cells counted under a fluorescent microscope. Cells homing to the lung and liver are shown (right and quantified left). Scale bars: 200 μm. Error bars represent mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001.
The cyclin D1b/Slug network is conserved in advanced cancers.
Previous reports have implicated Slug as a downstream target of both androgen signaling and TGF-β in hormone-dependent disease (39, 51–53), yet little is known about its regulation and function in CRPC. Data herein implicate cyclin D1b as a driver of Slug expression and aggressive phenotypes in both hormone-sensitive and CRPC models. As CRPC represents the lethal form of PCa (54), cyclin D1b and Slug expression were assessed in 118 samples of CRPC obtained from transurethral resection (representing 108 patients). Notably, a majority of the samples demonstrated intermediate-to-intense nuclear cyclin D1b staining (∼89%), with only a smaller fraction exhibiting low-to-no cyclin D1b (∼10%; Figure 8A). Previous studies analyzing cyclin D1b in primary, hormone-dependent disease reported that only approximately one-third of tumors expressed cyclin D1b, indicating that cyclin D1b expression is induced as a function of disease progression. While Slug staining was overall less extensive than that of cyclin D1b, similar trends were observed, whereby nearly 70% of samples demonstrated intermediate-to-high nuclear staining with, approximately 30% exhibiting low-to-no staining (Figure 8A). Correlation analyses were performed between the 2 matched data sets, which uncovered a strong, positive association between cyclin D1b and Slug expression (Spearman correlation, r = 0.61; P < 0.0001, r2 = 0.39l Figure 8B). Interestingly, while cyclin D1b alone was not indicative of Ki67 levels, high Slug levels did predict for heightened Ki67, mimicking the in vivo data presented previously (Figure 6C). Taken together, the data indicate that cyclin D1b is enriched in clinical samples of CRPC, whose expression highly correlates with Slug status. Collectively these data identify evidence of the convergence of the cyclin D1b–AR and Slug networks in human disease and implicate aberrant AR transcriptional programs in the progression of lethal tumor phenotypes.
Figure 8. The cyclin D1b–Slug network is conserved in advanced cancers.

(A) A panel of 108 CRPC cores were stained for cyclin D1b and Slug, and distribution of cyclin D1b and Slug staining as a function of intensity is plotted as a percentage of all samples (n = 118). Pathological determination of low-to-no staining was calculated as harboring an intensity score of 0–40, intermediate staining a score of 41–120, and high staining as greater than 120. (B) Left: representative staining of low (top) and high (bottom) cyclin D1b and Slug from matched samples of CRPC. Original magnification, ×5 (left); ×20 (right). (B) Right: correlation analyses plotted with Slug expression as a function of cyclin D1b intensity score was conducted using a 2-tailed Spearman’s correlation, and a line of best fit was generated using linear regression software. (C) Slug intensity scores were divided into the bottom and top 50% and matched Ki67 (percentage positive obtained from University of Tampere) scores are reported. Statistical analysis utilized Students 2-tailed t test to calculate differences between the 2 groups. *P < 0.05.
Discussion
The present study demonstrates cooperation between 2 major oncogenic pathways that results in acquisition of aggressive tumor phenotypes. Data shown identify AR-dependent upregulation of Slug as a major downstream consequence of cyclin D1b induction and provide robust evidence that the cyclin D1b/Slug axis confers proinvasive and prometastatic phenotypes in vitro and in vivo. Moreover, analyses of aggressive PCa confirmed the hypothesis that cyclin D1b and Slug expression are tightly linked and concomitantly induced in advanced disease. Together, these data establish a mechanism by which cyclin D1b functions to promote tumor growth and progression and identify the cyclin D1b/Slug axis as a target for development of therapeutic intervention.
The concept that cyclin D1b promotes Slug-dependent tumor phenotypes via AR regulation reveals an unexpected role for cyclin D1b function in hormone-dependent cancers as a context-specific cofactor. Findings herein describe what we believe to be a novel function of cyclin D1b as an AR coregulator, which facilitates increased AR association with the SNAI2 regulatory locus and, consonantly, upregulation of Slug expression. The finding that cyclin D1b exerts oncogenic phenotypes via transcriptional regulatory functions synergizes with recent findings associated with cyclin D1a, wherein unbiased proteomic analyses from several tissue types revealed the majority of the cyclin D1a interactome belongs to transcriptional family regulators (12); as such, it is tempting to speculate that cyclin D1b may induce tissue- and tumor-type–specific alterations in transcriptional regulatory programs that facilitate disease development and progression. Indeed, gene expression profiling revealed that cyclin D1b is a major modulator of gene expression and suggests that cyclin D1b likely hijacks the function of multiple transcription factors to promote disease phenotypes in a manner distinct from that of cyclin D1a. Since cyclin D1b also interacts with ERα in luminal breast cancer cells (13, 14) and can promote resistance to ERα-directed therapeutics (20), ongoing studies are focused on discerning the downstream impact on cellular outcomes and tumor behavior. Moreover, since cyclin D1b also likely interacts with multiple transcription factors relevant to cancer (i.e., STAT3, refs. 55, 56; PPARϒ, ref. 57; and TR, ref. 58), it is probable that cyclin D1b has an impact on the pathobiology of tumors other than those driven by nuclear receptor action. Combined, these observations are the first, to our knowledge, to establish the transcriptional modulatory functions of cyclin D1b as a major means by which the protein exerts oncogenic potential and highlight the importance of discerning the mechanisms regulating cyclin D1b–mediated transcriptional outcomes in human disease.
Identification of Slug as a key effector of cyclin D1b function provides a substantial advance in understanding aggressive cyclin D1b–induced tumor phenotypes and provides strong evidence linking 2 major oncogenic pathways. Slug is postulated to promote invasive phenotypes via transcriptional inhibition of the epithelial marker E-cadherin (CDH1) and indirect induction of mesenchymal markers (i.e., Vimentin and N-cadherin) (38, 39) in vitro. Whereas these previous findings suggest that Slug can promote changes associated with the epithelial-mesenchymal transition (EMT), no such alterations were observed in the context of PCa (in our model systems). Such results are not unexpected, as previous findings in multiple cancer lines suggest that Snail (SNAI1), a family member of Slug, is a much more potent repressor of E-cadherin expression and resultant inducer of EMT (59). Indeed, reporter assays utilizing tandem E-BOX elements of the E-cadherin promoter found that, while exogenous expression of Slug can repress the activity of isolated promoter elements, expression of the CDH1 gene itself remained unaltered, implicating Slug as a weak inducer of EMT-like signatures (60). Parallel studies in colon cancer describe weak inhibition of the vitamin D receptor expression by Slug alone, a phenotype that is reversed if Slug is coexpressed with family member Snail (61). Such results suggest that efficient regulation of EMT targets (i.e., E-cadherin) by Slug requires cooperation with other transcriptional repressors, such as Snail. Importantly, while Slug alone was unable to promote EMT-like signatures in these models, Slug expression did correlate with markers of de-differentiation, indicating independent Slug expression likely promotes aggressive cancer phenotypes independently of classic EMT signatures in these models (60). Similar results were observed with independent Slug induction in PCa models. First, despite marked upregulation of Slug as a consequence of cyclin D1b, unbiased gene expression analyses revealed no concomitant loss of E-cadherin. Similar results were observed when Slug was independently introduced, supporting the notion that Slug does not exert prometastatic phenotypes though a classical EMT process. Further underscoring this conclusion, induction of Slug in cells of prostatic origin showed no change of E-cadherin protein levels or localization (Figure 5 and Supplemental Figures 4 and 5), yet cells expressing Slug maintained an enhanced capacity for growth and organ homing in vivo. It will thus be crucial to identify Slug-regulated transcriptional networks in AR-positive PCa that promote prometastatic phenotypes and define those functions of Slug that are cyclin D1b dependent.
The finding that Slug expression is critical for AR-mediated metastatic phenotypes is of critical importance, as few downstream targets of the AR signaling axis that contribute to metastatic progression have been identified. Interestingly, induction of Slug has been shown to be requisite in multiple model systems of metastatic progression for homing and colonization to distant metastatic sites (40). Data herein are concordant with such conclusions and validate the role of Slug in the progression to metastatic PCa. However, as the progression to advanced metastatic disease involves many complex biological processes (62), it is likely that Slug-driven programs cooperate with other oncogenic pathways to promote distal metastatic growth. Indeed, numerous pathways, some independent of AR signaling, that enhance invasion and growth of cancer cells at metastatic sites have been identified. Most recently, ETS-TMPRSS2 fusions have been identified in an overwhelming majority of PCa patients and have been shown to promote metastatic growth in response to androgen (63). Additionally, in fusion-negative patients, SPINK1 has been identified as a marker of aggressive disease and is associated with enhanced invasive potential of cancer cells (64). Thus, it is postulated that Slug-driven networks work in concert with such pathways to promote both homing and growth at distal metastatic sites, and future efforts will focus on identifying these collaborative networks.
Finally, the findings herein provide needed insight into the mechanisms by which nuclear receptors facilitate acquisition of prometastatic phenotypes in hormone-dependent cancers. In PCa, progression to the lethal phase of disease (metastatic, CRPC) is known to be reliant on AR signaling (54, 65), and identification of AR as a key mediator of the cyclin D1b/Slug axis is likely of high translational and disease relevance. Consistent with this concept, the present study demonstrates a strong link between cyclin D1b and Slug in AR-positive, castration-resistant cancers. Additionally, genome-wide analyses of AR occupancy in multiple cancer models driven by AR suggest that AR occupancy at SNAI2 regulatory loci is preserved (Supplemental Figure 3C), indicating the link between cyclin D1b and Slug is likely present across multiple cancer types. Thus, while the current studies highlight AR function as a key mediator of cyclin D1b/Slug cooperation, it will be important to ascertain the tissue specificity of this process and to determine what oncogenic signals and pathways might regulate this functional interaction.
In summary, the present study identifies Slug as a critical mediator and effector of cyclin D1b–induced oncogenic activity and demonstrates that cyclin D1b elicits protumorigenic functions associated with lethal disease via transcriptional modulatory functions. These data highlight the convergence of the oncogenic cyclin D1b/Slug and AR networks to promote phenotypes implicated in metastatic progression, providing insight into the mechanisms of aberrant AR signaling pathways in CRPC. This unexpected pathway provides the basis for development of novel nodes of therapeutic intervention, so as to oppose development of metastatic disease.
Methods
Cell culture and treatment.
See Supplemental Methods.
Microarray data analysis.
Gene set enrichment analysis (GSEA) (66) was performed on ranked gene lists, ordered by their SAM standardized distance metric, to identify significant enrichment among Gene Ontology Biological Process terms. When ranking genes represented on the HG-U133 plus 2 microarray with multiple probe sets, only the probe set with the maximum absolute standardized distance metric was used. All data have been uploaded to the GEO database (GSE40794). Additional information regarding microarray processing, normalization, and analysis can be found in the supplemental material.
Generation of constructs.
See supplemental materials.
Gene expression, RNA interference, and morpholinos.
A complete list of primer sequences utilized in this study can be found in Supplemental Table 1. See also Supplemental Methods.
Chromatin tethering and immunoprecipitation.
Indicated cell lines were plated at a density of 1.4 × 106 cells in 10 cm poly-l-lysine–coated dishes in complete medium for 24 hours. Chromatin tethering was then performed as previously described (31, 67). Briefly, cells were washed in buffer A (150 mM NaCl, 20 mM HEPES [pH 7.8], and 10% glycerol in water) and then lysed in 200 μl buffer B (150 mM NaCl, 0.5 mM MgCl2, 20 mM HEPES [pH 7.8], 10% glycerol, 0.5% Triton-X 100, and 1 mM DTT in water). Lysates were centrifuged at 16,300 g for 20 minutes at 4°C to separate chromatin bound versus soluble complexes. Where indicated, immunoprecipitation of AR (sc-816; Santa Cruz Biotechnology Inc.) or 3× Flag-cyclin D1b (F3165; Sigma-Aldrich) was performed as previously described (19) from soluble and chromatin-tethered fractions using 100 μg of fractionated protein and 3 μg of antibody per IP.
Immunoblotting and immunohistochemistry.
A detailed list of all antibodies and conditions used within the manuscript can be found in Supplemental Methods.
Soft agar assay.
A total of 10,000 single cells (of indicated cell lines) were resuspended in 0.3% Bacto Agar (214050; BD) in 5% FBS- or charcoal dextran treated–containing (CDT-containing) medium. The cell/agar suspension was plated on top of a basement layer of 0.6% Bacto Agar (in 5% FBS- or CDT-containing medium) and allowed to solidify at room temperature. Medium was replaced every 5 days, and individual colonies were allowed to grow for 4 weeks. Plates were then stained with 0.1% crystal violet and colonies greater than 75 μm in diameter were counted.
Matrigel invasion chamber assays.
Indicated cell lines were cultured in complete medium for 24 hours after plating. A total of 20,000 cells were plated in the upper chamber of an invasion chamber (354483; BD Biosciences) in serum-free medium. A chemoattractant gradient was established in the lower chamber with 5% FBS- or CDT-containing medium, and cells were allowed to invade through the Matrigel-fibronectin matrix for 24 hours. Individual chambers were then washed 3× with 1× PBS, DAPI stained (Invitrogen D130), and total invading cell number was counted under a fluorescent microscope at ×20 magnification.
3C (chromosome conformation capture).
LN-Vec or LN-D1b cells were starved of hormone for 72 hours (5% CDT) and stimulated with 10 nM DHT or EtOH for 3 hours. Cells were fixed, restriction digested using the HindIII enzyme, and processed and analyzed using specific TaqMan primer/probe conditions, as previously described (36). All ligated PCR products were run on an agarose gel, and single bands were excised, purified, and sequenced to ensure appropriate product generation.
ChIP.
ChIP analyses for AR, Flag (F3165; Sigma-Aldrich), RNA polymerase II (AC-005-100; Diagenode), and acetylated histone H4 (06-866; Millipore) were preformed as previously described (32). Cells were treated as above except cells were supplemented with 10 nM DHT for 3 hours where indicated. Genomic DNA was purified and quantitative PCR performed for the indicated loci as previously described (68). Data were analyzed as a percentage of input of total samples calculated as previously described (69).
Xenografts.
Three million cells were injected subcutaneously in 6- to 8-week-old athymic nude male mice (01B70; NCI Frederick) as previously described (69). After injection, tumor volume was measured twice weekly using luminescence (100 mg/kg luciferin IP injected, visualized using an IVIS In Vivo imaging system) and electronic calipers. Where indicated, week 1 refers to a luminescence signal above background. All mice were sacrificed 8 weeks after tumor formation due to ethical concerns related to tumor volume.
CAM assay.
An in-depth description of the CAM assay is provided in Supplemental Methods.
Cardiac and tail-vein injections.
Cardiac injections of indicated cell lines were performed as previously described (47) with the following exceptions: AR-positive PCa cell lines were injected at a concentration of 2 × 105 in 100 μl of sterile 1× PBS along with fluorescent tracking beads (F8824; Invitrogen) diluted 1:100 (49). After injection, animals were sacrificed and whole tissues harvested. Tissues were processed and cryofrozen as described (47), and total GFP/RFP-positive cells were counted for each tissue type (n = at least 3 for each cell type and organ). Cell counts for each organ began approximately 320 μm into each tissue and ended approximately 240 μm before the end of the tissue (to account for positioning and sectioning bias). Total sections counted were similar between all groups and organs (lung, P = 0.354; liver, P = 0.411). GFP-labeled LN-vec or LN-SLUG cells were injected via the tail vein (150,000/mouse) and allowed to circulate for 1 hour. Organs were then harvested and cells counted as above.
Clinical samples and analysis.
CRPC TMAs were obtained from Tampere University Hospital from transurethral resection and contained 118 samples of CRPC as described (52). Individual TMAs were scanned using the Aperio scope AT system, and scoring was conducted using spectrum software and validated by a clinical pathologist. Staining intensity ranged from a score of 3 (highest intensity) to 0 (no staining), and intensity scores for each core were calculated using the following formula: Σ(staining intensity × nuclear percentage). Intensity scores were then plotted as a function of cyclin D1b staining vs. Slug staining, and Spearman’s correlation and linear regression analyses performed using Graph Pad Prism 4 software.
Statistics.
All results were analyzed using the 2-tailed Student’s t test (adjusted for variance) or Mann-Whitney test. For all analyses, P < 0.05 was deemed significant.
Study approval.
The use of clinical material was approved by the ethical committee of Tampere University Hospital and the National Authority for Medicolegal Affairs (Helsinki, Finland). Written informed consent was not obtained, as according to Finnish law, in cases in which informed consent cannot be obtained (e.g., large retrospective materials or patients who had died prior to the study), permission to use samples can be given by the National Authority for Medicolegal Affairs. All animal studies were conducted within the guidelines of animal protocols approved by IACUC.
Supplementary Material
Acknowledgments
This work was supported by NIH grants (R01 CA116777-05, R01 CA099996-09, and R01 ES016675-11 to K.E. Knudsen; K01 DK084205 to D.E. Frigo), a Commonwealth of Pennsylvania grant (SAP# 41000548782 to K.E. Knudsen), and a predoctoral fellowship from the Department of Defense (DOD) (PC094596 to M.A. Augello). We thank the members of the K.E. Knudsen lab for insightful discussions and critical reading of the manuscript, R. Schrecengost for technical assistance with the cardiac injections, the D. Merry lab for use of their equipment, W.K. Kelly for expert assistance and feedback on clinical topics, and E. Schade for graphical and technical support.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Note regarding evaluation of this manuscript: Manuscripts authored by scientists associated with Duke University, The University of North Carolina at Chapel Hill, Duke-NUS, and the Sanford-Burnham Medical Research Institute are handled not by members of the editorial board but rather by the science editors, who consult with selected external editors and reviewers.
Citation for this article: J Clin Invest. 2013;123(1):493–508. doi:10.1172/JCI64750.
References
- 1.Knudsen KE. The cyclin D1b splice variant: an old oncogene learns new tricks. Cell Div. 2006;1:15. doi: 10.1186/1747-1028-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer. 2011;11(8):558–572. doi: 10.1038/nrc3090. [DOI] [PubMed] [Google Scholar]
- 3.Lu F, Gladden AB, Diehl JA. An alternatively spliced cyclin D1 isoform, cyclin D1b, is a nuclear oncogene. Cancer Res. 2003;63(21):7056–7061. [PubMed] [Google Scholar]
- 4.Kim CJ, et al. Cyclin D1b variant promotes cell invasiveness independent of binding to CDK4 in human bladder cancer cells. Mol Carcinog. 2009;48(10):953–964. doi: 10.1002/mc.20547. [DOI] [PubMed] [Google Scholar]
- 5.Millar EK, et al. Cyclin D1b protein expression in breast cancer is independent of cyclin D1a and associated with poor disease outcome. Oncogene. 2009;28(15):1812–1820. doi: 10.1038/onc.2009.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Comstock CE, et al. Cyclin D1 splice variants: polymorphism, risk, and isoform-specific regulation in prostate cancer. Clin Cancer Res. 2009;15(17):5338–5349. doi: 10.1158/1078-0432.CCR-08-2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abramson VG, et al. Cyclin D1b in human breast carcinoma and coexpression with cyclin D1a is associated with poor outcome. Anticancer Res. 2010;30(4):1279–1285. [PMC free article] [PubMed] [Google Scholar]
- 8.Solomon DA, et al. Cyclin D1 splice variants. Differential effects on localization, RB phosphorylation, and cellular transformation. J Biol Chem. 2003;278(32):30339–30347. doi: 10.1074/jbc.M303969200. [DOI] [PubMed] [Google Scholar]
- 9.Betticher DC, Thatcher N, Altermatt HJ, Hoban P, Ryder WD, Heighway J. Alternate splicing produces a novel cyclin D1 transcript. Oncogene. 1995;11(5):1005–1011. [PubMed] [Google Scholar]
- 10.Olshavsky NA, et al. Identification of ASF/SF2 as a critical, allele-specific effector of the cyclin D1b oncogene. Cancer Res. 2010;70(10):3975–3984. doi: 10.1158/0008-5472.CAN-09-3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paronetto MP, et al. Alternative splicing of the cyclin D1 proto-oncogene is regulated by the RNA-binding protein Sam68. Cancer Res. 2010;70(1):229–239. doi: 10.1158/0008-5472.CAN-09-2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bienvenu F, et al. Transcriptional role of cyclin D1 in development revealed by a genetic-proteomic screen. Nature. 2010;463(7279):374–378. doi: 10.1038/nature08684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neuman E, et al. Cyclin D1 stimulation of estrogen receptor transcriptional activity independent of cdk4. Mol Cell Biol. 1997;17(9):5338–5347. doi: 10.1128/mcb.17.9.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zwijsen RM, Buckle RS, Hijmans EM, Loomans CJ, Bernards R. Ligand-independent recruitment of steroid receptor coactivators to estrogen receptor by cyclin D1. Genes Dev. 1998;12(22):3488–3498. doi: 10.1101/gad.12.22.3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burd CJ, Petre CE, Moghadam H, Wilson EM, Knudsen KE. Cyclin D1 binding to the androgen receptor (AR) NH2-terminal domain inhibits activation function 2 association and reveals dual roles for AR corepression. Mol Endocrinol. 2005;19(3):607–620. doi: 10.1210/me.2004-0266. [DOI] [PubMed] [Google Scholar]
- 16.Comstock CE, et al. Cyclin D1 is a selective modifier of androgen-dependent signaling and androgen receptor function. J Biol Chem. 2011;286(10):8117–8127. doi: 10.1074/jbc.M110.170720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Petre-Draviam CE, et al. A central domain of cyclin D1 mediates nuclear receptor corepressor activity. Oncogene. 2005;24(3):431–444. doi: 10.1038/sj.onc.1208200. [DOI] [PubMed] [Google Scholar]
- 18.Reutens AT, et al. Cyclin D1 binds the androgen receptor and regulates hormone-dependent signaling in a p300/CBP-associated factor (P/CAF)-dependent manner. Mol Endocrinol. 2001;15(5):797–811. doi: 10.1210/me.15.5.797. [DOI] [PubMed] [Google Scholar]
- 19.Burd CJ, et al. Cyclin D1b variant influences prostate cancer growth through aberrant androgen receptor regulation. Proc Natl Acad Sci U S A. 2006;103(7):2190–2195. doi: 10.1073/pnas.0506281103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Y, et al. Cyclin D1b is aberrantly regulated in response to therapeutic challenge and promotes resistance to estrogen antagonists. Cancer Res. 2008;68(14):5628–5638. doi: 10.1158/0008-5472.CAN-07-3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Petre-Draviam CE, Cook SL, Burd CJ, Marshall TW, Wetherill YB, Knudsen KE. Specificity of cyclin D1 for androgen receptor regulation. Cancer Res. 2003;63(16):4903–4913. [PubMed] [Google Scholar]
- 22.Knudsen KE, Cavenee WK, Arden KC. D-type cyclins complex with the androgen receptor and inhibit its transcriptional transactivation ability. Cancer Res. 1999;59(10):2297–2301. [PubMed] [Google Scholar]
- 23.Yang J, Zhang G, Chen J. CCND1 G870A polymorphism is associated with increased risk of colorectal cancer, especially for sporadic colorectal cancer and in Caucasians: A meta-analysis. Clin Res Hepatol Gastroenterol. 2012;36(2):169–177. doi: 10.1016/j.clinre.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 24.Xu Y, Chen SY, Ross KN, Balk SP. Androgens induce prostate cancer cell proliferation through mammalian target of rapamycin activation and post-transcriptional increases in cyclin D proteins. Cancer Res. 2006;66(15):7783–7792. doi: 10.1158/0008-5472.CAN-05-4472. [DOI] [PubMed] [Google Scholar]
- 25.Kumar AS, Naruszewicz I, Wang P, Leung-Hagesteijn C, Hannigan GE. ILKAP regulates ILK signaling and inhibits anchorage-independent growth. Oncogene. 2004;23(19):3454–3461. doi: 10.1038/sj.onc.1207473. [DOI] [PubMed] [Google Scholar]
- 26.Frigo DE, et al. Induction of Kruppel-like factor 5 expression by androgens results in increased CXCR4-dependent migration of prostate cancer cells in vitro. Mol Endocrinol. 2009;23(9):1385–1396. doi: 10.1210/me.2009-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alves CC, Carneiro F, Hoefler H, Becker KF. Role of the epithelial-mesenchymal transition regulator Slug in primary human cancers. Front Biosci. 2009;14:3035–3050. doi: 10.2741/3433. [DOI] [PubMed] [Google Scholar]
- 28.Yang HW, Menon LG, Black PM, Carroll RS, Johnson MD. SNAI2/Slug promotes growth and invasion in human gliomas. BMC Cancer. 2010;10:301. doi: 10.1186/1471-2407-10-301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang K, et al. Slug enhances invasion ability of pancreatic cancer cells through upregulation of matrix metalloproteinase-9 and actin cytoskeleton remodeling. Lab Invest. 2011;91(3):426–438. doi: 10.1038/labinvest.2010.201. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30.Burd CJ, Morey LM, Knudsen KE. Androgen receptor corepressors and prostate cancer. Endocr Relat Cancer. 2006;13(4):979–994. doi: 10.1677/erc.1.01115. [DOI] [PubMed] [Google Scholar]
- 31.Lake RJ, Geyko A, Hemashettar G, Zhao Y, Fan HY. UV-induced association of the CSB remodeling protein with chromatin requires ATP-dependent relief of N-terminal autorepression. Mol Cell. 2010;37(2):235–246. doi: 10.1016/j.molcel.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Q, et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell. 2009;138(2):245–256. doi: 10.1016/j.cell.2009.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wyce A, Bai Y, Nagpal S, Thompson CC. Research Resource: The androgen receptor modulates expression of genes with critical roles in muscle development and function. Mol Endocrinol. 2010;24(8):1665–1674. doi: 10.1210/me.2010-0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ni M, et al. Targeting androgen receptor in estrogen receptor-negative breast cancer. Cancer Cell. 2011;20(1):119–131. doi: 10.1016/j.ccr.2011.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Massie CE, et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011;30(13):2719–2733. doi: 10.1038/emboj.2011.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hagege H, et al. Quantitative analysis of chromosome conformation capture assays (3C-qPCR). Nat Protoc. 2007;2(7):1722–1733. doi: 10.1038/nprot.2007.243. [DOI] [PubMed] [Google Scholar]
- 37.Guo W, et al. Slug and sox9 cooperatively determine the mammary stem cell state. Cell. 2012;148(5):1015–1028. doi: 10.1016/j.cell.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shirley SH, Hudson LG, He J, Kusewitt DF. The skinny on Slug. Mol Carcinog. 2010;49(10):851–861. doi: 10.1002/mc.20674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cesi V, Casciati A, Sesti F, Tanno B, Calabretta B, Raschella G. TGFbeta-induced c-Myb affects the expression of EMT-associated genes and promotes invasion of ER+ breast cancer cells. Cell Cycle. 2011;10(23):4149–4161. doi: 10.4161/cc.10.23.18346. [DOI] [PubMed] [Google Scholar]
- 40.Tanno B, et al. Expression of Slug is regulated by c-Myb and is required for invasion and bone marrow homing of cancer cells of different origin. J Biol Chem. 2010;285(38):29434–29445. doi: 10.1074/jbc.M109.089045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bermejo-Rodriguez C, Perez-Caro M, Perez-Mancera PA, Sanchez-Beato M, Piris MA, Sanchez-Garcia I. Mouse cDNA microarray analysis uncovers Slug targets in mouse embryonic fibroblasts. Genomics. 2006;87(1):113–118. doi: 10.1016/j.ygeno.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 42.Perez-Caro M, Bermejo-Rodriguez C, Gonzalez-Herrero I, Sanchez-Beato M, Piris MA, Sanchez-Garcia I. Transcriptomal profiling of the cellular response to DNA damage mediated by Slug (Snai2). Br J Cancer. 2008;98(2):480–488. doi: 10.1038/sj.bjc.6604084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Uygur B, Wu WS. SLUG promotes prostate cancer cell migration and invasion via CXCR4/CXCL12 axis. Mol Cancer. 2011;10:139. doi: 10.1186/1476-4598-10-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sobel RE, Sadar MD. Cell lines used in prostate cancer research: a compendium of old and new lines--part 1. J Urol. 2005;173(2):342–359. doi: 10.1097/01.ju.0000141580.30910.57. [DOI] [PubMed] [Google Scholar]
- 45.Sobel RE, Sadar MD. Cell lines used in prostate cancer research: a compendium of old and new lines--part 2. J Urol. 2005;173:360–372. doi: 10.1097/01.ju.0000149989.01263.dc. [DOI] [PubMed] [Google Scholar]
- 46.Brenner JC, et al. Mechanistic rationale for inhibition of poly(ADP-ribose) polymerase in ETS gene fusion-positive prostate cancer. Cancer Cell. 2011;19(5):664–678. doi: 10.1016/j.ccr.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Russell MR, Liu Q, Lei H, Kazlauskas A, Fatatis A. The alpha-receptor for platelet-derived growth factor confers bone-metastatic potential to prostate cancer cells by ligand- and dimerization-independent mechanisms. Cancer Res. 2010;70(10):4195–4203. doi: 10.1158/0008-5472.CAN-09-4712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eckhardt BL, Francis PA, Parker BS, Anderson RL. Strategies for the discovery and development of therapies for metastatic breast cancer. Nat Rev Drug Discov. 2012;11(6):479–497. doi: 10.1038/nrd2372. [DOI] [PubMed] [Google Scholar]
- 49.Jamieson-Gladney WL, Zhang Y, Fong AM, Meucci O, Fatatis A. The chemokine receptor CX(3)CR1 is directly involved in the arrest of breast cancer cells to the skeleton. Breast Cancer Res. 2011;13(5):R91. doi: 10.1186/bcr3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kelly WK, et al. Liver metastases (LM) to predict for short overall survival (OS) in metastatic castration-resistant prostate cancer (mCRPC) patients (pts). J Clin Oncol. 2012;30 (suppl; abstr 4655). [Google Scholar]
- 51.Liu YN, et al. Critical and reciprocal regulation of KLF4 and SLUG in transforming growth factor β-initiated prostate cancer epithelial-mesenchymal transition. Mol Cell Biol. 2012;32(5):941–953. doi: 10.1128/MCB.06306-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Urbanucci A, et al. Overexpression of androgen receptor enhances the binding of the receptor to the chromatin in prostate cancer. Oncogene. 2012;31(17):2153–2163. doi: 10.1038/onc.2011.401. [DOI] [PubMed] [Google Scholar]
- 53.Chen M, Chen LM, Chai KX. Androgen regulation of prostasin gene expression is mediated by sterol-regulatory element-binding proteins and SLUG. Prostate. 2006;66(9):911–920. doi: 10.1002/pros.20325. [DOI] [PubMed] [Google Scholar]
- 54.Knudsen KE, Penning TM. Partners in crime: deregulation of AR activity and androgen synthesis in prostate cancer. Trends Endocrinol Metab. 2010;21(5):315–324. doi: 10.1016/j.tem.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bienvenu F, Barre B, Giraud S, Avril S, Coqueret O. Transcriptional regulation by a DNA-associated form of cyclin D1. Mol Biol Cell. 2005;16(4):1850–1858. doi: 10.1091/mbc.E04-08-0654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bienvenu F, Gascan H, Coqueret O. Cyclin D1 represses STAT3 activation through a Cdk4-independent mechanism. J Biol Chem. 2001;276(20):16840–16847. doi: 10.1074/jbc.M100795200. [DOI] [PubMed] [Google Scholar]
- 57.Wang C, et al. Cyclin D1 repression of peroxisome proliferator-activated receptor gamma expression and transactivation. Mol Cell Biol. 2003;23(17):6159–6173. doi: 10.1128/MCB.23.17.6159-6173.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lin HM, Zhao L, Cheng SY. Cyclin D1 is a ligand-independent co-repressor for thyroid hormone receptors. J Biol Chem. 2002;277(32):28733–28741. doi: 10.1074/jbc.M203380200. [DOI] [PubMed] [Google Scholar]
- 59.Cano A, et al. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2(2):76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- 60.Come C, et al. Snail and slug play distinct roles during breast carcinoma progression. Clin Cancer Res. 2006;12(18):5395–5402. doi: 10.1158/1078-0432.CCR-06-0478. [DOI] [PubMed] [Google Scholar]
- 61.Larriba MJ, et al. Snail2 cooperates with Snail1 in the repression of vitamin D receptor in colon cancer. Carcinogenesis. 2009;30(8):1459–1468. doi: 10.1093/carcin/bgp140. [DOI] [PubMed] [Google Scholar]
- 62.Nguyen DX, Bos PD, Massague J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer. 2009;9(4):274–284. doi: 10.1038/nrc2622. [DOI] [PubMed] [Google Scholar]
- 63.Tomlins SA, et al. Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia. 2008;10(2):177–188. doi: 10.1593/neo.07822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tomlins SA, et al. The role of SPINK1 in ETS rearrangement-negative prostate cancers. Cancer Cell. 2008;13(6):519–528. doi: 10.1016/j.ccr.2008.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Knudsen KE, Scher HI. Starving the addiction: new opportunities for durable suppression of AR signaling in prostate cancer. Clin Cancer Res. 2009;15(15):4792–4798. doi: 10.1158/1078-0432.CCR-08-2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bourgo RJ, et al. RB restricts DNA damage-initiated tumorigenesis through an LXCXE-dependent mechanism of transcriptional control. Mol Cell. 2011;43(4):663–672. doi: 10.1016/j.molcel.2011.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Comstock CE, et al. Cyclin D1 is a selective modifier of androgen-dependent signaling and androgen receptor function. J Biol Chem. 2011;286(10):8117–8127. doi: 10.1074/jbc.M110.170720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sharma A, et al. The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. J Clin Invest. 2010;120(12):4478–4492. doi: 10.1172/JCI44239. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.