

Abstract
Neurofibromatosis type 1 (NF1) predisposes individuals to the development of juvenile myelomonocytic leukemia (JMML), a fatal myeloproliferative disease (MPD). In genetically engineered murine models, nullizygosity of Nf1, a tumor suppressor gene that encodes a Ras-GTPase–activating protein, results in hyperactivity of Raf/Mek/Erk in hematopoietic stem and progenitor cells (HSPCs). Activated Erk1/2 phosphorylate kinases and transcription factors with myriad mitogenic roles in diverse cell types. However, genetic studies examining Erk1/2’s differential and/or combined control of normal and Nf1-deficient myelopoiesis are lacking. Moreover, prior studies relying on chemical Mek/Erk inhibitors have reached conflicting conclusions in normal and Nf1-deficient mice. Here, we show that while single Erk1 or Erk2 disruption did not grossly compromise myelopoiesis, dual Erk1/2 disruption rapidly ablated granulocyte and monocyte production in vivo, diminished progenitor cell number, and prevented HSPC proliferation in vitro. Genetic disruption of Erk1/2 in the context of Nf1 nullizygosity (Mx1Cre+Nf1flox/floxErk1–/–Erk2flox/flox) fully protects against the development of MPD. Collectively, we identified a fundamental requirement for Erk1/2 signaling in normal and Nf1-deficient hematopoiesis, elucidating a critical hematopoietic function for Erk1/2 while genetically validating highly selective Mek/Erk inhibitors in a leukemia that is otherwise resistant to traditional therapy.
Introduction
Neurofibromatosis type 1 (NF1) is a common genetic disorder predisposing patients to multiple malignancies, including malignant peripheral nerve sheath tumors, plexiform neurofibromas, and juvenile myelomonocytic leukemia (JMML) (1), a universally fatal myeloproliferative disease (MPD) arising from deregulated Ras signaling in GM-CSF, SCF, and IL-3–responsive hematopoietic stem and progenitor cells (HSPCs) (2–5). NF1 results from autosomal dominant mutations in the NF1 tumor suppressor gene, the protein product of which accelerates the intrinsic hydrolysis of active Ras-GTP to inactive Ras-GDP (6). NF1 nullizygosity in HSPCs engenders leukemogenesis, as evidenced in studies of humans with NF1 and in mouse models of Nf1-deficient MPD (1, 3). Specifically, hyperactivation of the Raf/Mek/Erk cascade downstream of deregulated Ras appears to modulate NF1-associated leukemogenesis (3, 7, 8). Moreover, a recent study by Chung et al. demonstrated that constitutively active Mek, independent of Nf1 mutations, induces MPD (9). However, it remains unknown whether Nf1-deficient MPD progression requires Mek/Erk signals, and some data have suggested Mek/Erk’s dispensability in the context of cooperating mutations (7).
The coordinated differentiation and proliferation of HSPCs toward mature myeloid cells require multiple cytokines transducing signals via JAK-STAT and receptor tyrosine kinase networks. These networks near-ubiquitously activate Erk1 and Erk2, proline-directed mitogenic kinases that share 83% sequence homology and function to phosphorylate mitogens and transcription factors broadly implicated in cell-cycle control, protein synthesis, survival, and differentiation (10). While prior hematopoietic studies have examined Erk1 and/or Erk2 disruption in lymphocytes (11, 12), no study has examined Erk1/2 disruption in normal or Nf1-deficient myelopoiesis, even though pathologically important cytokines implicated in JMML, including GM-CSF, activate this pathway (4, 5). Given the recent development of pharmacological grade Mek/Erk inhibitors and the unresponsiveness of JMML to traditional chemotherapy, these genetic studies inform future translational studies in children with this malignancy.
Here, we show that normal production of granulocytes and monocytes requires either Erk1 or Erk2. Concomitant Nf1 deficiency cannot overcome the requirement for Mek/Erk signals in driving myelopoiesis. Thus, we clarify critical functions for Erk1/2 in myelopoiesis while genetically validating highly selective new generation Mek/Erk inhibitors for the attenuation of leukemia dependent on hyperactive Ras.
Results and Discussion
Following Cre induction, 4 genotypes were initially examined: Erk2flox/flox (WT), Erk1–/–Erk2flox/flox (Erk1-KO), Mx1Cre+Erk2flox/flox (Erk2-KO), and Mx1Cre+Erk1–/–Erk2flox/flox (Erk1/2-KO) (see Methods). From previous lymphocyte-focused studies of Erk1/2 disruption in the hematopoietic system (including excision driven by Mx1Cre in ref. 12), we did not anticipate a gross myeloid phenotype. However, following poly(I:C) induction, Erk1/2-KO marrow showed a 2-fold reduction in total marrow cellularity, which corresponded histologically to vacant marrow space (Figure 1A). Erk1/2-KO marrow demonstrated a prominent reduction in the characteristic myeloid FSChiSSChi population, indicating loss of monocyte and/or granulocyte progenitors, both in frequency and total cell number (Figure 1B). Further flow cytometry–based analyses of the Erk1/2-KO bone marrow demonstrated prominent reductions in the frequency and absolute number of mixed marrow granulocytes and their immediate progenitors (CD3–B220–Gr1+Mac1+) (Figure 1, C and D). None of these deficiencies manifested in Erk1-KO or Erk2-KO animals.
Figure 1. Erk1/2 disruption diminishes myeloid cellularity.

(A and B) Immediately following poly(I:C), Erk1/2-KO bone marrow shows a 2-fold reduction in total cellularity (**P < 0.01 Erk1/2-KO vs. all groups, 1-way ANOVA with Bonferroni’s correction), which corresponds histologically to vacant marrow spaces and a prominent shift from the characteristic myeloid (FSChiSSChi) population to lymphoid (FSCloSSClo), shown by frequency and total cell number. n = 4–6, **P < 0.01; ***P < 0.001, Erk1/2-KO vs. all groups, 2-way ANOVA with Bonferroni’s correction. Original magnification, ×100 (top panels); ×400 (bottom panels). (C and D) Representative flow pictograph and quantitative data demonstrate the reduction in frequency and total cell number of CD3–B220–Gr1+Mac1+ myeloid cells in the bone marrow of Erk1/2-KO mice. ***P < 0.001, Erk1/2-KO vs. WT within each subgroup, 2-way ANOVA with Bonferroni’s correction.
To ascertain whether this defect originates in primitive progenitor cells, we established low and high proliferative potential cell colony forming cell assays (LPPC-CFU and HPPC-CFU) with saturating concentrations of multiple hematopoietic cytokines in semisolid medium. While progenitor numbers from Erk1-KO and Erk2-KO marrow were comparable to WT marrow, Erk1/2-KO marrow produced few colonies (Figure 2, A and B). To test the possibility that Erk1/2 disruption induces a differentiation block (but permits proliferation), single, primitive HSPCs (CD150+CD48–41–lin–sca1+) were sorted into cytokine-enriched methylcellulose in a single well of a 96-well plate. Erk1/2-KO cells produced no colonies (Figure 2C) or appreciable cell proliferation on Hoechst staining of individual wells (not shown), suggesting, in conjunction with LPPC/HPPC data, that HSPC proliferation in these assays requires Erk1/2.
Figure 2. Granulocytopoiesis and monocytopoiesis require Erk.

(A and B) Erk1/2-KO cells form few colonies in LPPC-HPPC assays. **P < 0.01; ***P < 0.001, Erk1 /2-KO vs. all, 1-way ANOVA with Bonferroni’s correction. (C) Single Erk1/2-KO SLAM-LS cells fail to produce colonies and show no evidence of expansion using Hoechst-based cell detection (not shown). *P < 0.05 Erk1/2-KO vs. all, 1-way ANOVA with Bonferroni’s correction. (D) Lethally irradiated CD45.1/2+ mice received mixed CD45.2+ (Erk mutant) and CD45.1+ (WT) marrow cells at a one-to-one ratio. (E and F) After Cre induction (4 months following transplantation), the WT CD45.2+ recipients demonstrated stable chimerism, but the Erk1/2-KO recipients experienced rapid and progressive loss of CD45.2+ cells, as measured in the peripheral blood (for WT vs. Erk1/2-KO 45.2+ chimerism, data not significant at –0.5 months, P < 0.001 at 1 and 6 months, 2-way ANOVA with Bonferroni’s correction). (G–I) Peripheral blood flow cytometric analyses after Cre induction demonstrate ablated Erk1/2-KO-derived CD45.2+ circulating nonlymphoid cells (CD3–B220–), granulocytes (CD3–B220–Gr1+Mac1+), and monocytes (CD3–B220–Gr1–Mac1+). P < 0.0001, WT vs. Erk1/2-KO, Student’s 2-tailed unpaired t test.
To test marrow-autonomous Erk deletion in vivo, we transplanted CD45.2+ Erk mutant bone marrow cells mixed with isogenic CD45.1+ WT BoyJ cells into lethally irradiated CD45.1/2+ hosts (Figure 2D), a strategy allowing discrimination among donor, competitor, and residual recipient cell populations. Before poly(I:C) injection, Erk2floxflox and Erk1–/–Erk2flox/floxMx1Cre+ (i.e., essentially Erk1-KO) recipients showed similar chimerism (Figure 2, E and F, –0.5 month). However, Erk1/2-KO CD45.2+ chimerism fell dramatically following Cre induction (Figure 2F). In a separate experiment, secondary transplantation of Erk1/2-KO bone marrow accelerated chimerism loss (Supplemental Figure 1A; supplemental material available online with this article; doi: 10.1172/JCI66167DS1). In contrast, mice reconstituted with Erk1-KO or Erk2-KO bone marrow showed long-term stability and production of both myeloid and lymphoid lineages, and mice competitively reconstituted with Mx1Cre+Erk2flox/flox marrow showed no significant chimerism loss following poly(I:C) injection, thus controlling for possible Cre-related alterations (Supplemental Figure 1, B–E).
In the Erk1/2-KO cohort, flow cytometric analyses of the peripheral blood revealed rapid, near-complete ablation of circulating nonlymphoid cells (CD45.2+CD3–B220–), granulocytes (CD45.2+CD3–B220–Gr1+Mac1+), and monocytes (CD45.2+CD3–B220–Gr1–Mac1+) (Figure 2, G–I). As early as 2 weeks after Cre induction, the majority of the few remaining Erk1/2-KO CD45.2+ cells were T or B cells (not shown). However, given the diminution in overall CD45.2+ chimerism, the Erk1/2-KO peripheral blood showed relatively few lymphocytes, concordant with previous lymphocyte-focused studies (12).
Consistent with initial clonogenic assays, Erk1/2-KO bone marrow demonstrated diminution in the frequency and total number of phenotypically defined multipotent and lineage-restricted progenitors (Table 1). Intriguingly, WT and Erk1/2-KO bone marrow contained the same frequency of phenotypically defined primitive HSPCs (SLAM-LSK: CD150+CD48–41–lin–sca1+c-kit+), revealing a possible linkage between HSPC number and bone marrow cellularity, thus reinforcing a hypothesis of impaired proliferation in the Erk1/2-KO HSPCs. Similar to a recent study relying on Erk1/2 interference in keratinocytes (13), Erk1/2-KO HSPCs showed a nucleic acid profile most consistent with G2/M arrest.
Table 1.
Comparison of WT and Erk1/2-KO phenotypically defined progenitor populations
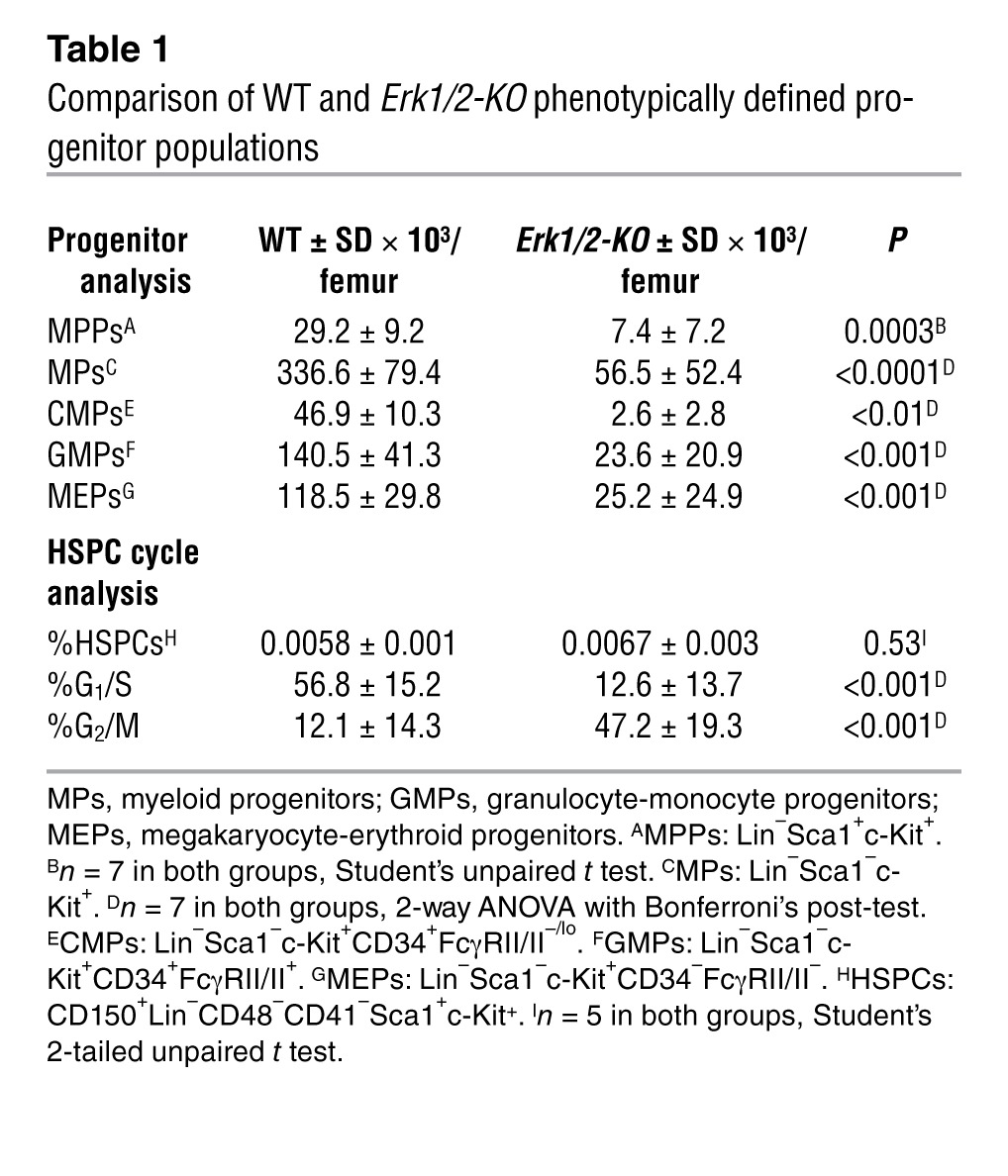
This genetic finding that granulocytopoiesis and monocytopoiesis require Erk clarifies recent conflicting studies reliant on chemical Mek/Erk inhibitors, some arguing Erk promotes monocytopoiesis while suppressing granulocytopoiesis (14, 15), while others showing that Erk directly regulates granulocyte development (10). Another argued that Erk regulates GM colony formation from HSCs/multipotent progenitors (MPPs) but not from common myeloid progenitors (CMPs), implying its dispensability in later precursors (16). In comparison, our in vivo genetic findings indicate a critical role for Erk1/2 in both granulocytopoiesis and monocytopoiesis.
To test whether Nf1 deficiency and consequent constitutive Ras hyperactivation bypass this Erk requirement, we transplanted CD45.1/2+ mice with CD45.1+ WT and CD45.2+ Nf1flox/flox (WT), Mx1Cre+Nf1flox/flox (Nf1-KO), Mx1Cre+Erk1–/–Erk2flox/flox (Erk1/2-KO), or Mx1Cre+Nf1flox/floxErk1–/–Erk2flox/flox (Nf1-Erk1/2-KO) HSCs (Figure 3A). After 4 months, we injected the recipients with poly(I:C). At sacrifice, Nf1-KO recipient mice had enlarged spleens (Figure 3B), demonstrating myeloid cell invasion and disruption of the normal splenic architecture, histological features consistent with MPD (Figure 3C). In contrast, Nf1-Erk1/2-KO recipient mice had a spleen size and architecture indistinguishable from the WT and Erk1/2-KO recipient control groups (Figure 3, B and C).
Figure 3. Nf1-deficient MPD requires Erk.

(A) CD45.2+ bone marrow cells harvested from Mx1Cre+Nf1flox/floxErk1–/–Erk2flox/flox mice were mixed with CD45.1+ WT bone marrow cells and transplanted into lethally irradiated CD45.1/2+ recipients, alongside control groups, as shown. After 4 months, all mice received poly(I:C) and were then killed approximately 6 months later. (B and C) Erk1, Erk2, and Nf1 disruption in the hematopoietic system protects against the phenotype observed in Nf1-KO recipients, which have enlarged spleens with effacement of the regular white pulp cellular architecture. *P < 0.05; **P < 0.01; ***P < 0.001, all vs. Nf1-KO; all other comparisons P > 0.05, 1-way ANOVA with Bonferroni’s correction. Original magnification, ×100 (top panels); ×300 (bottom panels). (D) Similarly, the Nf1-Erk1/2-KO recipients show ablated CD45.2+ populations in the peripheral blood, similar to the Erk1/2-KO cohort. **P < 0.01; ***P < 0.001, all vs. Nf1-KO, 1-way ANOVA with Dunnett’s correction. In comparison, the Nf1-KO recipients show expansion of total CD45.2+ cells, total CD45.2+ myeloid cells, and total CD45.2+ granulocytes in the peripheral blood. (E) In the bone marrow, the Nf1-Erk1/2-KO recipients show few CD45.2+ myeloid cells, including both CD45.2+CD3–B220–Gr1+Mac1+ and CD45.2+CD3–B220–Gr1–Mac+ populations, which contrasts with the increased frequency observed in the Nf1-KO cohort. *P < 0.05; **P < 0.01; ***P < 0.001, all vs. Nf1-KO, 1-way ANOVA with Dunnett’s correction.
Consistent with previous studies, Nf1-KO peripheral blood showed increased total CD45.2+ cells, total CD45.2+ nonlymphoid cells, and total CD45.2+ granulocytes (Figure 3D). Similarly, Nf1-KO bone marrow showed increased frequency of total myeloid cells, granulocyte precursors, and monocyte precursors (Figure 3E). In contrast, Nf1-Erk1/2-KO recipients showed a rapid and sustained loss of total CD45.2+ blood and marrow cells, a finding analogous to Erk1/2-KO recipients (Figure 3, D and E). In short, Nf1-KO recipient myeloid outgrowth resembles JMML, while combined Nf1, Erk1, and Erk2 disruption parallels the Erk1/2 disrupted hematopoietic phenotype.
JMML is a highly aggressive malignancy found in infants and young children with NF1. Biochemically, JMML is characterized by constitutive Ras hyperactivation. In this genetically engineered murine model of MPD resembling JMML, we provide genetic evidence indicating a requirement for Erk1/2 in the disease’s genesis. Importantly, NF1-associated JMML phenotypically resembles MPD observed in patients with other genetic developmental disorders that involve aberrations in Ras signaling, including mutations in the genes encoding PTPN11, NRAS, and KRAS, collectively accounting for up to 90% of all known JMML cases. Thus, this fundamental requirement for Erk signals in Nf1-associated MPD genesis may inform investigations into other hyperactive Ras-dependent hematological malignancies.
This translational study provides strong rationale for pursuing pharmacologic Mek/Erk inhibitors in MPD treatment. Recent preclinical studies have reached contradictory conclusions regarding the efficacy of chemical Mek/Erk inhibition in JMML models. Lauchle et al. found that the Mek inhibitor CI-1040 failed to attenuate Nf1-deficient MPD (7). However, in a related K-ras–dependent model of JMML, a different Mek inhibitor demonstrated short-term in vivo efficacy (17). Though this disparity could result from differences between K-ras mutant and Nf1-deficient myeloid malignancies, it is plausible that the different pharmacokinetics of the 2 tested drugs have an impact on the efficacy of target inhibition. Alternatively, nonselective, off-target effects in one of the drugs may have led to variable efficacy in each model. Clearly, our genetic data in this preclinical model support a hypothesis that Mek inhibitors could efficaciously treat JMML, although our data cannot address the amount of Mek inhibition required for pharmacologic efficacy in the hematopoietic system, especially as clinical Mek inhibitor trials for other malignancies have not revealed profound hematopoietic effects. Regardless, these findings warrant preclinical testing in murine MPD models using newer, orally bioavailable compounds that inhibit Erk1/2 phosphorylation at nanomolar concentrations.
Methods
Mice, genotyping, and Mx1Cre induction.
Previously described Erk1–/– (18) and Erk2flox//flox mice (11) were bred with Mx1Cre transgenic mice, each other, and with Nf1flox/flox mice (19) and genotyped as described (11, 18, 19). In each experiment, all mice (Mx1Cre+ or Mx1Cre–) received 7 i.p. injections of (poly[I:C]; Sigma-Aldrich) dissolved in sterile PBS, using graded doses from 15 to 30 μg/g body weight. Transplanted mice received 6 doses, to approximately 15 μg/g body weight. Erk2 protein levels were detected by Western blot of peripheral blood and/or bone marrow, and Nf1 recombination was detected by PCR of peripheral blood (Supplemental Figure 2).
Marrow isolation and colony assays.
After Cre induction, femoral marrow was flushed and mononuclear cells isolated, as described (3). LPPC-HPPC assays were performed as described (3) (details in Supplemental Methods). For single HSPC colony assays, cells were stained with HSPC surface markers and sorted by FACS (FACS Aria; BD) into a 96-well flat bottom plate containing 100 μl of 1% methylcellulose with 30% FBS, 100 ng/ml SCF, 10 ng/ml IL-3, 10 ng/ml Flt3-ligand, and 4 U/ml EPO (Amgen) and allowed to grow for 10 days in 5% CO2 at 37°C.
Transplantation and blood isolation.
For competitive transplantation, CD45.2+ Erk mutant and CD45.1+ WT BoyJ marrow cells were mixed at indicated ratios and injected i.v. into lethally irradiated (1100 cGy, split dose) CD45.1/2+ WT mice, each receiving 2 × 106 cells in 200 μl PBS. At indicated times after transplantation, peripheral white blood cells were isolated by incubating (15 minutes, 23°C) 50–100 μl of blood extracted from the tail veins of transplant recipients in rbc lysis buffer (QIAGEN), followed by washing twice in PBS.
Flow cytometry.
Peripheral white blood cells or bone marrow cells were incubated at 4°C for 45 minutes with saturating concentrations of anti-mouse antibodies in approximately 100 μl of 3% FBS/0.09% NaN3 in PBS with 0.25 μg anti-mouse CD16/CD32 (Fc Block). All antibodies were purchased from BD Biosciences or eBiosciences (see Supplemental Methods). Data were acquired on an LSR II 407 flow cytometer outfitted with red 633 nm, 488 nm, and 407 nm lasers. Single-color compensation controls were obtained during each experiment using polystyrene microbeads (BD Biosciences). Compensation matrices were calculated and analyses performed with FlowJo 7.6.3 software (TreeStar). Gates were determined using fluorescence minus-one controls. Cell-cycle analysis staining protocol can be found in the Supplemental Methods.
Statistics.
All analyses were performed with GraphPad Prism 5.0. Data represent mean ± SEM. One- or 2-way ANOVA was performed, as appropriate, with Bonferroni’s or Dunnett’s post-hoc corrections. For 2 variable comparisons, unpaired 2-tailed Student’s t tests were used. Specific tests and significance levels are found in figure legends. P < 0.05 was considered significant.
Study approval.
Mice were maintained at Indiana University School of Medicine according to the Institutional Animal Care and Use Committee and Institutional Review Board guidelines. The IACUC and the IRB approved all animal methods used in this study.
Supplementary Material
Acknowledgments
This work was supported, in part, by NIH-NCI/RO1 CA074177-11A1/D and P50 NS052606-04 (to D.W. Clapp), with additional support by predoctoral fellowships from the Howard Hughes Medical Institute and NIH T32 CA111198 (to K. Staser) and a KL2 TR000163 award from the NIH/NCATS (to G. Nalepa). We also thank Julie Mund, Eddie Srour, and Jamie Case, and the Indiana University School of Medicine Flow Cytometry Facility for invaluable cytometry assistance. We also thank Merv Yoder for critiquing our manuscript.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(1):329–334. doi:10.1172/JCI66167.
References
- 1.Side L, et al. Homozygous inactivation of the NF1 gene in bone marrow cells from children with neurofibromatosis type 1 and malignant myeloid disorders. N Engl J Med. 1997;336(24):1713–1720. doi: 10.1056/NEJM199706123362404. [DOI] [PubMed] [Google Scholar]
- 2.Ingram DA, Wenning MJ, Shannon K, Clapp DW. Leukemic potential of doubly mutant Nf1 and Wv hematopoietic cells. Blood. 2003;101(5):1984–1986. doi: 10.1182/blood-2002-08-2635. [DOI] [PubMed] [Google Scholar]
- 3.Zhang YY, et al. Nf1 regulates hematopoietic progenitor cell growth and ras signaling in response to multiple cytokines. J Exp Med. 1998;187(11):1893–1902. doi: 10.1084/jem.187.11.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Birnbaum RA, et al. Nf1 and Gmcsf interact in myeloid leukemogenesis. Mol Cell. 2000;5(1):189–195. doi: 10.1016/S1097-2765(00)80415-3. [DOI] [PubMed] [Google Scholar]
- 5.Kim A, et al. Beta common receptor inactivation attenuates myeloproliferative disease in Nf1 mutant mice. Blood. 2007;109(4):1687–1691. doi: 10.1182/blood-2006-05-025395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bourne HR, Sanders DA, McCormick F. The GTPase superfamily: a conserved switch for diverse cell functions. Nature. 1990;348(6297):125–132. doi: 10.1038/348125a0. [DOI] [PubMed] [Google Scholar]
- 7.Lauchle JO, et al. Response and resistance to MEK inhibition in leukaemias initiated by hyperactive Ras. Nature. 2009;461(7262):411–414. doi: 10.1038/nature08279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bollag G, et al. Loss of NF1 results in activation of the Ras signaling pathway and leads to aberrant growth in haematopoietic cells. Nat Genet. 1996;12(2):144–148. doi: 10.1038/ng0296-144. [DOI] [PubMed] [Google Scholar]
- 9.Chung E, Hsu CL, Kondo M. Constitutive MAP kinase activation in hematopoietic stem cells induces a myeloproliferative disorder. PLoS One. 2011;6(12):e28350. doi: 10.1371/journal.pone.0028350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Geest CR, Coffer PJ. MAPK signaling pathways in the regulation of hematopoiesis. J Leukoc Biol. 2009;86(2):237–250. doi: 10.1189/jlb.0209097. [DOI] [PubMed] [Google Scholar]
- 11.Fischer AM, Katayama CD, Pages G, Pouyssegur J, Hedrick SM. The role of erk1 and erk2 in multiple stages of T cell development. Immunity. 2005;23(4):431–443. doi: 10.1016/j.immuni.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 12.Yasuda T, et al. Erk kinases link pre-B cell receptor signaling to transcriptional events required for early B cell expansion. Immunity. 2008;28(4):499–508. doi: 10.1016/j.immuni.2008.02.015. [DOI] [PubMed] [Google Scholar]
- 13.Dumesic PA, Scholl FA, Barragan DI, Khavari PA. Erk1/2 MAP kinases are required for epidermal G2/M progression. J Cell Biol. 2009;185(3):409–422. doi: 10.1083/jcb.200804038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jack GD, Zhang L, Friedman AD. M-CSF elevates c-Fos and phospho-C/EBPalpha(S21) via ERK whereas G-CSF stimulates SHP2 phosphorylation in marrow progenitors to contribute to myeloid lineage specification. Blood. 2009;114(10):2172–2180. doi: 10.1182/blood-2008-11-191536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ross SE, et al. Phosphorylation of C/EBPalpha inhibits granulopoiesis. Mol Cell Biol. 2004;24(2):675–686. doi: 10.1128/MCB.24.2.675-686.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hsu CL, Kikuchi K, Kondo M. Activation of mitogen-activated protein kinase kinase (MEK)/extracellular signal regulated kinase (ERK) signaling pathway is involved in myeloid lineage commitment. Blood. 2007;110(5):1420–1428. doi: 10.1182/blood-2007-02-071761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lyubynska N, et al. A MEK inhibitor abrogates myeloproliferative disease in Kras mutant mice. Sci Transl Med. 2011;3(76):76ra27. doi: 10.1126/scitranslmed.3001069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pages G, et al. Defective thymocyte maturation in p44 MAP kinase (Erk 1) knockout mice. Science. 1999;286(5443):1374–1377. doi: 10.1126/science.286.5443.1374. [DOI] [PubMed] [Google Scholar]
- 19.Zhu Y, Ghosh P, Charnay P, Burns DK, Parada LF. Neurofibromas in NF1: Schwann cell origin and role of tumor environment. Science. 2002;296(5569):920–922. doi: 10.1126/science.1068452. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.