

Abstract
N-formyl peptide receptors (FPRs) are critical regulators of host defense in phagocytes and are also expressed in epithelia. FPR signaling and function have been extensively studied in phagocytes, yet their functional biology in epithelia is poorly understood. We describe a novel intestinal epithelial FPR signaling pathway that is activated by an endogenous FPR ligand, annexin A1 (ANXA1), and its cleavage product Ac2-26, which mediate activation of ROS by an epithelial NADPH oxidase, NOX1. We show that epithelial cell migration was regulated by this signaling cascade through oxidative inactivation of the regulatory phosphatases PTEN and PTP-PEST, with consequent activation of focal adhesion kinase (FAK) and paxillin. In vivo studies using intestinal epithelial specific Nox1–/–IEC and AnxA1–/– mice demonstrated defects in intestinal mucosal wound repair, while systemic administration of ANXA1 promoted wound recovery in a NOX1-dependent fashion. Additionally, increased ANXA1 expression was observed in the intestinal epithelium and infiltrating leukocytes in the mucosa of ulcerative colitis patients compared with normal intestinal mucosa. Our findings delineate a novel epithelial FPR1/NOX1-dependent redox signaling pathway that promotes mucosal wound repair.
Introduction
The intestinal epithelium forms a selective barrier that separates luminal contents from underlying tissue compartments, thereby playing a pivotal role in regulating exposure of the mucosal immune system to luminal microorganisms and their products. Pathologic states associated with epithelial injury and ulceration result in compromised integrity of the epithelial barrier. Repair of epithelial discontinuities and ultimate restoration, termed restitution, involve induced and coordinated migration of epithelial cells (1). Restitution is stimulated by a cascade of pro-resolving mediators in the inflamed mucosa (2–8). An important pro-resolving mediator is the calcium-dependent phospholipid-binding protein annexin A1 (ANXA1). Previous studies have reported that ANXA1 induced in endotoxemia, peritonitis, and arthritis exerts anti-inflammatory properties by suppression of leukocyte activation and transmigration (9–12). Such anti-inflammatory properties of ANXA1 are mediated by autocrine and paracrine signaling of a 26-amino-acid ANXA1 N-terminus peptide, Ac2-26, that is released from the full-length protein through regulated proteolysis (2, 13–16).
The biological effects of ANXA1 and its cleavage product Ac2-26 peptide are mediated by formyl peptide receptors (FPRs) (2, 17, 18). In humans 3 FPRs (FPR1, FPR2, and FPR3) regulate innate inflammatory responses. Their function has been most extensively investigated in the context of leukocyte recruitment and activation, where FPRs promote cell motility (chemotaxis) and microbicidal respiratory burst (19, 20). Neutrophil FPR activation with the bacterial peptide fMLF (formyl-methionyl-leucyl phenylalanine) induces Rac2-dependent NADPH oxidase–2 (NOX2) activation and microbicidal levels of ROS generation, which in turn mediate host defense (21). Additionally, ROS generated by non-phagocyte NOX1 in epithelial cells mediates important signaling events, and its levels are up to 2 orders of magnitude lower than during phagocyte microbicidal burst (22). While leukocyte FPR function has been well studied, it was only recently appreciated that FPRs are also expressed in the intestinal epithelium, where NOX1 function has remained enigmatic.
In this report we use complementary in vitro and in vivo approaches to delineate a novel signaling pathway by which ANXA1 promotes intestinal epithelial migration through activation of FPR1-, Rac1-, and NOX1-dependent redox signaling, leading to the oxidative inactivation of regulatory phosphatases, with subsequent modification of focal adhesion proteins involved in regulating cell migration. Furthermore, we demonstrate a therapeutic effect of ANXA1 mediated by NOX1 in promoting intestinal epithelial cell (IEC) wound closure in vivo. These findings elucidate a novel pro-resolution function of ANXA1 in mucosal wound repair.
Results
ANXA1 promotes IEC wound closure and cell-matrix adhesion.
We have previously reported that the endogenous FPR ligand ANXA1 promotes epithelial cell motility and wound closure, but the underlying mechanism remains poorly understood (23). During inflammation and epithelial wound healing, ANXA1 is cleaved, generating an active peptide, Ac2-26. While the full-length ANXA1 protein binds leukocyte FPR2, its cleavage peptide activates FPR1 at a physiological concentration (3–10 μM) as used in our previous studies (24, 25). Using a model intestinal epithelial cell line, SK-CO15, and a well-characterized in vitro scratch wound assay, we observed that exposure to the ANXA1 mimetic peptide Ac2-26 (3 μM) enhanced wound closure in IECs compared with control cells (68.72% ± 1.03% vs. 35.79% ± 0.78% at 24 hours, P < 0.0001) (Figure 1, A and B). The specificity of Ac2-26–induced effects on wound closure was confirmed by the FPR1 receptor antagonist cyclosporin H (CsH) (26), which blocked the pro-migratory effects of Ac2-26. Similarly, the pan-FPR antagonist Boc2 (27) reversed the effects of Ac2-26 on wound closure (Supplemental Figure 2A; supplemental material available online with this article; doi: 10.1172/JCI65831DS1). Boc2 is selective for FPR1 at a low concentration (1 μM), but this selectivity is lost when the concentration is higher. However, the peptide WRW4, known to antagonize FPR2 and FPR3, did not influence Ac2-26–mediated wound closure (28). These findings suggest that the pro-restitution effects of Ac2-26 are not likely mediated by FPR2 and FPR3. To confirm the inhibitory effects of WRW4 on FPR2 and FPR3 in IECs, we incubated cells with selective FPR2 and FPR3 agonists, MMK1 (29) and F2L (30, 31), respectively, and examined their influence on wound closure. As shown in Supplemental Figure 1, WRW4 inhibited the effects of MMK1 and F2L on wound closure. These data support a role of FPR1 in mediating Ac2-26 effects on IEC wound closure. To determine the relevance of the in vitro results, we assessed intestinal epithelial wound recovery in Anxa1-null (AnxA1–/–) mice. We determined the rate of colonic wound closure in AnxA1–/– and control mice by generating defined mechanical colonic mucosal wounds using a mouse colonoscopy equipped with biopsy forceps. Images captured from the wound sites were used to quantify wound restitution at days 2 and 4 after injury. Representative images of wounds in control mice (BALB/c) and AnxA1–/– are shown in Figure 1C. Mice lacking ANXA1 demonstrated a significant delay in wound healing at day 4 as indicated by 29.8% ± 0.9% wound closure compared with 75.3% ± 1.57% in control normal BALB/c mice (P = 0.023). Collectively, these in vitro and in vivo results are consistent with a major function of ANXA1 in promoting IEC wound closure. Regulated turnover of cell matrix adhesions plays an important role in promoting cell migration. Thus, to investigate the mechanisms by which ANXA1 promotes cell motility, we analyzed the influence of Ac2-26 on IEC-matrix adhesion. SK-CO15 adhesion assays were performed using surfaces coated with ECL Cell Attachment Matrix (Millipore). As shown in Figure1D, 15 minutes’ exposure to Ac2-26 increased the number of adherent cells compared with control cells (38.6 ± 0.3 vs. 17.5 ± 0.2, P < 0.0001). We next explored potential signaling pathways to identify the mechanism by which Ac2-26 enhances IEC-matrix adhesion and cell migration. Tyrosine phosphorylation and activation of focal adhesion proteins paxillin (Pax) and focal adhesion kinase (FAK) regulate cell matrix adhesion turnover and cell migration (32, 33). Thus, we analyzed the tyrosine phosphorylation of FAK and Pax by immunoblotting and confocal microscopy. Indeed, immunoblot analysis demonstrated that migrating cells incubated with Ac2-26 had increased tyrosine phosphorylation of Pax (Y118, 2.4-fold) and FAK (Y861, 2.2-fold) (Figure 1E). These findings were corroborated by confocal microscopy. As shown in Figure 1F, increased Pax p-Y118 and FAK p-Y861 were observed in basal “punctate” structures corresponding to focal adhesions in migrating IECs exposed to Ac2-26, as compared with control IECs. The FAK p-Y861 labeling was more prominent at the leading edge of migrating IECs. These results are consistent with increased cell-matrix adhesion/turnover and consequent IEC motility in cells exposed to Ac2-26.
Figure 1. ANXA1 regulates wound healing via FPR1 signaling in IECs and stimulates phosphorylation of focal adhesion proteins (FAK and Pax).

(A) SK-CO15 monolayers were subjected to scratch wound assay in the presence of Ac2-26 peptide (3 μM). Wound widths were determined at 0 and 24 hours. Photomicrograph shows representative results for control (Ctl) and Ac2-26–treated cells. Scale bar: 200 μm. (B) SK-CO15 cells were also incubated with Ac2-26 and CsH (1 μM) or WRW4 (10 μM). The ANXA1 cleavage product Ac2-26 alone significantly enhanced wound closure (*P < 0.0001). The increase in wound closure was inhibited in the presence of CsH (#P < 0.0001) but not WRW4. The experiment was repeated 3 times, and results of 1 representative experiment done with 5 parallel samples are shown. (C) Endoscopic images of colonic mucosal wounds in mice (AnxA1–/– and control BALB/c) at days 2 and 4 after injury. Quantification of wound repair is shown in the graph (mean ± SEM, *P = 0.023, n = 11 mice/group). (D) SK-CO15 cells were plated on ECL gel, and non-adherent cells were removed by washing at 1 hour after plating. Adhesion increased within 15 minutes of Ac2-26 exposure compared with non-stimulated cells (*P < 0.0001). (E) Immunoblot of SK-CO15 cells revealed a significant increase in Pax phosphorylation (Y118, 2.4-fold) and FAK phosphorylation (Y861, 2.2-fold) in cells stimulated with Ac2-26 for 15 minutes compared with unstimulated cells (0.1% DMSO). Normalized signal intensity is indicated above the blots. (F) Laser confocal micrographs of Pax p-Y118 (white) or FAK p-Y861 (white) and F-actin (red) in migrating SK-CO15 cells with or without Ac2-26 (3 μM) for 15 minutes. Photomicrographs are representative of 3 independent experiments done in triplicate. Scale bars: 10 μm.
Nox1 is a major determinant of Ac2-26/FPR1–induced ROS in IECs.
Recently, we reported that IEC migration stimulated by enteric commensal microbiota is linked to induction of ROS (34). Since FPR activation in phagocytes induces ROS, via activation of the NOX2 NADPH oxidase (21), we investigated whether Ac2-26 induces intracellular ROS generation in epithelial cells. IECs express NOX1 NADPH oxidase, a ROS-generating enzyme and paralog of leukocyte NOX2 (35). We first confirmed NOX1 expression in cells of the model IEC line SK-CO15 by RT-PCR (Supplemental Figure 4C). IEC exposure to Ac2-26 induced a rapid increase in fluorescence of a stable intracellular redox-sensitive dye, Hydro-Cy3, within 15 minutes, and this effect was inhibited with the FPR1 antagonist CsH (Figure 2A). Importantly, Ac2-26–induced ROS generation was abrogated in cells pretreated with pertussis toxin (PTx; Supplemental Figure 3), a known Gi/Go protein inhibitor (36). Pretreatment of stimulated IECs with a general NADPH oxidase inhibitor, diphenyleneiodonium chloride (DPI), and the ROS scavenger N-acetyl cysteine (NAC), but not the mitochondrial electron transport inhibitor rotenone, abrogated Ac2-26–induced ROS generation (Figure 2A). Interestingly, ROS generation was most prominent at the wound edge, and this effect was accentuated by 15 minutes exposure to Ac2-26 (Figure 2B), consistent with an analogous increase in active focal adhesion proteins in the vicinity of wounds (Figure 1F). Furthermore, preincubation with DPI inhibited wound healing in IECs treated with Ac2-26 (Figure 2C). Together, these data are consistent with a role of NADPH oxidase in mediating ROS generation and wound closure, downstream of FPR1 activation by Ac2-26.
Figure 2. Ac2-26 peptide induces rapid ROS generation in IECs via FPR1 and NOX1.

(A) SK-CO15 cells were incubated with Ac2-26 peptide for 15 minutes, and ROS generation was detected by confocal microscopy using the fluorescent hydro-Cy3 dye. Ac2-26 treatment increased fluorescence intensity, which was inhibited by CsH, DPI, and NAC, but not by rotenone. Summarized data for hydro-Cy3 fluorescence intensity are presented in the graph (mean ± SEM,*P < 0.05 vs. control, #P < 0.05 vs. Ac2-26, n = 3). (B) ROS generation in scratch-wounded SK-CO15 monolayer was increased in cells adjoining the wound. Confocal micrographs are representative of 3 independent experiments (C) SK-CO15 monolayers were subjected to scratch wound assay in the presence of Ac2-26 (3 μM) with or without DPI. Wound widths were determined at 0 and 24 hours (mean ± SEM, *P < 0.05 vs. Ctl, #P < 0.05 vs. Ac2-26). (D) Whole mount preparations of colon taken from WT (D), Nox1–/–IEC (E), and Nox2–/– (F) mice injected with hydro-Cy3 (i.p., 30 minutes) were mechanically wounded ex vivo. Inset in D highlights ROS generation at the leading edge of migrating cells. Mucosa was luminally treated for 15 minutes with vehicle (Ctl) or Ac2-26 with or without preincubation of the tissue samples with CsH. Confocal micrographs are representative of 3 independent experiments (5 mice/group). Scale bars: 40 μm.
For further determination of the relevance of these in vitro results, mice were pretreated with hydro-Cy3, mechanical wounds were generated in exteriorized colonic mucosa, and the hydro-Cy3 fluorescence signal was detected by confocal microscopy. Of interest was the observation that a 15-minute exposure of IECs to the Ac2-26 peptide enhanced ROS generation in wounded colonic mucosa from WT mice (Figure 2D). Furthermore, increased ROS was largely abrogated by the FPR1 inhibitor CsH, further supporting a role of FPR1 in mediating this cellular response. Interestingly, compared with the striking ROS generation in control mice, colonic epithelial cells in IEC-specific Nox1-null (Nox1–/–IEC) mice revealed minimal ROS generation that was not increased following treatment with Ac2-26 (Figure 2E). These Nox1–/–IEC mice were produced as shown in Supplemental Figure 4, A and B. To show that the oxidant production was specific to NOX1, we examined phagocytic ROS generation in wounded epithelium by testing Nox2–/– mice. As shown in Figure 2F, ROS generation was detected in colonic epithelial cells adjoining wounds of Nox2–/– mice to a level comparable to that in WT mice. These results highlight a novel role of intestinal epithelial NOX1 in mediating ROS generation in response to Ac2-26.
Ac2-26 promotes oxidative inactivation of PTP-PEST and PTEN.
Intracellular ROS signaling modulates a subset of regulatory proteins by rapid and reversible oxidation of redox-reactive cysteine residues that results in their catalytic inactivation (Figure 3A and refs. 37–40). Such regulatory proteins include phosphatases that modulate the phosphorylation status of focal adhesion proteins such as FAK and Pax, important in controlling cell movement (41). Thus, to determine whether Ac2-26–induced ROS could influence IEC motility and therefore wound closure by a redox-dependent mechanism, we analyzed the oxidation of cysteine residues in candidate relevant phosphatases such as PTP-PEST, PTEN, and SHP2. ROS-induced oxidation of cysteine residues was determined using the thiol-reactive agent biotin-N-maleimide (biotin-NM), which irreversibly alkylates thiol-anionic -SH groups of active redox-sensitive cysteines. Capture of associated phosphatases by streptavidin precipitation and immunoblotting thus reflects the amount of active protein. As shown in Figure 3B, exposure of migrating cells to Ac2-26 (3 μM) for 15 minutes oxidized reactive cysteines in PTP-PEST and PTEN but not SHP2. H2O2 (15 μM) was used as a positive control to demonstrate global nonspecific cysteine oxidation and, as expected, induced loss of maleimide modification of these phosphatases. These observations support a role for ROS-induced oxidative inactivation of PTP-PEST and PTEN that, in turn, increases phosphorylation and activation of downstream focal adhesion proteins, promoting IEC migration (42, 43).
Figure 3. Ac2-26 oxidizes PTP-PEST and PTEN and activates Rac1-GTPases.

(A) ROS oxidize thiols in redox-sensitive phosphatases (PTPs). Maleimide groups react efficiently and specifically with reduced sulfhydryls (R-S–) of PTPs to form stable thioester bonds but do not react and bind R-SOH oxidized sulfenic acid (thioester) forms. (B) Cell were treated with vehicle, H2O2 (15 μM), or Ac2-26 (3 μM) for 15 minutes and subjected to labeling with biotin-NM. Biotinylated proteins were precipitated with SA beads and subjected to immunoblot analysis using PTEN, PTP-PEST, and SHP2 antibodies. β-Actin was used as a loading control. Immunoblots presented are representative of 3 independent experiments. Normalized signal intensity is indicated above the blots. (C) SK-CO15 cells were incubated for 30 minutes at room temperature with 100 μM GTPγS, vehicle, or Ac2-26 peptide for 15 minutes. Active Rac1 (Rac1-GTP) was pulled down using agarose bead–conjugated GST-PAK-RBD. Total (Rac1) and active Rac1 (Rac1-GTP) were analyzed by immunoblot. Immunoblots are representative of 3 independent experiments. (D) Active Rac1 pull-down samples were subjected to immunoblot analysis with antibodies against Vav2, p120, and NOX1. Immunoblots are representative of 3 independent experiments. (E) ROS generation was analyzed in SK-CO15 cells, treated with hydro-Cy3 dye, by confocal microscopy. Cells were incubated with vehicle, Ac2-26 (3 μM), or Ac2-26 plus NSC23766. Micrographs are representative of 3 independent experiments. Scale bar: 20 μm. (F) SK-CO15 cells were transfected with siRNA targeting p120 catenin. Three days after siRNA transfection, cells were treated with Ac2-26 for 15 minutes, and ROS generation was detected. Confocal micrographs are representative of 4 independent experiments. Scale bar: 20 μm. Summarized data for hydro-Cy3 fluorescence intensity are presented in the graphs (mean ± SEM,*P < 0.05 vs control, #P < 0.05 vs. Ac2-26, n = 3).
Ac2-26–induced FPR1 stimulation promotes formation of a Rac1 signaling complex linked to ROS generation.
We next sought to determine the signaling events that regulate ROS production downstream of Ac2-26. Given the relationship of the GTPase Rac and NOX2/ROS in leukocytes, we next examined Ac2-26 promotion of ROS generation by activation of Rac1 GTPase in epithelial cells. Ac2-26–induced activation of epithelial Rac1 was followed by measurement of the specific interaction of Rac1-GTP with its effector protein p21-activated kinase (PAK) (44, 45). Ac2-26 induced a robust and rapid activation of Rac1 within 15 minutes of peptide exposure (3-fold increase, Figure 3C) that was inhibited by the FPR1 antagonist CsH (Figure 3D). Analogous inhibition of Rac1 activity was confirmed with another classical FPR inhibitory peptide, Boc2 (Supplemental Figure 2B).
Activation of Rac1 is catalyzed by guanine nucleotide exchange factors (GEFs) (46). We therefore determined the influence of Ac2-26 in promoting association of key candidate epithelial Rac-specific GEFs (Tiam1 and Vav2) and, as negative control, the Cdc42-specific GEF Tuba with active Rac1. Ac2-26 increased the CsH-sensitive association of epithelial Vav2 with the active Rac1 complex (Figure 3D). However, Tiam1 and Tuba GEFs were not identified in the complex with active Rac1 (data not shown). Interestingly, a previous study reported an association of Vav2 with p120 catenin and regulation of Rho GTPases and F-actin cytoskeletal restructuring (47). Thus, to investigate a possible link between Ac2-26–induced epithelial FPR1 signaling and Rac1 activation, we determined whether p120 associated with active Rac1. Indeed, Ac2-26 mediated p120 catenin association with active Rac1 (Figure 3D). Since Rac is an essential regulator of a multi-component NOX1 (48), we determined whether Ac2-26 stimulation also promoted association of epithelial NOX1 with active Rac1. Indeed, Ac2-26 exposure rapidly induced the association of NOX1 with active Rac1 (Figure 3D). Such protein complex formation was decreased by preexposure of cells to CsH. To further confirm a role of Rac1 signaling in mediating Ac2-26–induced ROS, we evaluated ROS generation in IECs incubated with NSC23766, which inhibits Rac1 activity (49). As shown in Figure 3E, confocal microscopy revealed inhibition of ROS generation following NSC23766 treatment. Since we had observed that Ac2-26 promoted p120 catenin association with the active Rac1 complex in a FPR1-dependent manner, we determined whether downregulation of p120 catenin might influence ROS generation in response to Ac2-26. Indeed, ROS generation in response to Ac2-26 was inhibited in cells with siRNA-induced downregulation of p120 catenin (Figure 3F). Successful depletion of p120 after 3 days was confirmed by immunoblotting (Supplemental Figure 5). Taken together, these results suggest that Ac2-26–induced ROS generation in IECs is mediated by a Rac1 signaling complex containing Vav2, NOX1, and p120 catenin.
Ac2-26 enhances Src kinase activity.
Since we observed that p120 mediates Ac2-26–induced ROS generation, further experiments were performed to identify mechanisms of p120 activation and ROS generation. Previous reports demonstrated that p120 was first identified as an Src substrate (50). Additionally, Src has been reported to increase NOX1-dependent ROS generation (51). We therefore investigated whether Src activation mediates Ac2-26–induced ROS generation by analyzing Src phosphorylation at the Y418 residue (52). Indeed, immunoblot analysis revealed a significant increase in Ac2-26–induced Src phosphorylation (8-fold increase compared with unstimulated cells). This effect was abrogated by CsH (Figure 4A), while the total Src protein remained unchanged. To test the functional relevance of Src activation in IEC ROS generation following Ac2-26 treatment, we incubated IECs with Ac2-26 in the presence of pharmacologic Src inhibitors (PP2 and AZM475271) (53–55). As shown in Figure 4B, a significant decrease in ROS generation was observed following treatment of cells with the Src inhibitors. These findings suggest a novel role for Src kinase in mediating FPR1-induced ROS generation following stimulation of epithelial cells with Ac2-26.
Figure 4. Ac2-26 peptide activates Src kinase.

(A) Immunoblots of phospho-Src (Src p-Y418) and total Src (c-Src) expression in SK-CO15 cells stimulated with control (0.1% DMSO) and Ac2-26 (3 μM) for 15 minutes. Immunoblots are representative of 3 independent experiments. Normalized signal intensity is indicated above the blots. (B) Confocal images of ROS-sensitive hydro-Cy3 in SK-CO15 cells treated with vehicle, Ac2-26 (3 μM), or Ac2-26 (3 μM) after preincubation with PP2 and AZM. Confocal micrographs are representative of 4 independent experiments. Scale bar: 20 μm. Summarized data for hydro-Cy3 fluorescence intensity are presented in the graph (mean ± SEM,*P < 0.05 vs. control, #P < 0.05 vs. Ac2-26 n = 4).
In vivo administration of ANXA1 promotes recovery of colonic mucosal wounds.
Since our in vitro data suggested that ANXA1 facilitates epithelial wound closure, thereby serving as a pro-resolution factor, we analyzed the influence of ANXA1 administration on colonic mucosal wound healing in vivo using recombinant ANXA1 protein. After generating mucosal wounds with mouse colonoscopy, we investigated wound closure following ANXA1 i.p. systemic administration versus PBS delivery (control) in WT and Nox1–/–IEC mice. Consistent with the in vitro wound closure results, ANXA1 administration promoted mucosal wound repair (64.57% wound closure in ANXA1-treated mice vs. 40.69% wound closure in WT control mice, P < 0.001). Interestingly, ANXA1 did not augment wound healing in the absence of NOX1, and recovery in NOX1-deficient mice treated with PBS was indistinguishably from that in WT mice (Figure 5A). To further confirm our results, we additionally analyzed ANXA1 pro-resolution activity in mice with DSS-induced colonic mucosal injury. Analysis of disease activity index revealed that ANXA1 administration (5 μg, twice per day) improved clinical recovery from colitis (P < 0.001, Figure 5B). Interestingly, ANXA1 administration was associated with reduced mucosal ulceration (Figure 5C and Supplemental Figure 5B). Even a lower dose of ANXA1 (1 μg, twice per day) afforded significant mucosal healing in WT mice (Supplemental Figure 6A). To further analyze the role of NOX1 in mediating the pro-resolution activity of ANXA1 during mucosal repair following colitis, we examined the effect of ANXA1 administration in Nox1–/–IEC mice. Figure 5, B and C, show that ANXA1 administration did not improve the clinical outcome of mice recovering from DSS-induced colitis in the absence of epithelial NOX1 enzyme. Since ANXA1 promoted colonic mucosal wound recovery in vivo, we analyzed ANXA1 expression in the vicinity of mucosal wounds. Mouse colonic wounds recovering from biopsy-induced injury were harvested and ANXA1 immunolocalized by confocal microscopy (Figure 6A, green). At the time of injury (day 0), baseline ANXA1 expression was observed in the intestinal epithelium and in scattered mucosal inflammatory cells. However, the expression of ANXA1 in the epithelium and in infiltrating leukocytes during wound recovery was markedly increased (days 1 and 6 after injury). Given these observations in mice, we assessed ANXA1 expression in human intestinal mucosa of patients with ulcerative colitis (UC). As shown in Figure 6B, ANXA1 expression increased in the intestinal epithelium and infiltrating leukocytes in the injured mucosa of individuals with UC compared with normal intestinal mucosa. These findings are consistent with a recent study that reported increased ANXA1 receptor FPR2 expression in IBD patients (56).
Figure 5. ANXA1 accelerates in vivo intestinal mucosal wound healing and recovery via NOX1.

(A) Endoscopic images of healing colonic mucosal wounds 2 or 4 days after biopsy-induced injury in WT and Nox1–/–IEC mice treated with i.p. injections of PBS or ANXA1 (5 μg, twice/day). Quantification of wound repair is shown in the graph. ANXA1 administration promoted recovery of wounds in WT mice, but in the absence of NOX1 resulted in a significant delay in wound healing. (B) Clinical disease activity index (DAI) of mice subjected to DSS colitis for 7 days, followed by recovery from colitis for 6 days. ANXA1 was administered i.p. (5 μg, twice/day) during recovery. Significantly decreased DAI was observed in WT mice treated with ANXA1 (red line) compared with control mice treated with PBS (black line). However, ANXA1 failed to enhance recovery in Nox1–/–IEC mice (blue line). (C) Representative photomicrographs of H&E-stained histological sections. Boxed areas are magnified in insets. The total magnification of the photomicrographs is ×2 and in the insets it is ×40. Data in all graphs are presented as mean ± SEM; *P < 0.05 and **P < 0.001, WT ANXA1 vs. WT PBS, n = 12 mice/group; #P < 0.05 and ##P < 0.01, Nox1–/–IEC ANXA1 vs. Nox1–/–IEC PBS, n = 12 mice/group.
Figure 6. Mucosal ANXA1 expression is increased in injured murine and human colonic tissue.
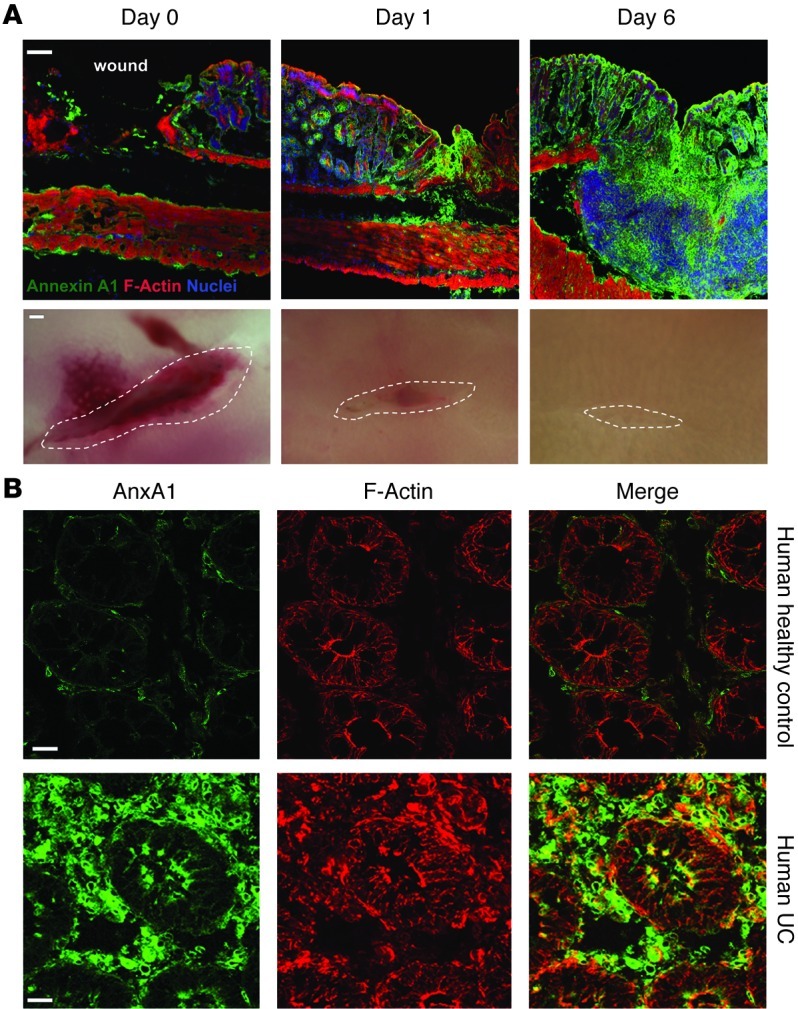
(A) Representative images of colonic mucosa from WT mice 0, 1, and 6 days after biopsy-induced injury. Frozen sections were stained with antibodies against ANXA1 (green), F-actin (phalloidin, red), and nuclei (TO-PRO-3, blue). Scale bar: 100 μm. The bottom row shows photomicrographs of colonic mucosa wounds on days 0, 1, and 6; n = 5 mice/group. (B) Sections of human colonic tissue from healthy control and UC patients were stained with ANXA1 (green) and F-actin (red). Scale bars: 20 μm.
Inflammation is characteristic of IBD, and proinflammatory NF-κB signaling is reflective of mucosal injury (57, 58). Recently, Ouyang et al. showed that MC-12, an ANXA1-based peptide exerts protective effects in experimental colitis by inhibiting the NF-κB pathway (59). Since the extent of p65 translocation into the nucleus qualitatively reveals NF-κB activation, we analyzed p65 localization in human IECs by immunofluorescence labeling and confocal microscopy. Ac2-26 inhibited p65 nuclear translocation induced by a combination of cytokines, TNF-α and IFN-γ (Supplemental Figure 7). Furthermore, to mechanistically demonstrate that ROS production is important in Ac2-26 inhibitory effects, we preincubated IECs with the NOX1 inhibitor DPI to block ROS generation. Ac2-26 failed to inhibit p65 nuclear translocation under these conditions. Taken together, these results suggest a role of endogenous ANXA1 and its mimetic peptide Ac2-26 in promoting epithelial barrier recovery via inhibition of proinflammatory events regulated by ROS generation.
Discussion
Mucosal inflammation compromises the epithelial barrier, resulting in exposure of systemic tissues to luminal bacterial products and antigens. In response, the epithelium has evolved a remarkable capacity to efficiently reseal the barrier defects. Such important reparative responses are modulated by mucosal mediators, such as anti-inflammatory protein ANXA1 (60–63). The beneficial effects of ANXA1 are exerted by a unique N-terminal peptide that is released by proteolysis from the full-length protein (2, 13, 14, 64). The importance of the N-terminal domain in mediating anti-inflammatory activity is further supported by studies wherein deletion of the N terminus induced inactivation of the protein, while use of synthetic N terminus peptides exerted pharmacologic activity (65, 66). Our in vivo studies demonstrated increased ANXA1 expression in the injured intestinal epithelium as well as in infiltrating leukocytes. These findings suggest that increased ANXA1 derived from distinct cell populations within wounds exerts paracrine/autocrine effects on epithelial FPR to facilitate wound closure and epithelial barrier recovery. Congruently, extravasated leukocytes synthesize ANXA1 (11, 12), which provides resolution signals to promote mucosal repair. In support of our experimental in vitro and in vivo observations in mice, we also observed increased ANXA1 protein in inflamed intestinal mucosa of patients with UC. These findings are in keeping with a previous published study showing that ANXA1 was endogenously secreted in the colon of patients with UC (67).
ANXA1 is an endogenous ligand for FPR. Classically, FPR signaling during phagocyte response to bacterial products results in a high level of ROS production, the microbiocidal “oxidant burst.” In this report, we demonstrate a beneficial role of FPR signaling in mediating ANXA1-induced epithelial ROS generation that exerts a novel pro-resolution function in vivo. Our study demonstrating the importance of FPR signaling in promoting epithelial wound closure supports previous reports by Gronert et al. (68–70). These studies highlighted a critical role of another pro-resolving FPR2 ligand, LXA4, in facilitating recovery of corneal epithelial wounds. Topical treatment with LXA4 not only accelerated wound closure, but also attenuated chemokine release. Furthermore, LXA4-dependent protective effects were sex specific, as estradiol was able to downregulate LXA4 formation (68).
Therapeutic effects of ANXA1 on intestinal epithelial wound closure were observed both following biopsy of the colonic mucosa and in recovery from DSS-induced colitis. Our studies provide a detailed understanding of the mechanism by which ANXA1-dependent ROS generation promotes wound repair. The novelty of our work is the identification of a central role of intestinal epithelial FPR and NOX1 in mediating the downstream beneficial effects of ANXA1 in facilitating mucosal repair. AnxA1–/– and newly generated Nox1–/–IEC mice were instrumental in demonstrating the beneficial effects and signaling pathways by which ANXA1 promotes mucosal barrier recovery. ANXA1 administration in Nox1–/–IEC mice failed to have pro-resolution effects on wound closure as observed in WT mice. Additionally, delayed intestinal mucosal wound healing was observed in AnxA1–/– mice. Analysis of the signaling pathway revealed a role of Src and Vav2 in mediating NOX1-dependent ROS generation induced by ANXA1. The downstream beneficial effects of NOX1-dependent ROS signaling in turn are mediated by oxidative inactivation of key regulatory phosphatases that regulate actin cytoskeletal and focal adhesion dynamics (71–73). We have recently demonstrated oxidative inactivation of low-molecular-weight protein tyrosine phosphatase (LMW-PTP) that was induced by bacterially stimulated ROS to promote wound closure (34). Consistent with these studies, we report here that PTEN and PTP-PEST are rapidly and transiently inactivated by ROS in cells treated with Ac2-26. Additionally, the link between inactivation of these phosphatases and focal adhesion protein dynamics is supported by previous studies demonstrating that PTP-PEST and PTEN can directly regulate the tyrosine phosphorylation of FAK and Pax and therefore influence cell motility (42, 43).
In the context of resolution of mucosal inflammation, epithelial repair represents an important final event after modulation of immune cell recruitment and clearance of extravasated cells. ANXA1 has been shown to dampen excessive immune cell trafficking by distinct mechanisms that include inhibition of neutrophil influx (11, 74) and promotion of neutrophil apoptosis at the site of resolving inflammation (75, 76). Intact ANXA1 and its derived peptides have been shown to mediate suppression of the proinflammatory NF-κB pathway (9, 77, 78). Recently, Ouyang et al. showed that MC-12, an ANXA1-based peptide, also inhibits NF-κB, thereby exerting a protective effect in experimental colitis (59). Similarly, we show that the ANXA1 mimetic peptide Ac2-26 can inhibit NF-κB in a redox-dependent manner (Supplemental Figure 7). These findings are consistent with our past observations that ROS induced by exogenous agents serves to suppress NF-κB activation by the same biochemical mechanism shown herein: namely, oxidative inactivation of catalytic cysteines in the active site of enzymes involved in processing ubiquitin-like proteins necessary for NF-κB activation (79, 80). In the context of wound injury, the ROS-mediated activation of pro-restitution enzymes such as FAK that stimulate epithelial cell migration would be effectively linked and coordinated with blockade of NF-κB and consequent cellular inflammation that would impede optimal wound healing.
In summary, the current study has uncovered a novel FPR1/NOX1-dependent ROS signaling pathway in IECs and has identified key regulatory components that mediate tissue repair activity of ANXA1 (Figure 7). This cascade involves key signaling proteins including Src, p120, Vav2, Rac1, and Nox1 and culminates in ROS generation, oxidation/inactivation of phosphatases, PTP-PEST, and PTEN, which in turn promotes activation of focal cell matrix adhesion proteins, cell motility, and ultimately wound closure. Taken together, the in vitro and in vivo studies (epithelial wound repair/recovery) demonstrate an influence of ANXA1 in the “final act” during resolution of inflammation, which requires efficient tissue repair and restoration of mucosal homeostasis. Last, these studies suggest that ANXA1 could potentially be used as a therapeutic agent to facilitate wound repair and barrier recovery in the intestine.
Figure 7. Model for the molecular mechanism of ANXA1-induced NOX1-dependent redox signaling during epithelial wound repair.

Ac2-26 binds to FPR1, which induces Src activation (Src p-Y418), resulting in the formation of a complex comprising active Rac1, p120 catenin, Vav2, and NOX1. ROS generated by NOX1 induces oxidative inactivation of PTEN and PTP-PEST, leading to increased phosphorylation of the focal adhesion proteins FAK and Pax, thereby promoting epithelial cell movement and wound repair.
Methods
Supplemental Methods are available online with this article; doi:10.1172/JCI65831DS1.
Mice.
WT female BALB/c mice and Nox2-null mice were purchased from the Jackson Laboratory. AnxA1–/– mice were a gift from R.J. Flower (Barts and The London School of Medicine, London, United Kingdom) (81). Nox1–/–IEC mice were generated as described in Supplemental Figure 3. Animals were housed under a standard day/night cycle, with free access to food and water.
Antibodies and reagents.
Primary antibodies to SHP-2, PTP-PEST, Pax p-Y118, and Vav2 were purchased from Cell Signaling Technology; p120, Rac1, p65, and FAK from BD Biosciences; FAK p-Y861 from Calbiochem; PTEN from Abcam; Src (N-terminal region) from ECM Biosciences; Src p-Y418 from Millipore; and β-actin and ANXA1 from Invitrogen. Secondary antibodies for Western blot analysis were obtained from Jackson ImmunoResearch Laboratories Inc. The chemical compound F2L was purchased from Phoenix Pharmaceuticals; NSC23766 was obtained from EMD; NAC, H2O2, and biotin-NM from Sigma-Aldrich; ECL gel from Invitrogen; peptide Ac2–26 (acetyl-AMVSEFLKQAWFIENEEQEYVVQTVK), MMK1, DPI, WRW4, PP2, and AZM475271 from Tocris; rotenone from Ultra Scientific; Boc2 from MP Biomedicals; PTx from Calbiochem; CsH from Enzo Life Sciences; and Rac-Assay reagent from Millipore. Hydro-Cy3 dye was synthesized in our laboratory.
ANXA1 recombinant protein purification.
cDNA of human ANXA1 carrying a cleavable N-terminal poly-His tag was expressed in E. coli. Recombinant protein was purified on IMAC (GE Healthcare), and the poly-His tag was subsequently removed. Purity of recombinant ANXA1 was confirmed by SDS-PAGE and the 4800 MALDI-TOF/TOF (82) (Applied Biosystems), revealing a 38.6-kDa protein that was >95% pure.
In vivo ANXA1 treatment.
Intraperitoneal injections of 1 or 5 μg ANXA1 in 100 μl PBS twice a day were performed. This dosing schedule was chosen based on published literature (65, 83, 84).
Cell lines and culture conditions.
Human IECs (SK-CO15) were grown in high-glucose (4.5 g/liter) Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 15 mM HEPES (pH 7.4), 2 mM l-glutamine, and 1% non-essential amino acids, as previously described (85, 86).
Immunoblot.
Epithelial cell monolayers were harvested in RIPA lysis buffer (150 mM NaCl, 1% NP-40, 0.5% deoxycholic acid, 0.1% SDS, 50 mM Tris [pH 8.0]) containing protease and phosphatase inhibitors (Sigma-Aldrich) and sonicated, and insoluble material was removed by centrifugation (16,000 g/10 min/4°C). Protein concentrantion was determined with a BCA assay, and samples were boiled in SDS buffer. Equal amounts of protein were separated by SDS-PAGE and transferred onto nitrocellulose membranes. Membranes were blocked for 1 hour with 3% wt/vol BSA in Tris-buffered saline containing 0.1% vol/vol Tween-20 and incubated with primary antibodies in blocking buffer overnight at 4°C, followed by 1-hour secondary antibody incubation. Densitometer analysis was performed using the ImageJ analysis program (NIH).
Transfection of siRNA.
The p120 siRNA sequence pair (5′-GCUAUGAUGACCUGGAUUA and 5′-UAAUCCAGGUCAUCAUAGC) was purchased from Sigma-Aldrich (87 SASI_Hs02_00317795) and used for transient knockdown of p120. Transient transfections were performed with LipofectAMINE 2000 (Invitrogen) according to the manufacturer’s instructions.
Cell adhesion assays.
96-well plates were incubated with ECM gel (5 μg/ml) overnight at 37°C. 5 × 104 cells were labeled with bis-carboxymethyl-carboxyfluorescein (BCECF) and added to the wells. Cells were allowed to adhere for 2 hours at 37°C, and non-adherent cells were removed by washing the plates 3 times. Absorbance of adherent cells was measured at 488 nm in a microtiter plate reader (Multiskan plus version 2.03, Labsystems).
Wound healing assay.
Cell migration was assessed using a scratch wound healing assay as previously published (87).
Intracellular ROS generation experiments.
ROS production was visualized by using a stable and nontoxic redox-sensitive hydrocyanine dye, hydro-Cy3 (88). Epithelial cells treated with Ac2-26 for the indicated times were incubated with 15 μM hydro-Cy3 for 30 minutes at 37°C. Epithelial cells were pretreated with NAC (20 mM), DPI (10 μM), rotenone (10 μM), PTx (1 μg/ml), CsH (1 μM) or Boc2 (1 μM), PP2 (10 μM), and NSC23766 (50 μM) 30 minutes prior to Ac2-26 treatment. ROS was detected by confocal laser scanning microscopy (Zeiss LSM 510) at ×40 magnification. Quantification of fluorescence intensity of ROS was determined using ImageJ software. For in vivo ROS measurements, 0.1 ml PBS containing hydro-Cy3 (40 μM) was intrarectally administered with flexible no. 4 catheters inserted 3–4 cm into the distal colon of a mouse. After 30 minutes of dye administration, the colon was exteriorized, epithelial tissue was wounded, and HBSS (control) or Ac2-26 was added to the tissue for 15 minutes; in the same experiments, some mice were preincubated with CsH for 30 minutes before Ac2-26 administration. Mice were sacrificed; colon tissue was fixed in PFA 3.7%, sectioned, and mounted with Vectashield; and fluorescence was analyzed by confocal microscopy.
Analysis of protein oxidation.
Phosphatase oxidation was monitored by labeling with the thiol-modifying reagent biotin-NM. Cells were treated with H2O2 (15 μM) or Ac2-26 (3 μM) biotinylated by incubation for 30 minutes at 50°C with a solution containing 50 mM HEPES-NaOH, pH 7.5, 1 mM EDTA, 2% SDS, and protease inhibitors with 12 μM biotin-NM. The reaction was stopped by the addition of DTT to a final concentration of 100 nM, and biotinylated proteins were then precipitated by incubation overnight at 4°C with 50 μl streptavidin-agarose (SA) beads. The beads were washed 5 times with a solution containing 20 mM HEPES-NaOH (pH 7.7), 200 mM NaCl, 1 mM EDTA, and 0.5% SDS. Biotinylated proteins were released from the beads by boiling in SDS/PAGE sample buffer and subjected to immunoblot analysis.
Rac1 activation assay.
SK-CO15 cells were washed in cooled TBS before lysis in supplied magnesium lysis buffer (MLB) containing protease inhibitors. Lysates were normalized for protein concentrations using a bicinchoninic acid assay (Pierce) and incubated with recombinant PAK1-GST coupled to agarose beads (45 minutes, at 4°C with rotation). Beads were washed with MLB and resuspended in SDS sample buffer for immunoblot analysis.
Colonoscopy in live mice.
Mice were anesthetized by i.p. injection of a ketamine (100 mg/kg)/xylazine (10 mg/kg) solution. To create intestinal mucosal wounds and monitor wound closure, we used a high-resolution colonoscopy system equipped with biopsy forceps (89, 90). This system consisted of a miniature rigid endoscope (1.9-mm outer diameter), a xenon light source, a triple-chip high-resolution CCD camera, and a 3 Fr operating sheath. Endoscopic procedures were viewed with high-resolution (1,024 × 768 pixels) images on a flat-panel color monitor. Wound size averaged approximately 1 mm2, which is equivalent to removal of approximately 250–300 crypts (91). In each experiment, 10–15 lesions from 5–6 mice per group were examined (92).
Immunofluorescence microscopy.
Cells grown on coverslips were fixed with 3.7% wt/vol PFA for 15 minutes, followed by 0.5 % vol/vol Triton X-100 for 5 minutes. Tissue sections were fixed in 100% vol/vol ethanol. Samples were then blocked with 3% wt/vol BSA for 1 hour and incubated with primary antibody overnight at 4°C, washed, and incubated for 1 hour with fluorophore-labeled secondary antibodies, then mounted in p-phenylene. Images were taken on an LSM 510 confocal microscope (Zeiss) with Plan-NEOFLUAR ×100/1.3 oil, ×40/1.3 oil, and ×20/0.5 dry objectives, with software supplied by the vendor.
Induction of colitis.
3% (wt/vol) DSS (molecular mass, 36–50 kDa; MP Biomedicals) was dissolved in purified water and orally administered to mice as previously described (82). Mice were allowed free access to food and drinking water containing 3% DSS from day 0 until day 7, and DSS was withdrawn to allow recovery from colitis for an additional 6 days. Mice were sacrificed on day 13. Daily clinical assessment of DSS-treated animals included evaluation of stool consistency, detection of blood in stool, and body weight loss measurements. An individual score (ranging from 0 to 4) was attributed for each one of these parameters, and a disease activity index ranging from 0 to 4 was calculated by combining all three scores (82).
Statistics.
Quantitative data are expressed as mean ± SEM for each treatment group. Statistical comparisons were performed by either 2-tailed Student’s t test or ANOVA with Tukey’s multiple comparison post-test (GraphPad Prism; GraphPad Software). P values less than 0.05 were considered significant.
Study approval.
All procedures using animals were reviewed and approved by the Emory University IACUC and were performed according to NIH criteria.
Supplementary Material
Acknowledgments
The authors would like to thank R.J. Flower (Barts and The London School of Medicine) for providing the AnxA1–/– mouse; Attila E. Farkas and Al Jesaitis for critically reviewing the manuscript; and Courtney S. Ardita for Nox1–/–IEC mouse genotyping and technical help. This work was supported by R01DK089763 and R01DK055679 to A. Nusrat; R01AI64462 to A.S. Neish; DK061379, DK072564, and DK079392 to C.A. Parkos; R01HL096796-3 to N. Murthy; and grants from the William Harvey Research Foundation to M. Perretti, the German Research Foundation (Deutsche Forschungsgemeinschaft, NE1834/1-1) to P.-A. Neumann, and the Emory Digestive Diseases Research Development Center Core Grant (DK064399).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(1):443–454. doi:10.1172/JCI65831.
References
- 1.Lotz MM, Nusrat A, Madara JL, Ezzell R, Wewer UM, Mercurio AM. Intestinal epithelial restitution. Involvement of specific laminin isoforms and integrin laminin receptors in wound closure of a transformed model epithelium. Am J Pathol. 1997;150(2):747–760. [PMC free article] [PubMed] [Google Scholar]
- 2.Perretti M, et al. Endogenous lipid- and peptide-derived anti-inflammatory pathways generated with glucocorticoid and aspirin treatment activate the lipoxin A4 receptor. Nat Med. 2002;8(11):1296–1302. doi: 10.1038/nm786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Serhan CN. The resolution of inflammation: the devil in the flask and in the details. FASEB J. 2011;25(5):1441–1448. doi: 10.1096/fj.11-0502ufm. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Serhan CN. Systems approach to inflammation resolution: identification of novel anti-inflammatory and pro-resolving mediators. J Thromb Haemost. 2009;7(suppl 1):44–48. doi: 10.1111/j.1538-7836.2009.03396.x. [DOI] [PubMed] [Google Scholar]
- 5.Serhan CN, et al. Resolution of inflammation: state of the art, definitions and terms. FASEB J. 2007;21(2):325–332. doi: 10.1096/fj.06-7227rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Norling LV, Spite M, Yang R, Flower RJ, Perretti M, Serhan CN. Cutting edge: humanized nano-proresolving medicines mimic inflammation-resolution and enhance wound healing. J Immunol. 2011;186(10):5543–5547. doi: 10.4049/jimmunol.1003865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Serhan CN, Levy B. Novel pathways and endogenous mediators in anti-inflammation and resolution. Chem Immunol Allergy. 2003;83:115–145. doi: 10.1159/000071558. [DOI] [PubMed] [Google Scholar]
- 8.Levy BD, et al. Multi-pronged inhibition of airway hyper-responsiveness and inflammation by lipoxin A(4). Nat Med. 2002;8(9):1018–1023. doi: 10.1038/nm748. [DOI] [PubMed] [Google Scholar]
- 9.Perretti M, D’Acquisto F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat Rev Immunol. 2009;9(1):62–70. doi: 10.1038/nri2470. [DOI] [PubMed] [Google Scholar]
- 10.Yang YH, et al. Modulation of inflammation and response to dexamethasone by Annexin 1 in antigen-induced arthritis. Arthritis Rheum. 2004;50(3):976–984. doi: 10.1002/art.20201. [DOI] [PubMed] [Google Scholar]
- 11.Damazo AS, Yona S, Flower RJ, Perretti M, Oliani SM. Spatial and temporal profiles for anti-inflammatory gene expression in leukocytes during a resolving model of peritonitis. J Immunol. 2006;176(7):4410–4418. doi: 10.4049/jimmunol.176.7.4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Damazo AS, Yona S, D’Acquisto F, Flower RJ, Oliani SM, Perretti M. Critical protective role for annexin 1 gene expression in the endotoxemic murine microcirculation. Am J Pathol. 2005;166(6):1607–1617. doi: 10.1016/S0002-9440(10)62471-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perretti M. Lipocortin 1 and chemokine modulation of granulocyte and monocyte accumulation in experimental inflammation. Gen Pharmacol. 1998;31(4):545–552. doi: 10.1016/s0306-3623(98)00039-1. [DOI] [PubMed] [Google Scholar]
- 14.La M, et al. Annexin 1 peptides protect against experimental myocardial ischemia-reperfusion: analysis of their mechanism of action. FASEB J. 2001;15(12):2247–2256. doi: 10.1096/fj.01-0196com. [DOI] [PubMed] [Google Scholar]
- 15.Liu L, Zimmerman UJ. An intramolecular disulfide bond is essential for annexin I-mediated liposome aggregation. Biochem Mol Biol Int. 1995;35(2):345–350. [PubMed] [Google Scholar]
- 16.Oliani SM, Paul-Clark MJ, Christian HC, Flower RJ, Perretti M. Neutrophil interaction with inflamed postcapillary venule endothelium alters annexin 1 expression. Am J Pathol. 2001;158(2):603–615. doi: 10.1016/S0002-9440(10)64002-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perretti M. The annexin 1 receptor(s): is the plot unravelling? Trends Pharmacol Sci. 2003;24(11):574–579. doi: 10.1016/j.tips.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 18.Walther A, Riehemann K, Gerke V. A novel ligand of the formyl peptide receptor: annexin I regulates neutrophil extravasation by interacting with the FPR. Mol Cell. 2000;5(5):831–840. doi: 10.1016/S1097-2765(00)80323-8. [DOI] [PubMed] [Google Scholar]
- 19.Ye RD, et al. International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol Rev. 2009;61(2):119–161. doi: 10.1124/pr.109.001578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Le Y, Murphy PM, Wang JM. Formyl-peptide receptors revisited. Trends Immunol. 2002;23(11):541–548. doi: 10.1016/S1471-4906(02)02316-5. [DOI] [PubMed] [Google Scholar]
- 21.Bylund J, Samuelsson M, Collins LV, Karlsson A. NADPH-oxidase activation in murine neutrophils via formyl peptide receptors. Exp Cell Res. 2003;282(2):70–77. doi: 10.1016/S0014-4827(02)00010-1. [DOI] [PubMed] [Google Scholar]
- 22.Hurd TR, DeGennaro M, Lehmann R. Redox regulation of cell migration and adhesion. Trends Cell Biol. 2012;22(2):107–115. doi: 10.1016/j.tcb.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Babbin BA, et al. Annexin I regulates SKCO-15 cell invasion by signaling through formyl peptide receptors. J Biol Chem. 2006;281(28):19588–19599. doi: 10.1074/jbc.M513025200. [DOI] [PubMed] [Google Scholar]
- 24.Ernst S, Lange C, Wilbers A, Goebeler V, Gerke V, Rescher U. An annexin 1 N-terminal peptide activates leukocytes by triggering different members of the formyl peptide receptor family. J Immunol. 2004;172(12):7669–7676. doi: 10.4049/jimmunol.172.12.7669. [DOI] [PubMed] [Google Scholar]
- 25.Hayhoe RP, Kamal AM, Solito E, Flower RJ, Cooper D, Perretti M. Annexin 1 and its bioactive peptide inhibit neutrophil-endothelium interactions under flow: indication of distinct receptor involvement. Blood. 2006;107(5):2123–2130. doi: 10.1182/blood-2005-08-3099. [DOI] [PubMed] [Google Scholar]
- 26.Wenzel-Seifert K, Seifert R. Cyclosporin H is a potent and selective formyl peptide receptor antagonist. Comparison with N-t-butoxycarbonyl-L-phenylalanyl-L-leucyl-L-phenylalanyl-L- leucyl-L-phenylalanine and cyclosporins A, B, C, D, and E. J Immunol. 1993;150(10):4591–4599. [PubMed] [Google Scholar]
- 27.Stenfeldt AL, Karlsson J, Wenneras C, Bylund J, Fu H, Dahlgren C. Cyclosporin H, Boc-MLF and Boc-FLFLF are antagonists that preferentially inhibit activity triggered through the formyl peptide receptor. Inflammation. 2007;30(6):224–229. doi: 10.1007/s10753-007-9040-4. [DOI] [PubMed] [Google Scholar]
- 28.Bae YS, et al. Identification of peptides that antagonize formyl peptide receptor-like 1-mediated signaling. J Immunol. 2004;173(1):607–614. doi: 10.4049/jimmunol.173.1.607. [DOI] [PubMed] [Google Scholar]
- 29.Hu JY, et al. Synthetic peptide MMK-1 is a highly specific chemotactic agonist for leukocyte FPRL1. J Leukoc Biol. 2001;70(1):155–161. [PubMed] [Google Scholar]
- 30.Migeotte I, et al. Identification and characterization of an endogenous chemotactic ligand specific for FPRL2. J Exp Med. 2005;201(1):83–93. doi: 10.1084/jem.20041277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Devosse T, et al. Processing of HEBP1 by cathepsin D gives rise to F2L, the agonist of formyl peptide receptor 3. J Immunol. 2011;187(3):1475–1485. doi: 10.4049/jimmunol.1003545. [DOI] [PubMed] [Google Scholar]
- 32.Ridley AJ, et al. Cell migration: integrating signals from front to back. Science. 2003;302(5651):1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- 33.Sieg DJ, Hauck CR, Schlaepfer DD. Required role of focal adhesion kinase (FAK) for integrin-stimulated cell migration. J Cell Sci. 1999;16(pt 16):2677–2691. doi: 10.1242/jcs.112.16.2677. [DOI] [PubMed] [Google Scholar]
- 34.Swanson PA 2nd, et al. Enteric commensal bacteria potentiate epithelial restitution via reactive oxygen species-mediated inactivation of focal adhesion kinase phosphatases. Proc Natl Acad Sci U S A. 2011;108(21):8803–8808. doi: 10.1073/pnas.1010042108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4(3):181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 36.Smigel M, Katada T, Northup JK, Bokoch GM, Ui M, Gilman AG. Mechanisms of guanine nucleotide-mediated regulation of adenylate cyclase activity. Adv Cyclic Nucleotide Protein Phosphorylation Res. 1984;17:1–18. [PubMed] [Google Scholar]
- 37.Jones DP. Radical-free biology of oxidative stress. Am J Physiol Cell Physiol. 2008;295(4):C849–868. doi: 10.1152/ajpcell.00283.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rhee SG, Chang TS, Bae YS, Lee SR, Kang SW. Cellular regulation by hydrogen peroxide. J Am Soc Nephrol. 2003;14(8 suppl 3):S211–S215. doi: 10.1097/01.ASN.0000077404.45564.7E. [DOI] [PubMed] [Google Scholar]
- 39.Terada LS. Specificity in reactive oxidant signaling: think globally, act locally. J Cell Biol. 2006;174(5):615–623. doi: 10.1083/jcb.200605036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boivin B, Yang M, Tonks NK. Targeting the reversibly oxidized protein tyrosine phosphatase superfamily. Sci Signal. 2010;3(137):pl2. doi: 10.1126/scisignal.3137pl2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Angers-Loustau A, Cote JF, Tremblay ML. Roles of protein tyrosine phosphatases in cell migration and adhesion. Biochem Cell Biol. 1999;77(6):493–505. doi: 10.1139/o99-064. [DOI] [PubMed] [Google Scholar]
- 42.Wade R, Brimer N, Lyons C, Vande Pol S. Paxillin enables attachment-independent tyrosine phosphorylation of focal adhesion kinase and transformation by RAS. J Biol Chem. 2011;286(44):37932–37944. doi: 10.1074/jbc.M111.294504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zheng Y, Yang W, Xia Y, Hawke D, Liu DX, Lu Z. Ras-induced and extracellular signal-regulated kinase 1 and 2 phosphorylation-dependent isomerization of protein tyrosine phosphatase (PTP)-PEST by PIN1 promotes FAK dephosphorylation by PTP-PEST. Mol Cell Biol. 2011;31(21):4258–4269. doi: 10.1128/MCB.05547-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bagrodia S, Taylor SJ, Creasy CL, Chernoff J, Cerione RA. Identification of a mouse p21Cdc42/Rac activated kinase. J Biol Chem. 1995;270(39):22731–22737. doi: 10.1074/jbc.270.39.22731. [DOI] [PubMed] [Google Scholar]
- 45.Bagrodia S, Taylor SJ, Jordon KA, Van Aelst L, Cerione RA. A novel regulator of p21-activated kinases. J Biol Chem. 1998;273(37):23633–23636. doi: 10.1074/jbc.273.37.23633. [DOI] [PubMed] [Google Scholar]
- 46.Takai Y, Sasaki T, Matozaki T. Small GTP-binding proteins. Physiol Rev. 2001;81(1):153–208. doi: 10.1152/physrev.2001.81.1.153. [DOI] [PubMed] [Google Scholar]
- 47.Noren NK, Liu BP, Burridge K, Kreft B. p120 catenin regulates the actin cytoskeleton via Rho family GTPases. J Cell Biol. 2000;150(3):567–580. doi: 10.1083/jcb.150.3.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheng G, Diebold BA, Hughes Y, Lambeth JD. Nox1-dependent reactive oxygen generation is regulated by Rac1. J Biol Chem. 2006;281(26):17718–17726. doi: 10.1074/jbc.M512751200. [DOI] [PubMed] [Google Scholar]
- 49.Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci U S A. 2004;101(20):7618–7623. doi: 10.1073/pnas.0307512101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reynolds AB, Daniel J, McCrea PD, Wheelock MJ, Wu J, Zhang Z. Identification of a new catenin: the tyrosine kinase substrate p120cas associates with E-cadherin complexes. Mol Cell Biol. 1994;14(12):8333–8342. doi: 10.1128/mcb.14.12.8333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gianni D, Taulet N, DerMardirossian C, Bokoch GM. c-Src-mediated phosphorylation of NoxA1 and Tks4 induces the reactive oxygen species (ROS)-dependent formation of functional invadopodia in human colon cancer cells. Mol Biol Cell. 2010;21(23):4287–4298. doi: 10.1091/mbc.E10-08-0685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu KP, Yin J, Yu FS. SRC-family tyrosine kinases in wound- and ligand-induced epidermal growth factor receptor activation in human corneal epithelial cells. Invest Ophthalmol Vis Sci. 2006;47(7):2832–2839. doi: 10.1167/iovs.05-1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yezhelyev MV, et al. Inhibition of SRC tyrosine kinase as treatment for human pancreatic cancer growing orthotopically in nude mice. Clin Cancer Res. 2004;10(23):8028–8036. doi: 10.1158/1078-0432.CCR-04-0621. [DOI] [PubMed] [Google Scholar]
- 54.Nam JS, Ino Y, Sakamoto M, Hirohashi S. Src family kinase inhibitor PP2 restores the E-cadherin/catenin cell adhesion system in human cancer cells and reduces cancer metastasis. Clin Cancer Res. 2002;8(7):2430–2436. [PubMed] [Google Scholar]
- 55.Hanke JH, et al. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J Biol Chem. 1996;271(2):695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- 56.Vong L, et al. Up-regulation of Annexin-A1 and lipoxin A(4) in individuals with ulcerative colitis may promote mucosal homeostasis. PLoS One. 2012;7(6):e39244. doi: 10.1371/journal.pone.0039244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Morise Z, Grisham MB. Molecular mechanisms involved in NSAID-induced gastropathy. J Clin Gastroenterol. 1998;27(suppl 1):S87–S90. doi: 10.1097/00004836-199800001-00014. [DOI] [PubMed] [Google Scholar]
- 58.Kosone T, et al. Integrative roles of transforming growth factor-alpha in the cytoprotection mechanisms of gastric mucosal injury. BMC Gastroenterol. 2006;6:22. doi: 10.1186/1471-230X-6-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ouyang N, et al. MC-12, an annexin A1-based peptide, is effective in the treatment of experimental colitis. PLoS One. 2012;7(7):e41585. doi: 10.1371/journal.pone.0041585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Babbin BA, et al. Annexin A1 regulates intestinal mucosal injury, inflammation, and repair. J Immunol. 2008;181(7):5035–5044. doi: 10.4049/jimmunol.181.7.5035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Martin GR, Perretti M, Flower RJ, Wallace JL. Annexin-1 modulates repair of gastric mucosal injury. Am J Physiol Gastrointest Liver Physiol. 2008;294(3):G764–G769. doi: 10.1152/ajpgi.00531.2007. [DOI] [PubMed] [Google Scholar]
- 62.Brancaleone V, Dalli J, Bena S, Flower RJ, Cirino G, Perretti M. Evidence for an anti-inflammatory loop centered on polymorphonuclear leukocyte formyl peptide receptor 2/lipoxin A4 receptor and operative in the inflamed microvasculature. J Immunol. 2011;186(8):4905–4914. doi: 10.4049/jimmunol.1003145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dalli J, Jones CP, Cavalcanti DM, Farsky SH, Perretti M, Rankin SM. Annexin A1 regulates neutrophil clearance by macrophages in the mouse bone marrow. FASEB J. 2012;26(1):387–396. doi: 10.1096/fj.11-182089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dalli J, Montero-Melendez T, McArthur S, Perretti M. Annexin A1 N-terminal derived Peptide ac2-26 exerts chemokinetic effects on human neutrophils. Front Pharmacol. 2012;3:28. doi: 10.3389/fphar.2012.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lim LH, Solito E, Russo-Marie F, Flower RJ, Perretti M. Promoting detachment of neutrophils adherent to murine postcapillary venules to control inflammation: effect of lipocortin 1. Proc Natl Acad Sci U S A. 1998;95(24):14535–14539. doi: 10.1073/pnas.95.24.14535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.John C, et al. Annexin 1-dependent actions of glucocorticoids in the anterior pituitary gland: roles of the N-terminal domain and protein kinase C. Endocrinology. 2002;143(8):3060–3070. doi: 10.1210/en.143.8.3060. [DOI] [PubMed] [Google Scholar]
- 67.Vergnolle N, et al. Annexin 1 is secreted in situ during ulcerative colitis in humans. Inflamm Bowel Dis. 2004;10(5):584–592. doi: 10.1097/00054725-200409000-00013. [DOI] [PubMed] [Google Scholar]
- 68.Wang SB, Hu KM, Seamon KJ, Mani V, Chen Y, Gronert K. Estrogen negatively regulates epithelial wound healing and protective lipid mediator circuits in the cornea. FASEB J. 2012;26(4):1506–1516. doi: 10.1096/fj.11-198036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gronert K, Maheshwari N, Khan N, Hassan IR, Dunn M, Laniado Schwartzman M. A role for the mouse 12/15-lipoxygenase pathway in promoting epithelial wound healing and host defense. J Biol Chem. 2005;280(15):15267–15278. doi: 10.1074/jbc.M410638200. [DOI] [PubMed] [Google Scholar]
- 70.Gronert K. Lipoxins in the eye and their role in wound healing. Prostaglandins Leukot Essent Fatty Acids. 2005;73(3–4):221–229. doi: 10.1016/j.plefa.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 71.Shinohara M, et al. Nox1 redox signaling mediates oncogenic Ras-induced disruption of stress fibers and focal adhesions by down-regulating Rho. J Biol Chem. 2007;282(24):17640–17648. doi: 10.1074/jbc.M609450200. [DOI] [PubMed] [Google Scholar]
- 72.Ostman A, Frijhoff J, Sandin A, Bohmer FD. Regulation of protein tyrosine phosphatases by reversible oxidation. J Biochem. 2011;150(4):345–356. doi: 10.1093/jb/mvr104. [DOI] [PubMed] [Google Scholar]
- 73.Chiarugi P, et al. Reactive oxygen species as essential mediators of cell adhesion: the oxidative inhibition of a FAK tyrosine phosphatase is required for cell adhesion. J Cell Biol. 2003;161(5):933–944. doi: 10.1083/jcb.200211118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chatterjee BE, et al. Annexin 1-deficient neutrophils exhibit enhanced transmigration in vivo and increased responsiveness in vitro. J Leukoc Biol. 2005;78(3):639–646. doi: 10.1189/jlb.0405206. [DOI] [PubMed] [Google Scholar]
- 75.Solito E, Kamal A, Russo-Marie F, Buckingham JC, Marullo S, Perretti M. A novel calcium-dependent proapoptotic effect of annexin 1 on human neutrophils. FASEB J. 2003;17(11):1544–1546. doi: 10.1096/fj.02-0941fje. [DOI] [PubMed] [Google Scholar]
- 76.Vago JP, et al. Annexin A1 modulates natural and glucocorticoid-induced resolution of inflammation by enhancing neutrophil apoptosis. J Leukoc Biol. 2012;92(2):249–258. doi: 10.1189/jlb.0112008. [DOI] [PubMed] [Google Scholar]
- 77.Zhang Z, Huang L, Zhao W, Rigas B. Annexin 1 induced by anti-inflammatory drugs binds to NF-kappaB and inhibits its activation: anticancer effects in vitro and in vivo. Cancer Res. 2010;70(6):2379–2388. doi: 10.1158/0008-5472.CAN-09-4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Perretti M, Gavins FN. Annexin 1: an endogenous anti-inflammatory protein. News Physiol Sci. 2003;18:60–64. doi: 10.1152/nips.01424.2002. [DOI] [PubMed] [Google Scholar]
- 79.Kumar A, et al. Commensal bacteria modulate cullin-dependent signaling via generation of reactive oxygen species. EMBO J. 2007;26(21):4457–4466. doi: 10.1038/sj.emboj.7601867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Collier-Hyams LS, Sloane V, Batten BC, Neish AS. Cutting edge: bacterial modulation of epithelial signaling via changes in neddylation of cullin-1. J Immunol. 2005;175(7):4194–4198. doi: 10.4049/jimmunol.175.7.4194. [DOI] [PubMed] [Google Scholar]
- 81.Hannon R, et al. Aberrant inflammation and resistance to glucocorticoids in annexin 1–/– mouse. . FASEB J. 2003;17(2):253–255. doi: 10.1096/fj.02-0239fje. [DOI] [PubMed] [Google Scholar]
- 82.Khounlotham M, et al. Compromised intestinal epithelial barrier induces adaptive immune compensation that protects from colitis. Immunity. 2012;37(3):563–573. doi: 10.1016/j.immuni.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Perretti M, Ahluwalia A, Harris JG, Goulding NJ, Flower RJ. Lipocortin-1 fragments inhibit neutrophil accumulation and neutrophil-dependent edema in the mouse. J Immunol. 1993;151(8):4306–4314. [PubMed] [Google Scholar]
- 84.Patel HB, et al. The impact of endogenous annexin A1 on glucocorticoid control of inflammatory arthritis. Ann Rheum Dis. 2012;71(11):1872–1880. doi: 10.1136/annrheumdis-2011-201180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lisanti MP, Caras IW, Davitz MA, Rodriguez-Boulan E. A glycophospholipid membrane anchor acts as an apical targeting signal in polarized epithelial cells. J Cell Biol. 1989;109(5):2145–2156. doi: 10.1083/jcb.109.5.2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mandell KJ, Babbin BA, Nusrat A, Parkos CA. Junctional adhesion molecule 1 regulates epithelial cell morphology through effects on beta1 integrins and Rap1 activity. J Biol Chem. 2005;280(12):11665–11674. doi: 10.1074/jbc.M412650200. [DOI] [PubMed] [Google Scholar]
- 87.Babbin BA, et al. Formyl peptide receptor-1 activation enhances intestinal epithelial cell restitution through phosphatidylinositol 3-kinase-dependent activation of Rac1 and Cdc42. J Immunol. 2007;179(12):8112–8121. doi: 10.4049/jimmunol.179.12.8112. [DOI] [PubMed] [Google Scholar]
- 88.Kundu K, Knight SF, Willett N, Lee S, Taylor WR, Murthy N. Hydrocyanines: a class of fluorescent sensors that can image reactive oxygen species in cell culture, tissue, and in vivo. Angew Chem Int Ed Engl. 2009;48(2):299–303. doi: 10.1002/anie.200804851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Becker C, et al. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity. 2004;21(4):491–501. doi: 10.1016/j.immuni.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 90.Becker C, et al. In vivo imaging of colitis and colon cancer development in mice using high resolution chromoendoscopy. Gut. 2005;54(7):950–954. doi: 10.1136/gut.2004.061283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Manieri NA, Drylewicz MR, Miyoshi H, Stappenbeck TS. Igf2bp1 is required for full induction of Ptgs2 mRNA in colonic mesenchymal stem cells in mice. Gastroenterology. 2012;143(1):110–121. doi: 10.1053/j.gastro.2012.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pickert G, et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med. 2009;206(7):1465–1472. doi: 10.1084/jem.20082683. [DOI] [PMC free article] [PubMed] [Google Scholar]