

Recent evidence suggests that glycogen synthase kinase 3s (GSK 3s) and their upstream and downstream regulators have key roles in many fundamental processes during neurodevelopment. Disruption of GSK3 signaling adversely affects brain development and is associated with several neurodevelopmental disorders. Here, we discuss the mechanisms by which GSK3 activity is regulated in the nervous system and provide an overview of the recent advances in understanding how GSK3 signaling controls neurogenesis, neuronal polarization, and axon growth during brain development. These studies suggest GSK3 as a major signaling node that mediate multiple signaling molecules regulating neurodevelopment, such as DISC1, Par3/6, and Wnts.
Glycogen synthase kinase 3s are serine/threonine kinases that were originally identified as key regulatory enzymes in glucose metabolism1, 2. There are two isoforms, GSK3α and GSK3β, encoded by separate genes, which are overall 85% homologous to each other, with 95% identity in the kinase domains3. In rodents and humans GSK3β2, an alternative splice variant of GSK3β has been reported, which contains a 13 amino acid insertion in an external loop near the catalytic domain4. In contrast to the ubiquitously expressed GSK3β1, GSK3β2 is expressed specifically in the nervous system, with the highest levels found during development4. Interestingly, recent studies suggest that GSK3β2 plays a specific part in neuronal morphogenesis in vitro5, 6, and it remains to be determined whether different isoforms have specific functions in vivo during brain development. Since their discovery, GSK3s have been shown to mediate various signaling pathways, among which the growth factor and Wnt signaling pathways are the most studied7. Consistent with the roles of growth factors and Wnt proteins in the nervous system, especially during neurodevelopment, emerging evidence points to GSK3s as key regulators in multiple neurodevelopmental processes, including neurogenesis, neuronal migration, neuronal polarization and axon growth and guidance.
How do GSK3s regulate such a wide spectrum of developmental events? The answer may lie in the broad range of GSK3 substrates. Among GSK3 substrates are many transcription factors, such as cAMP response element-binding protein (CREB)8, nuclear factor of activated T-cells (NFAT)9, 10, neurogenin 211, Smad112, c-Jun13, and β-catenin14, all of which play important parts in the regulation of gene expression throughout neurodevelopment. GSK3s regulate these transcription factors by controlling their protein levels, DNA binding activities, and/or nuclear localization. In addition to gene expression, cell morphogenesis requires reorganization of the cytoskeleton, especially microtubules. GSK3s regulate the activity of several microtubule-associated proteins (MAPs)15, and so might control mitotic spindle reorganization during cell division, coordinated movement of the leading process and soma during neuronal migration, and directed growth cone advancement during axon growth and guidance, all of which require coordinated control of microtubule dynamics. Changes in GSK3 activity have been associated with many psychiatric and neurodegenerative diseases, such as Alzheimer’s disease, schizophrenia, and autism spectrum disorders, and it has become increasingly apparent that GSK3 might be a common therapeutic target for different classes of psychiatric drugs16, 17. Indeed, lithium, a direct inhibitor of GSK318, has been used in humans as a mood stabilizer for over 50 years19. The hypothesis that disturbances of brain development play a part in the etiology of these disorders is further supported by the fact that several genetic susceptibility factors for psychiatric disorders have key roles in neurodevelopment. Intriguingly, many of the genes associated with schizophrenia encode proteins involved in GSK3 signaling, such as Disrupted-In-Schizophrenia 1 (DISC1), Neuregulin 1, and Frizzled-320. Genes associated with autism spectrum disorders also encode proteins that are involved in the modulation of or that are affected by GSK3 activity, including phosphatase and tensin homolog deleted on chromosome 10 (PTEN)21, DISC122, serotonin23, tuberous sclerosis complex 1/2 (TSC1/2)24, and adenomatous polyposis coli (APC)25, 26. Therefore, a better understanding of the role of GSK3 in neurodevelopment could provide insight into the etiology of these disorders and possibly open up the potential of a new library of therapeutic targets.
In this Review, we provide an overview of the involvement of GSK3 signaling in neurodevelopment, with a particular emphasis on neurogenesis, neuronal polarization, and axon growth. We will also discuss the potential crosstalk between GSK3 signaling and other pathways that are implicated in these developmental steps.
Regulation of GSK3 activity in the CNS
GSK3 is an unusual kinase, in the sense that it has a high basal activity in resting cells and is inactivated by upstream regulators in response to stimuli. Several signaling pathways regulate GSK3 activity (Fig. 1). Activation of the phosphatidylinositol 3-kinase (PI3K) pathway downstream of receptor tyrosine kinase signaling is thought to result in inactivation of GSK3 through phosphorylation of the N-terminal serine residue (S9 in GSK3β and S21 in GSK3α). It is widely accepted that AKT is the major mediator of this serine phosphorylation and subsequent inactivation of GSK3 (Fig. 1)27, 28. However, mutant mice (GSK3α-S21A/GSK3β-S9A double knock-in mice) with GSK3s that cannot be phosphorylated at their N-termini developed normally with no overt phenotype in the nervous system (although insulin-induced activation of glycogen synthase was disrupted in the muscle)29. This suggests that alternative pathways might regulate GSK3 activity, especially in the nervous system. Indeed, a recent study30 showed that p38 MAPK regulates the activity of GSK3β, but not GSK3α, by inducing an inhibitory phosphorylation on S389 (Fig. 1). However, p38 MAPK-mediated phosphorylation is insufficient to provide a regulatory mechanism of GSK3 in the nervous system because both isoforms of GSK3 are required for normal development31.
Figure 1. Proposed models of glycogen synthase kinase (GSK) 3 inactivation.
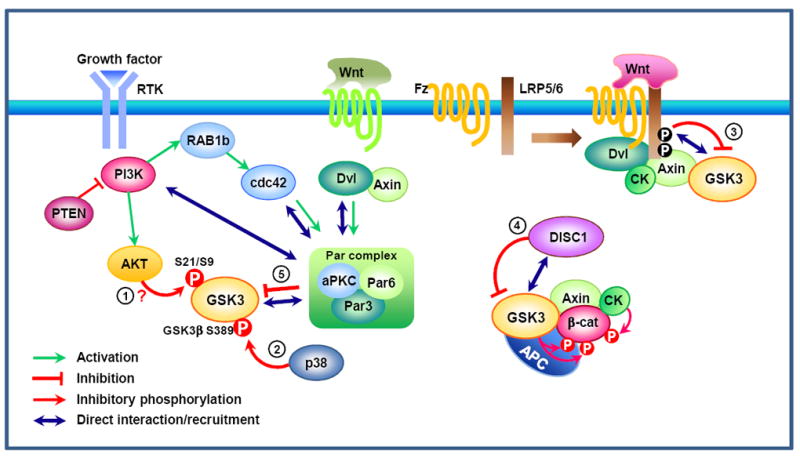
Upon activation of the phosphatidylinositol 3-kinase (PI3K) pathway downstream of receptor tyrosine kinase (RTK) signaling, AKT is thought to be the major kinase that inactivates GSK3 via phosphorylation of the N-terminal serine residue (S21 in GSK3α and S9 in GSK3β) (1). Phosphorylation of GSK3β (but not GSK3α) at S389 by p38 mitogen-activated protein kinase (MAPK) might be an alternative mechanism for inactivation30 (2). GSK3 regulation in the Wnt pathway is distinct from the way by which RTK signaling inhibits GSK3. In the canonical Wnt pathway, Wnt signaling recruits the destruction complex to the membrane, leading to the phosphorylation of the cytoplasmic tail of low-density lipoprotein receptor-related protein (LRP) 5/6 by casein kinase (CK) and GSK3β. Phosphorylated PPPSPXS motifs in the LRP5/6 intracellular domain directly inhibit GSK3’s activity towards β-catenin (3) 32, 33. In neuronal progenitor cells, a recent study showed that Disrupted in schizophrenia (DISC) 1 directly interacts with GSK3 and prevents it from phosphorylating β-catenin (4)22. Both in the Wnt37 and the RTK signaling35 pathways, GSK3 activity is also regulated by polarity proteins (5). GSK3 interacts with the Par complex — composed of Par3, Par6, and atypical protein kinase C (aPKC) — and so becomes phosphorylated and inactivated. Although phosphorylation of GSK3 occurs as a consequence, GSK3 phosphorylation is not required for its inactivation37. β-cat, β-catenin; Dvl, dishevelled; Fz, Frizzled; PTEN, phosphatase and tensin homologue deleted on chromosome 10; RAP1b, Ras-related protein 1b.
In the canonical Wnt pathway, Wnt-induced dissociation of GSK3 from its substrate β-catenin leads to the stabilization and activation of β-catenin. Upon Wnt stimulation, GSK3 is recruited to the membrane, where PPPSPXS motifs in the co-receptor low-density lipoprotein receptor-related protein (LRP) 5/6 associate with and inhibit GSK3 activity towards β-catenin (Fig. 1)32, 33. Regulation of GSK3 by LRP5/6 is achieved via protein-protein interaction, which sequesters GSK3 away from its substrate without affecting the kinase activity per se. Regulation of GSK3 activity towards a particular substrate rather than general inhibition of its kinase activity per se is also exemplified in a recent finding22, which suggested that by physically interacting with GSK3 DISC1 prevents GSK3 from phosphorylating β-catenin.
Both in response to Wnt34 and growth factors35 GSK3 activity is regulated by a conserved cell polarity pathway involving Cdc42-Par3/6-PKCς (Fig. 1). The regulation of GSK3 activity by the Par complex was first described in directed cell migration36. Specifically, localized activation of the small GTPase Cdc42 at the leading edge recruits the Par3/Par6/PKCς complex, which interacts with GSK3β, leading to its phosphorylation and inactivation36. Although GSK3 phosphorylation occurs as a consequence, PKCς is unable to directly phosphorylate GSK327 and a study using fibroblasts derived from GSK3α-S21A/GSK3β-S9A double knock-in mice37 demonstrated that GSK3 phosphorylation is not required for the Par3/6-mediated GSK3 inhibition. Instead, dishevelled (Dvl) and axin have been suggested to mediate GSK3 inhibition through this pathway by inducing a physical association between Dvl and PKCς during directed cell migration (Fig. 1). Interestingly, a recent study38 shows that Par3 directly binds PI3K and enhances its activity, providing an alternative explanation for the increased GSK3 phosphorylation downstream of Par3. Together, these lines of evidence suggest protein-protein interaction as a recurrent theme in the regulation of GSK3 activity, which in many cases does not require changes in the phosphorylation status of GSK3 (Box 1).
Box 1. Current methods used to detect GSK3 activity.
The most widely used method to establish GSK3 activity is to examine S9/S21 phosphorylation. However, results from GSK3α-S21A/GSK3β-S9A double knock-in mice29 raise the question of whether this serine phosphorylation of GSK3 is the major regulatory mechanism in the CNS. More importantly, it is becoming increasingly apparent that changes in GSK3 activity are not always accompanied by changes in their phosphorylation status. Thus, serine phosphorylation of GSK3 might not be used as the sole indicator of GSK3 activity.
The second approach is the in vitro kinase assay. Endogenous GSK3 can be immunoprecipitated from cell lysates to detect its activity towards its substrates. However, because the interaction of GSK3 with their regulators in cells might be transient and only a small percentage of total GSK3 in cells interacts with a particular regulator118, it is possible that the changes in GSK3 activity are not preserved in the in vitro kinase assay or are less likely to be detected.
The third approach is to examine the tyrosine phosphorylation in GSK3 (Y279 in GSK3α and Y216 in GSK3β), which facilitates its activity by promoting substrate accessibility119. Although there is evidence for signal-dependent regulation of tyrosine phosphorylation22, 120, 121, tyrosine phosphorylation of GSK3 is thought to occur through a post-translational and intramolecular autophosphorylation event119, 121, which in many cases is not dynamically regulated upon external stimuli.
The last approach is to examine changes in the phosphorylation status of known GSK3 substrates. Because GSK3 might not have equal access to all potential substrates in vivo owing to subcellular localization, this method allows us to detect more physiologically relevant changes in GSK3 activity. However, when this method is applied to assess GSK3 activation/inactivation in a pathway that does not have known GSK3 substrates, one should consider multiple potential substrates that can represent a diverse family of proteins, for instance, representing primed versus unprimed substrates with different subcellular localizations.
GSK3 signaling in neurogenesis
Recent evidence22, 39, 40 suggests that GSK3 signaling is essential for the coordination of progenitor proliferation and differentiation during brain development. Because the developmental process of mouse neocortex (Fig. 2) has been well-documented, we will use it as a model system to discuss the role of GSK3 signaling in neural development.
Figure 2. Neural development of the mammalian neocortex.
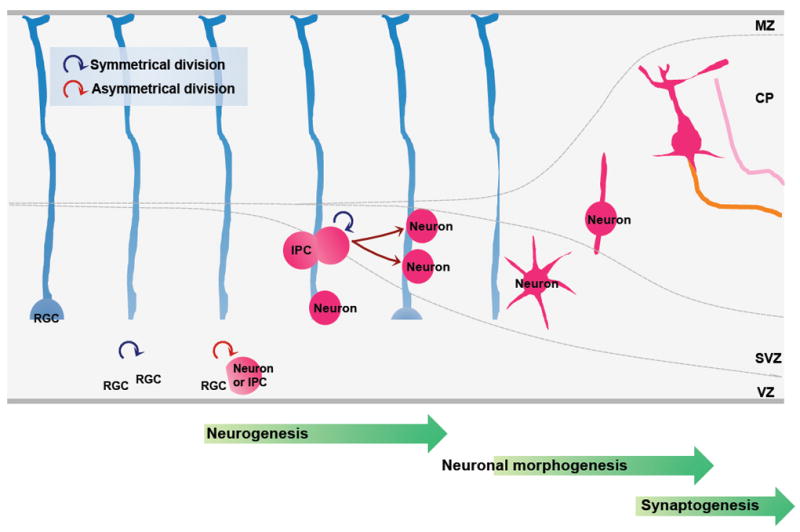
The formation of neural circuits during development involves the coordination of multiple cellular events, which can be divided into three steps. The first step is neurogenesis, during which a cohort of neural progenitors undergoes cycles of proliferation and differentiation to generate neurons in a highly regulated manner. The second step is neuronal morphogenesis, during which differentiated neurons migrate towards their final destination. While migrating, these neurons polarize to form axons and dendrites, which then grow and reach their specific target fields under the control of a myriad of guidance cues. The third step is synaptogenesis, during which axons cease growing and form synapses with their innervating targets.
Radial glial cells (RGS) are progenitors that generate the majority of neurons in the developing neocortex, either directly or indirectly, by undergoing two phases of coordinated cell division. In the first, proliferative phase, which occurs early in development, they mainly divide symmetrically in the ventricular zone (VZ) to generate two similar progenitor cells, which can further self-renew to expand the progenitor pool. In the second, neurogenic phase, most of the radial glial cells divide asymmetrically to generate one self-renewing progenitor and one post-mitotic neuron or one intermediate progenitor cells (IPCs). The IPCs do not self-renew but migrate to the subventricular zone (SVZ) and divide symmetrically to form neurons. At the end of neurogenesis, some radial glial cells undergo a terminal symmetrical division to generate two neurons. The transition of progenitors from symmetrical to asymmetrical division and the asymmetrical division per se are tightly regulated to produce the final total number of neurons. Perturbation in any of these processes will cause defects in neurogenesis and cortical development. MZ, marginal zone; CP, cortical plate.
GSK3 inactivation promotes progenitor proliferation
A recent study39 revealed a pivotal role of GSK3 signaling in the developing nervous system by selectively deleting both Gsk3α and Gsk3β in neural progenitors. The double knock-out mice had a significantly increased cortical surface area with a convoluted shape, due to an over-expansion of the neural progenitor cells. However, the cortex was thinner than that of control littermates, indicating a reduced number of neurons. Indeed, further analysis using various markers of progenitors, intermediate progenitor cells (IPCs) and post-mitotic neurons showed that deletion of GSK3s markedly enhanced the proliferation of progenitor cells while suppressing neuronal differentiation, indicating that spatiotemporal regulation of GSK3 activity is required for appropriate transition from the proliferative to the neurogenic phase during brain development.
Various signaling pathways have been identified to regulate neural progenitor proliferation, including Wnt41, sonic hedgehog (Shh)42, fibroblast growth factor (FGF)43, and Notch signaling pathway44. Intriguingly, GSK3 is implicated in the regulation of each of these pathways. In the canonical Wnt pathway, inhibition of GSK3 is crucial for the stabilization and nuclear translocation of β-catenin that leads to the subsequent activation of the T-cell factor (TCF) 4-dependent gene transcription. As discussed above, although the regulatory mechanism is not clear, GSK3 is a well-known downstream effector of PI3K, which provides an explanation for the disruption of FGF signaling in GSK3-deleted brains39. The mechanism by which Shh signaling was elevated in Gsk3-deleted brains39 is unclear, but a study in Drosophila45 suggests that GSK3 controls the stability of key transcriptional effectors (Ci/Gli proteins) in the Hedgehog pathway in a similar way as it regulates β-catenin in the Wnt pathway. There are contradicting results of how GSK3 regulates Notch signaling. In one study46 Notch protein was stabilized by GSK3-mediated phosphorylation46, whereas in another study the transcriptional activity of Notch was enhanced by inhibition of GSK347. However, the fact that Notch signaling was hyperactivated in Gsk3-deficient brains39 suggests that GSK3 negatively regulates Notch signaling in neural progenitors. Together, these lines of evidence suggest that GSK3 might function as a node molecule of multiple signaling pathways and thereby coordinate the proliferation and differentiation of neural progenitors.
The role of GSK3 signaling in neurogenesis is further supported by two recent studies22, 40, in which DISC1 or Par3 — upstream regulators of GSK3 — were manipulated in neural progenitors. Ectopic expression of either DISC1 or Par3 increased progenitor proliferation and suppressed neuronal differentiation, reminiscent of Gsk3 knock-out mice. One study22 showed that DISC1 directly interacts with GSK3, which seemed to prevent GSK3 from phosphorylating β-catenin and that over-expression of DISC1 led to the activation of canonical Wnt signaling. The other study40 did not directly test the involvement of GSK3 but showed that over-expression of Par3 led to the activation of Notch signaling, which was also observed in Gsk3 knock-out mice39. As Wnt and Notch signaling independently promote the proliferation of progenitors in Gsk3 knock-out mice39, it could be suggested that DISC1 and Par3 regulate GSK3 activity in the Wnt and the Notch pathway, respectively, allowing pathway-specific control of GSK3. These lines of evidence suggest that inhibition of GSK3 signaling, by either deleting Gsk3s39 or over-expressing their negative regulators22, 40, promotes the proliferation of progenitor cells while suppressing their differentiation into neurons.
GSK3 activation promotes neuronal differentiation
Kim et al.39 found that phosphorylation levels of c-Myc and β-catenin, targets of GSK3, were increased at later stages in development when progenitor proliferation is subsiding and neuronal differentiation predominates. Because both c-Myc48 and β-catenin are pro-proliferation factors and phosphorylation by GSK3 leads to their degradation, this finding suggests that activation of GSK3 promotes neuronal differentiation. Supporting this notion, Disc1 knockdown in neural progenitors caused premature neuronal differentiation at the expense of the size of the progenitor pool22. When GSK3β signaling was examined, the levels of phosphorylated β-catenin and tyrosine-phosphorylated GSK3β, which is associated with increased kinase activity (see Box 1), were increased22. Moreover, it was shown that pharmacological inhibition of GSK3 prevented the progenitor proliferation defects induced by Disc1 knockdown22. These results suggest that the premature differentiation observed in DISC1-depleted cells might have been due to activation of GSK3 signaling, although future studies of Disc1 knock-out mice are needed to confirm this conclusion. Depletion of Par3 also increases the neuronal population with a concomitant decrease in the number of progenitor cells40. Future studies are required to determine if this defect is also attributed to GSK3 activation.
As mentioned above, GSK3 has high basal activity in resting cells, and signaling is usually initiated by GSK3 inactivation in response to extracellular factors. However, lysophosphatidic acid (LPA) appears to increase GSK3 activity above basal levels, and that this activation is in part mediated by the small GTPase RhoA49. Consistent with the idea that GSK3 activation promotes neuronal differentiation, treatment of ex vivo cultured cortical hemispheres with LPA promoted terminal mitosis of neural progenitors, leading to their differentiation into neurons50. Moreover, a recent study51 showed that Lfc, a guanine nucleotide exchange factor of RhoA, promotes neuronal differentiation51. Knocking down Lfc, which inhibits Rho activation, increased the pool of radial glial progenitors and prevented neurogenesis, whereas genetic silencing of Tctex-1, a negative regulator of Lfc, had the opposite effect51. Although these results are consistent with the hypothesis that activation and inactivation of GSK3 promote neuronal differentiation and progenitor proliferation, respectively, it remains to be determined if these effects are indeed mediated by regulation of GSK3 activity.
The conventional view is that GSK3 is constitutively active in resting cells. If this is the case, neural progenitors would undergo neuronal differentiation without any additional stimuli. However, a recent study52 showed that an extracellular cue, retinoic acid derived from forebrain meninges, is required for neuronal differentiation52. Interestingly, it has been shown that retinoic acid decreases S9 phosphorylation of GSK3β53 and increases GSK3 expression5, raising the intriguing possibility that retinoic acid signaling leads to the elevation of GSK3 activity above basal levels to control neurogenesis. Supporting this hypothesis, Par3 was significantly upregulated in neural progenitors in the absence of retinoic acid52. Collectively, these studies provide some indirect evidence that GSK3 activation might also be regulated by extracellular cues during neuronal differentiation. The most direct way to test if an increase in GSK3 activity promotes neuronal differentiation would be to over-express GSK3 in the developing brain. Unfortunately, the currently available GSK3 transgenic mice over-express GSK3β under the control of the Thy-1 promoter, which is specifically expressed in post-mitotic neurons54, rendering them unsuitable for examining the role of GSK3 in neurogenesis. A mouse model that over-expresses GSK3 specifically in neural progenitors is needed to test this idea.
GSK3 controls neurogenesis via coordinated regulation of protein degradation and microtubule reorganization
Emerging evidence suggests that GSK3 regulates the stability of a wide range of proteins via activation of the ubiquitin-proteasome system (UPS)55, 56. Thus, one plausible mechanism by which GSK3 regulates neurogenesis is by controlling the levels of molecules involved in neurogenesis. Prominent candidates are transcriptional regulators, such as β-catenin in the Wnt pathway, Gli in the Shh pathway, and c-Myc in the FGF pathway39, 45, 48, all of which control progenitor proliferation through regulation of gene transcription. Supporting this hypothesis, during mitotic cell division GSK3-phosphorylated β-catenin, which is destined for degradation, is inherited by only one daughter cell57. Thus, it is possible that during asymmetrical division of radial glial cells, two daughter cells have different GSK3 activity owing to asymmetrical segregation of upstream GSK3 regulators (e.g. Par3). The daughter cell with lower GSK3 activity will accumulate β-catenin (and perhaps other pro-proliferative proteins) and thus remains a progenitor. Conversely, the daughter cell with higher GSK3 activity will degrade these proteins through activation of the UPS and differentiate into either a neuron or an IPC (Fig. 3).
Figure 3. Proposed model for the role of GSK3 signaling during neurogenesis.
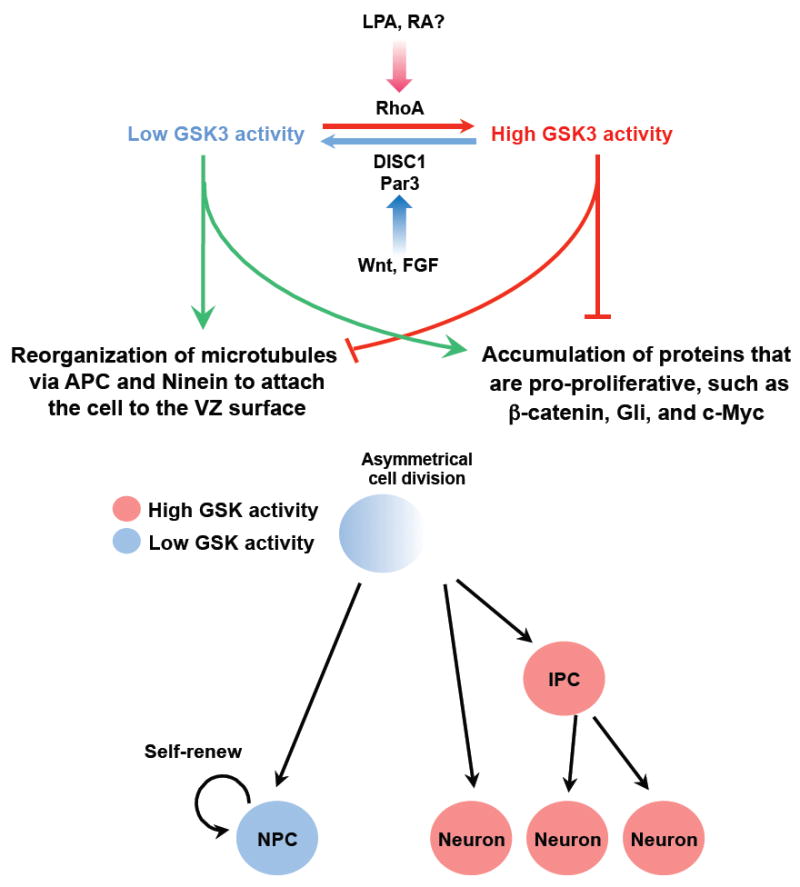
During asymmetrical division of radial glial cells, the daughter cells may have different GSK3 activities via asymmetrical inheritance of upstream regulators (e. g. Par336, Disrupted in schizophrenia (DISC1)22, or Rho49) under the control of extracellular factors (e. g. Wnt, fibroblast growth factor (FGF), lysophosphatidic acid (LPA50)50, retinoic acid (RA52)52). The daughter cell with lower GSK3 activity accumulates pro-proliferation factors39, such as β-catenin, Gli, and c-Myc. Microtubule-associated proteins adenomatosis polyposis coli (APC) and Ninein organize the astral microtubules to attach the cell to the ventricular zone (VZ) surface, and the cell adopts a progenitor fate. Conversely, the daughter cell with higher GSK3 activity removes pro-proliferative proteins and other proteins involved in microtubule assembly such as Ninein, presumably via the ubiquitin-proteasome system (UPS) mediated protein degradation. In addition, APC is unable to bind to the microtubules, disrupting the function of astral microtubules. As a result, the cell detaches from the VZ surface and become a neuron or an intermediate progenitor cell (IPC).
In addition to proteins regulating gene transcription, GSK3 could also regulate other molecules that are important for controlling proliferation or differentiation. A recent study58 has shown that during asymmetrical division of radial glial progenitors, daughter cells inheriting the centrosome that contains the new centriole migrate away from the ventricular zone (VZ) and differentiate into neurons. By contrast, daughter cells inheriting the centrosome that contains the original, mature centriole retain the capacity to self-renew and remain in the VZ. Removal of Ninein, which is mainly localized in the mature centriole, disrupts the asymmetric segregation of the centrosome and promotes neuronal differentiation. Intriguingly, Ninein is a substrate of GSK359-61 and its level is regulated by the UPS62. Therefore, during asymmetrical division Ninein would be accumulated in the daughter cell with low GSK3 activity. As Ninein is a microtubule minus end-anchoring protein involved in the formation of astral microtubules (which anchor cells to their niches) daughter cells with Ninein in their centrosome are more likely to be anchored to the apical surface of VZ and inherit a progenitor fate. On the other hand, daughter cells with high GSK3 activity will have little Ninein in their centrosome, leading to their detachment from the VZ surface and becoming neurons or IPCs.
Another major class of GSK3 substrates consists of MAPs, which control several aspects of microtubule dynamics. APC is not only a component of the Wnt pathway that regulates β-catenin stability but also a MAP that belongs to a family of microtubule plus end-binding proteins (+TIPs)63. When GSK3 activity is inhibited, APC binds to microtubule plus end and there, it anchors the spindle microtubules to the kinetochore or the astral microtubules to the cell cortex, both of which are important for (symmetrical and asymmetrical) cell division. APC has a key role in asymmetrical stem cell division by anchoring one daughter cell in the specific niche so that it retains self-renewal capacity64. In the Wnt signaling pathway, APC interacts with GSK3 to degrade β-catenin, and, similar to Gsk3 deletion, loss of APC leads to stabilization of β-catenin and subsequent activation of TCF465. However, in contrast to Gsk3 knock-out, removal of Apc in progenitors results in reduced rather than increased proliferation, which is probably due to the role of APC in regulating microtubules during cell division.
In summary, GSK3 signaling is emerging as a key regulator of neurogenesis; through its role in the regulation of protein stability and microtubule assembly, it coordinates proliferation and differentiation (Fig. 3).
GSK3 signaling in neuronal polarization
Studies in cultured hippocampal and cortical neurons have identified a number of molecules involved in neuronal polarization. These proteins, which include PI3K, AKT, GSK3, the small GTPases Rac1 and Cdc42, and the Par polarity complex66-76, might act in distinct neuronal populations at specific stages of embryonic development, but it is also possible that they form a network that controls neural development in a coordinated manner. In this regard, GSK3 is an appealing candidate ‘coordinator’ because it can respond to and integrate upstream signals, and downstream, it can phosphorylate various substrates involved in a wide array of cellular activities77. Below we discuss the evidence for a role of GSK3 signaling in neuronal polarization and explore the potential crosstalk of GSK3 signaling with other pathways involved in polarization (Fig. 4).
Figure 4. GSK3 in the regulation of neuronal polarization.
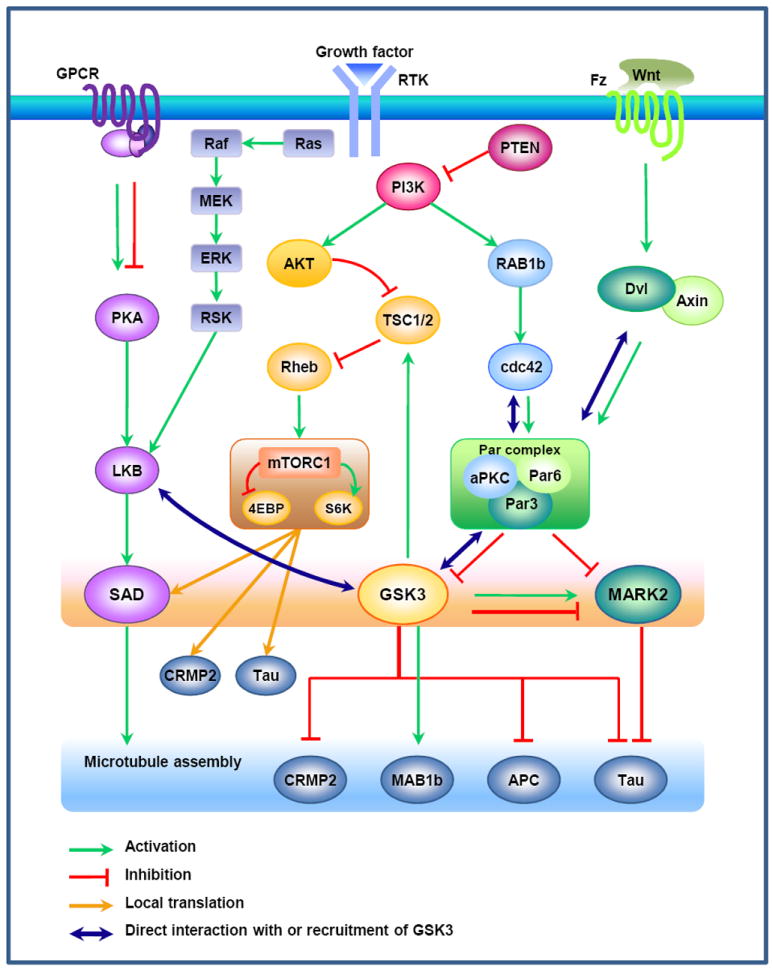
Local activation of phosphatidylinositol 3-kinase (PI3K) at the tip of the nascent axon is thought to function as a landmark for the induction of neuronal polarization. PI3K-mediated production of phosphatidylinositol-3,4,5-trisphosphate (PIP3) leads to the activation of AKT and Ras-related protein 1b (RAP1b). RAP1b activates Cdc42, which recruits and activates the Par3/Par6/atypical protein kinase C (aPKC) complex, leading to the inactivation of GSK336, 37. Microtubule affinity-regulating kinase (MARK) 2 has also been suggested to function downstream of the Par complex to control neuronal polarization68. aPKC in the Par complex associates with Dvl and mediates the Wnt5a-induced differentiation of axons94. On the other hand, activation of AKT induces inhibitory phosphorylation of tuberous sclerosis complex (TSC) 2, by which its GTPase activating protein (GAP) activity towards Rheb is reduced, subsequently leading to the activation of mammalian target of rapamycin (mTOR) signaling. Although phosphorylation of TSC2 by AKT inhibits TSC1/2 activity, phosphorylation of TSC2 by GSK3 has an opposite effect107. Thus, inhibition of GSK3 by PI3K or Wnt signaling would reduce GSK3-dependent stimulatory phosphorylation of TSC2 and thereby increase Rheb-GTP level, which in turn leads to the activation of mTOR-mediated translation. Activation of mTOR signaling in the axon induces local translation of collapsin response mediator protein (CRMP) 2 and Tau, substrates of GSK3106. Activation of LKB1 by protein kinase A (PKA) or ribosomal S6 kinase (RSK) leads to the activation of SAD kinases, which in turn phosphorylate MAPs such as Tau67, 72. The MAPs that are phosphorylated in response to LKB1-SAD kinase signaling to control neuronal polarization remain to be determined. A study in Xenopus shows that LKB1/XEEK1 physically associates with GSK3 and regulates its activity99. Together with GSK3, MARK2 and SAD control microtubule assembly and dynamics by regulating the phosphorylation status of several MAPs. APC, adenomatosis polyposis coli; Dvl, dishevelled; 4EBP, eukaryotic translation initiation factor 4E-binding protein; ERK, extracellular signal regulated kinase; Fz, frizzled; GPCR, G protein-coupled receptor; MAP1b, microtubule-associated protein 1b; MEK, mitogen-activated protein kinase kinase; PTEN, phosphatase and tensin homologue deleted on chromosome 10; RTK, receptor tyrosine kinase; S6K, protein S6 kinase
GSK3 regulates neuronal polarization by controlling microtubule dynamics
Several lines of evidence point to GSK3 as an essential regulator of neuronal polarity. Studies in hippocampal neurons show that the inactive form of GSK3β (phosphorylated at S9) is located at the tip of each neurite before polarization, but when neurons begin to polarize and one of these neurites develops into the axon, phospho-GSK3β becomes concentrated at the axonal tip, suggesting that maintaining local inactivation of GSK3β at the nascent axon is crucial for polarization69, 73, 76. Indeed, global inhibition of GSK3 by small-molecule inhibitors or knocking down Gsk3β induces the formation of multiple axons, whereas over-expressing GSK3β-S9A prevents axon formation69. Furthermore, local inhibition of GSK3 can convert dendritic processes into axons in already polarized neurons69, suggesting that localized inactivation of GSK3 is required for both the establishment and maintenance of neuronal polarity69, 76. To date, it is not clear how GSK3 inactivity becomes limited to one neurite during neuronal polarization.
Neuronal polarization requires major reorganization of the growth cone cytoskeleton. It has been proposed that a loose actin network at the tip of the future axon enables microtubules to selectively engorge into the axonal growth cone, forming the platform for subsequent axon elongation78-82. A recent study83 showed that microtubule stabilization in one neurite precedes axon formation, and that changing microtubule dynamics is sufficient to alter axon and dendrite specification83, pointing to the notion that regulation of local microtubule stability and dynamics has an instructive role in neuronal polarization84.
Several substrates of GSK3 are involved in neuronal polarization by regulating microtubule dynamics; these include collapsin response mediator protein 2 (CRMP2), APC, Tau, and microtubule-associated protein (MAP) 1b. Binding of CRMP2 to tubulin dimers, which is abolished by GSK3β phosphorylation76, promotes microtubule polymerization85. CRMP2 is enriched in the nascent axon and over-expression of CRMP2 is sufficient to induce the formation of multiple axons86. In hippocampal neurons, the microtubule +TIP protein APC localizes at the tip of immature neurites, and later becomes enriched in the growth cone of the nascent axon87. Similar to CRMP2, microtubule-binding activity of APC is abrogated by GSK3β phosphorylation88. Thus, GSK3β inactivation in the nascent axon promotes the association between APC and microtubule plus ends and thereby stabilizes the growing ends of axonal microtubules to support axon extension. As with CRMP2 and APC, Tau phosphorylation by GSK3β abolishes its binding to microtubules and thus impairs microtubule assembly89. GSK3 phosphorylation of MAP1b maintains microtubules in a dynamic state90. However, the lack of polarity defect in Tau/Map1b double knock-out mice suggest that they mainly function to regulate axon growth rather than neuronal polarization91.
GSK3 signaling and the PI3K/AKT pathway
Local activation of PI3K and accumulation of phosphatidylinositol-3,4,5-trisphosphate (PIP3) at the tip of the nascent axon is thought to act as a landmark for the induction of neuronal polarization. Several studies69, 73, 76 have proposed a model of neuronal polarization in which local activation of the PI3K/AKT pathway leads to the inactivation of GSK3β by inducing S9 phosphorylation. Supporting this model, the myristoylated, constitutively active form of Akt (Myr-AKT), but not the wild-type, induced the formation of multiple axons, which was partially blocked by over-expression of GSK3β-S9A. Moreover, ectopic expression of PTEN (a phosphatase that counteracts the actions of PI3K) prevented axon formation, an effect that was reversed by a GSK3 inhibitor69. Conversely, knocking down PTEN induced the formation of multiple axons, which was prevented by expression of GSK3β-S9A, leading the authors to conclude that GSK3β acts downstream of PI3K/AKT69. However, neurons from GSK3α-S21A/GSK3β-S9A double knock-in mice develop normal polarity in vivo and in vitro92. Importantly, these neurons still form multiple axons when treated with various GSK3 inhibitors. These results confirm that regulation of GSK3 activity is crucial for neuronal polarization, but suggest that there might be alternative mechanisms for GSK3 inactivation. Similarly, the finding that expression of Myr-AKT induces multiple axons suggests an alternative pathway downstream of AKT for regulating polarization.
GSK3 signaling and the Par3/Par6/PKCς polarity pathway
The Par3/Par6/PKCς complex is required for axon-dendrite specification of hippocampal neurons. Before polarization, Par3 and Par6 are localized at the tips of all processes but later on they become selectively enriched in the nascent axon and developing growth cone74, 93, which is consistent with their roles as negative regulators of GSK3. Localized inhibition of GSK3 at the nascent axon is also required for the polarized transport and proper localization of Par373, suggesting a feedback regulation between Par3 and GSK3.
Similar to the aforementioned mechanism that controls directed cell migration involving the Par3/6-GSK3 pathway37, in hippocampal neurons Dvl becomes enriched at the tip of the axon in response to Wnt5a and induces axon formation via activation of PKCς in the Par3/Par6/PKCς complex94. In these neurons, Dvl over-expression leads to the production of multiple axons but did not affect GSK3β-S9 phosphorylation, and Dvl was still able to induce multiple axon formation in the presence of GSK3β-S9A94. Based on these results, authors concluded that this pathway might not involve GSK3β, but might employ microtubule affinity-regulating kinase (MARK) 2/Par1 as an alternative downstream effector to control microtubule dynamics94. In this model, activation of PKCς in response to Wnt5a/Dvl leads to the inhibitory phosphorylation of MARK2, which in turn causes dephosphorylation of MAPs such as Tau (Fig. 4). Consistent with this, a previous observation showed that knocking down Mark2 decreased Tau phosphorylation and induced the formation of multiple axons, whereas ectopic expression of MARK2 increased Tau phosphorylation and blocked axon growth68. On the other hand, based on the remarkable similarity of the players (Wnt5a, Dvl and PKCς) involved in neuronal polarization94 and cell migration37, it could be suggested that GSK3 inactivation has a role downstream of Wnt/Dvl/PKCς to control neuronal polarization as well. Interestingly, in Drosophila, MARK acts as a priming kinase, triggering subsequent Tau phosphorylation by GSK3β95, suggesting that inactivation of MARK2 and GSK3β at the axonal tip together reduce Tau phosphorylation downstream of Par3/Par6/PKCς complex. Further complicating the situation are the findings that GSK3β can both directly activate96 and inhibit97 MARK2 via phosphorylation. The exact roles of MARK2 and GSK3β and their interplay during neuronal polarization remain to be fully defined (Fig. 4).
GSK3 signaling and the LKB1-SAD pathway
The SAD kinases, SAD-A and SAD-B, which are expressed in the developing nervous system, contain a kinase domain related to that of C. elegans PAR-1 and its vertebrate orthologs, MARK1–MARK470. In the brains of Sad-A, Sad-B double knock-out (Sad-null) mice, segregation of neuronal subtypes to sublayers within the cortical plate was disordered and major axonal tracts were missing, suggesting that SAD kinases are required for neural development in vivo. When cultured in vitro, hippocampal neurons from Sad-null mice develop processes of equivalent lengths, each of which contains both dephosphorylated Tau (Tau1), which is often used as an axonal marker, and MAP2, which is normally enriched in dendrites70. In Sad-null brains, Tau1 was found not only in the axon-rich intermediate zone but also in the cortical plate, where dendrites elaborate70, suggesting that SAD kinases play a pivotal role in neuronal polarization.
Similarly, neurons lacking Lkb1, the ortholog of Par4 in C. elegans, develop neurites of equivalent length containing both Tau1 and MAP267, 72. In the cortices of Lkb1 knock-out mice, levels of phosphorylated SAD-A and SAD-B were greatly reduced, and biochemical experiments revealed that LKB1 phosphorylated SAD kinases67. Phosphorylated forms of LKB1 and SAD kinases were concentrated in axons, and LKB1-mediated SAD phosphorylation correlated with Tau phosphorylation67, 72. Thus, once LKB1 is activated in axons, it activates SAD kinases, which in turn phosphorylate MAPs such as Tau. Although it seems somewhat contradictory that SAD kinases are active in axons, which seems to have low levels of phosphorylated Tau98, these studies66, 67, 70, 72 provide convincing evidence that LKB1 and SAD kinases are required for neuronal polarization in vivo. Interestingly, when Lkb1 was knocked down by in utero electroporation, S9-phosphorylated GSK3β was not detectable at the tip of any neurites in cortical neurons despite the uniform distribution of GSK3β66, suggesting that GSK3β could be a downstream target of LKB1 (Fig. 4). To date, it remains unclear if the defects in neuronal polarization observed in Lkb1-depleted neurons involve GSK3 signaling. It is, however, interesting to note that a study in Xenopus showed that LKB1/XEEK1 regulates GSK3 activity and physically associates with GSK3β and aPKC in vivo99. Future studies should examine whether LKB1 regulates axonal specification by inhibiting GSK3β in addition to activating SAD kinases.
GSK3 signaling and the TSC/mTOR pathway
Mutations in Tsc1 and Tsc2 cause tuberous sclerosis, a disease characterized by tumor predisposition and neurological abnormalities including epilepsy, mental retardation, and autism100. TSC1/TSC2 act as a heterodimer that negatively regulates mammalian target of rapamycin (mTOR) signaling101. Upon activation of the PI3K/AKT pathway, AKT induces inhibitory phosphorylation of TSC2, by which its GTPase activating protein (GAP) activity towards Rheb is reduced, subsequently leading to the activation of mTOR kinase (Fig. 4). mTOR in turn phosphorylates translational regulators, such as ribosomal S6 kinase (S6K) and eukaryotic translation initiation factor 4E-binding protein (4EBP), and thereby induces protein synthesis102-104. It has been suggested that inactivation of the TSC1/TSC2 complex and subsequent mTOR activation in a single neurite regulates axon formation by inducing the translation of polarity proteins such as SAD kinases105. Supporting this notion, phosphorylated forms of S6K and 4EBP were enriched in the axon during polarization, suggesting local activation of mTOR signaling105, 106. Together with the aforementioned elevation of SAD kinase activity by LKB167, the control of SAD protein levels by TSC/mTOR signaling might play a role in axon specification. Because GSK3α-S21A/GSK3β-S9A double knock-in mice show normal neuronal polarization92, AKT phosphorylation of TSC2 has been suggested as an alternative mechanism to convey PI3K/AKT signaling.
Notably, a direct interaction between TSC/mTOR signaling and GSK3 has been reported in the regulation of cell growth. Inoki et al.107 showed that inhibition of GSK3 is sufficient to activate mTOR-dependent protein translation through regulation of TSC2 phosphorylation. It is thus plausible that suppression of GSK3 activity in the axonal growth cone has a role in the activation of local translational machinery for the synthesis of key proteins for axonogenesis. Interestingly, mTOR activation in the axon leads to local translation of Crmp2 and Tau, both of which are GSK3 substrates106. It will be of particular interest to investigate whether the suppression of GSK3 activity at the tip of the axon controls neuronal development by regulating local translation of polarity genes, including its own substrates, in addition to directly controlling microtubule dynamics by regulating MAP phosphorylation.
A role of GSK3 activation in dendrites?
Local inhibition of GSK3 in the axon indicates higher GSK3 activity in dendrites. Does active GSK3 in dendrites play any role in polarization? Local protein degradation by activation of the UPS is an appealing mechanism to control asymmetrical accumulation of polarity proteins. Inhibition of the UPS with MG132 or lactacystin before or after the establishment of neuronal polarity leads to the formation of multiple axons108, suggesting that UPS activation is required for the establishment and maintenance of neuronal polarity. Interestingly, AKT, Par3 and aPKC are ubiquitinated at early stages of polarization before they become accumulated at axonal tips108. UPS inhibition restores the presence of AKT in all neurites, which is accompanied by multiple axon formation, suggesting that AKT is a target of the UPS for local degradation in dendrites108. UPS inhibition does not seem to alter the protein level of GSK3β, but an increase in GSK3β-S9 phosphorylation was observed108, suggesting changes in its activity. A recent study55 indicates that GSK3β regulates proteolytic degradation of a much broader range of proteins than was previously appreciated55. It will be interesting to investigate whether GSK3 activation in dendrites is involved in regulation of local protein degradation that contributes to redistribution of key molecules during polarization.
GSK3 signaling in axon outgrowth
It is well accepted that coordinated regulation of local axon assembly at the growth cone and gene transcription in the neuronal soma is required for efficient axon growth109. This section will discuss evidence for GSK3 signaling in those processes.
Differential regulation of GSK3 substrates to control local axon assembly
Local inhibition of GSK3 signaling is essential for promoting microtubule assembly at the growth cone. However, in seemingly discrepant studies, inhibition of GSK3 blocked axon growth. To reconcile this controversy, Kim et al31 suggested a model in which inhibition of GSK3 can both enhance and prevent axon growth depending on the substrates involved. An interesting feature of GSK3 is that, many substrates of GSK3s require phosphorylation by a distinct kinase — an event known as priming — before they can be phosphorylated by GSK3s (primed substrates), although GSK3s can also phosphorylate some substrates without priming (unprimed substrates). Among GSK3 substrates that regulate microtubule assembly at the growth cone, APC and CRMP2 are primed substrates, and GSK3 phosphorylation abrogates their microtubule-binding affinity76, 88. Consistent with local inhibition of GSK3 in the distal axon, dephosphorylated forms of CRMP2 and APC are enriched in the growth cone, promoting axon formation and mediating neurotrophin-induced axon growth76, 110. Inhibition of GSK3 activity specifically towards primed (but not unprimed) substrates (using a GSK3β-R96A construct)28, 111 results in reduced CRMP2 phosphorylation and increased axon outgrowth31. By contrast, MAP1b is an unprimed substrate, which can be phosphorylated by GSK3 on S1260 and T1265 without priming90. Phosphorylation of MAP1b at these sites renders microtubules more dynamic, so that they can efficiently probe the intracellular space and respond to extracellular signals, activities that are essential for axon growth15, 112. Therefore, to ensure efficient axon growth, GSK3 activity should be precisely controlled, so that its activity towards one subset of substrates is specifically blocked while its activity towards others is preserved (Fig. 5). Indeed, in growing axons phosphorylated MAP1b is enriched at the distal ends of axons90, where dephosphorylated forms of APC and CRMP2 are concentrated.
Figure 5. Differential regulation of GSK3 substrates during axon growth.
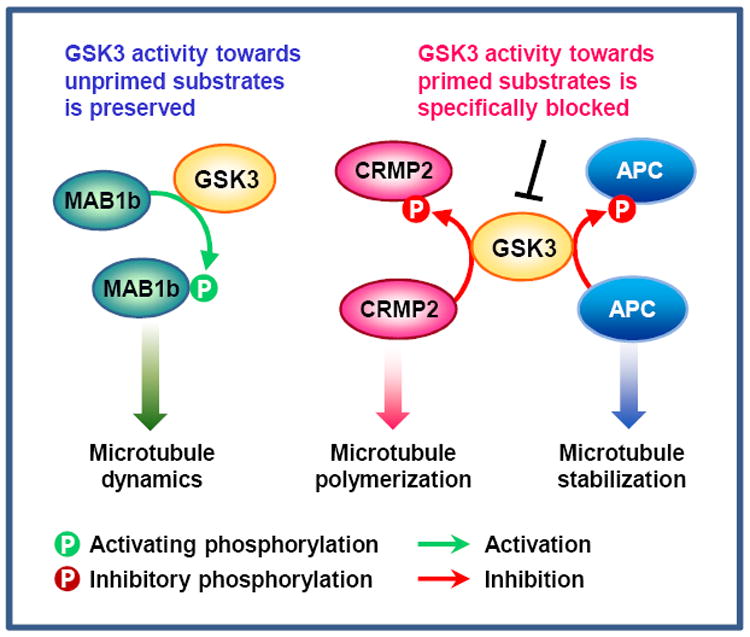
During rapid axon extension, GSK3 activity in the growth cone seems to be precisely controlled, so that its activity towards primed substrates is specifically blocked while its activity towards unprimed substrates is preserved31. Inhibition of GSK3 activity towards collapsin response mediator protein (CRMP) 2 and adenomatosis polyposis coli (APC) allows CRMP2 and APC to bind microtubules, thereby increasing microtubule polymerization and stability76, 85. By contrast, GSK3’s activity towards microtubule-associated protein (MAP) 1b is preserved in the growth cone. Phosphorylation of MAP1b maintains microtubules in a dynamic state, which is essential for axon growth15, 112. In this way, GSK3 can coordinate essential properties of axonal microtubules to ensure optimal microtubule assembly in axons.
These findings have led to a working model in which preferential suppression of GSK3 activity towards primed substrates promotes axon growth, whereas inhibition towards unprimed substrates blocks axon growth (Fig. 5)31. Identification of additional GSK3 substrates enriched in the growth cone will help to test this hypothesis and better understand how GSK3 signaling controls local axon assembly. Moreover, further studies are required to elucidate the molecular mechanisms by which GSK3 activity towards different substrates can be differentially regulated.
Transcriptional control of axon outgrowth by GSK3 signaling
In addition to stimulating the tips of axons for cytoskeletal regulation, GSK3 has been implicated in the transcriptional control of axon growth via regulation of β-catenin and NFAT transcription factors. In the canonical Wnt pathway, Wnt3a induces axon growth from developing sensory neurons via accumulation of β-catenin and subsequent activation of TCF4 (Fig. 6)113, providing evidence for GSK3/β-catenin signaling in axon growth. GSK3 could also play a part in the transcriptional control of neurotrophin-induced axon growth by phosphorylating CREB and NFAT proteins. CREB is a well-established transcription factor downstream of neurotrophins, and Creb null mice display impaired axon growth from sensory and sympathetic ganglion neurons114. Neurotrophins (and other stimuli) lead to the activation of CREB kinases, which phosphorylate CREB on the transcriptional regulatory site S133, thereby recruiting the transcriptional coactivator CREB-binding protein (CBP) and so promoting the assembly of basal transcriptional complex115. In developing neurons, the DNA binding activity of CREB is tightly controlled by extracellular stimuli via nitric oxide-dependent signaling, as opposed to the classical model in which CREB is constitutively bound to promoter sequences116. Given that CREB phosphorylation by GSK3 abrogates its DNA binding activity8, it is plausible that GSK3 and nitric oxide signaling work in concert to control the duration and intensity of CREB-dependent transcription (Fig. 6). Neurotrophins and netrins also induce transcription of genes that are essential for axon growth by triggering Ca2+/calcineurin-dependent nuclear translocation of NFAT, and mice lacking calcineurin/NFAT signaling display profound defects in sensory axon projection and commissural axon growth117. Because NFAT phosphorylation by GSK3 inhibits its DNA binding activity10 and is required for nuclear export9, GSK3 is likely to have a role in NFAT-mediated gene transcription during axon growth (Fig. 6). Given that there are many other transcription factors modulated by GSK3, additional evidence for the transcriptional control of axon growth by GSK3 signaling awaits to be determined.
Figure 6. Potential roles of GSK3 in the transcriptional regulation of axon growth.
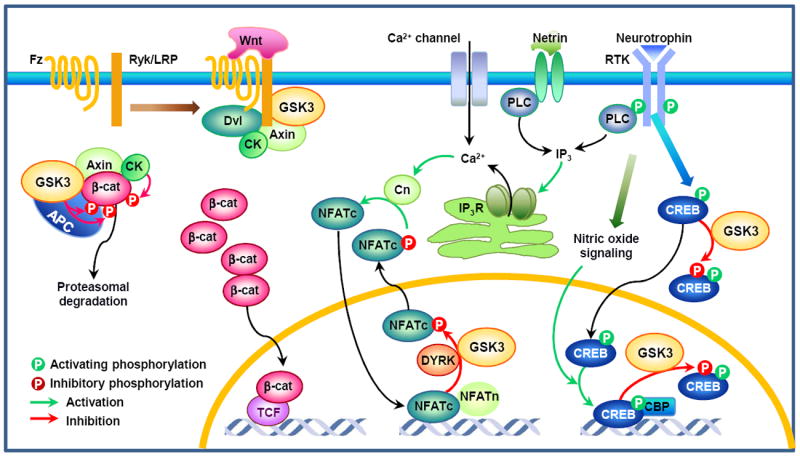
In the absence of Wnt, the transcriptional co-activator β-catenin (β-cat) is phosphorylated by GSK3 in the destruction complex, which tags β-cat for proteasomal degradation. Wnt promotes the association of Frizzled (Fz) receptor and the co-receptor low-density lipoprotein receptor-related protein (LRP) or Ryk113, leading to recruitment of the cytoplasmic dishevelled (Dvl) and the destruction complex to the membrane. Inhibition of GSK3 activity results in the accumulation of hypophosphorylated β-cat, which then can enter the nucleus and promote T-cell factor (TCF, also known as lymphoid enhancer-binding factor)-mediated gene transcription. Another family of transcription factors involved in axon growth is nuclear factor of activated T-cells (NFAT) proteins. Neurotrophins and netrins require calcineurin (Cn)/NFAT signaling for axon growth117. A rise in intracellular Ca2+ induced by neurotrophins, netrins, or calcium channels activates the serine/threonine phosphatase Cn, which dephosphorylates NFATc proteins. Dephosphorylation of NFATc triggers their nuclear translocation, where they form complexes with NFATn to induce gene transcription. NFATc proteins are rapidly excluded from the nucleus through sequential phosphorylation, first by a priming kinase such as protein kinase A (PKA) and dual-specificity tyrosine-phosphorylation regulated kinase (DYRK) 1A, then by GSK39. Because NFAT phosphorylation by GSK3 inhibits its DNA binding activity10 and is required for its nuclear export9, GSK3 could play a role in NFAT-mediated gene transcription downstream of neurotrophin and/or netrin signaling. cAMP response element binding protein (CREB) is a well-established transcription factor that mediates neurotrophin-induced axon growth114. Neurotrophins and other stimuli lead to the phosphorylation of CREB at S133, which allows CREB binding to the transcriptional co-activator CREB-binding protein (CBP). In developing neurons, DNA binding activity of CREB is tightly controlled via nitric oxide signaling, as opposed to the classical model in which CREB is constitutively bound to promoter sequences116. Similar to NFATc, GSK phosphorylation of CREB prevents it from binding to DNA8. Because GSK3 is found in the cytoplasm and the nucleus, CREB phosphorylation can occur in both places. However, it should be noted that GSK regulation of NFAT and CREB has not been directly tested in the context of axon growth. APC, adenomatous polyposis coli; CK, casein kinase; IP3, inositol-1,4,5-trisphosphate; IP3R, IP3 receptor.
Concluding remarks and future directions
An algorithm that integrates consensus sequence motifs with contextual information has predicted GSK3 to be one of the kinases with a high number of substrates77. Although further studies are required to validate the physiological relevance of this prediction, the large number of putative substrates suggests that GSK3 could affect a broad range of cellular activities (Fig. 7). Indeed, recent studies implicate GSK3 in many fundamental processes during neural development, and a number of neurological disorders are associated with deficits in GSK3 signaling. It is thus important to understand GSK3 signaling, which requires, first, a re-evaluation of the involvement of GSK3 based on more reliable methods to detect GSK activation/inactivation, and second, the identification of additional in vivo GSK3 substrates.
Figure 7. Representative substrates of GSK3 implicated in neural development.
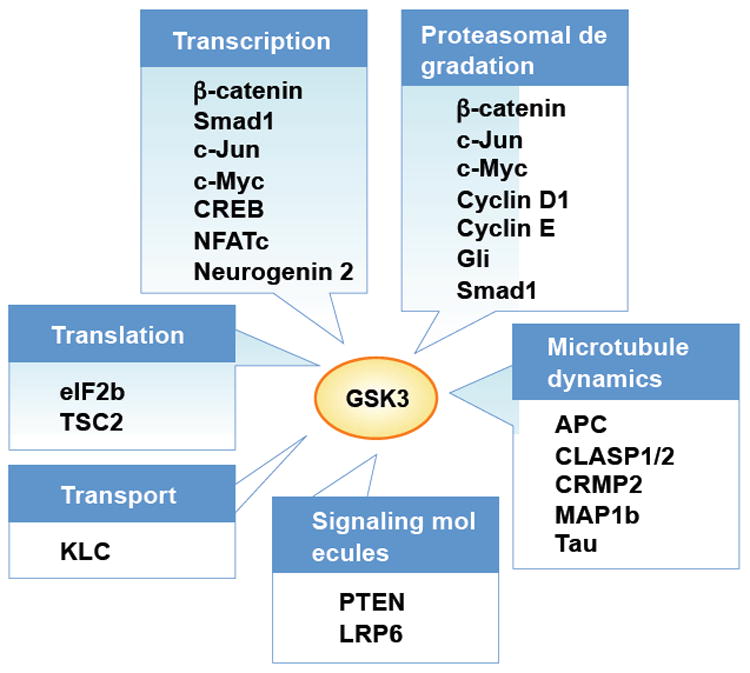
GSK3 regulation of neural development can be achieved through phosphorylation of proteins that control key steps in neurodevelopment. GSK3 phosphorylates several transcription factors, such as β-catentin122, Smad112, c-Jun123, c-Myc48, cAMP response element binding protein (CREB)8, nuclear factor of activated T-cells (NFATc)9, and neurogenin 211, many of which undergo proteasomal degradation after GSK3 phosphorylation. GSK3 phosphorylation mediates proteasomal targeting and degradation of other proteins as well, such as cyclin D1124 and cyclin E125. The most well-characterized substrates of GSK3 involved in neural development are microtubule-associated proteins that control neuronal polarization and axon growth. These substrates include adenomatosis polyposis coli (APC)88, CLIP-associated protein (CLASP) 1 and 2126, collapsin response mediator protein (CRMP) 276, MAP (microtubule-associated protein) 1b127 and Tau. Although CLASP is enriched in the growth cone and mediate Slit-induced axon repulsion during neural development in Drosophila128, its localization and role during neuronal polarization and/or axon growth in the mammalian nervous system remains to be determined. Signaling molecules such as phosphatase and tensin homologue deleted on chromosome 10 (PTEN)129 and Wnt co-receptor low-density lipoprotein receptor-related protein (LRP) 6 are also phosphorylated by GSK3. Other examples are kinesin light chain (KLC) that regulates selective transport130, and essential components of the translational machinery, such as eukaryotic initiation factor (eIF) 2b131 and tuberous sclerosis complex (TSC2)107.
The emerging picture is that GSK3 activity is controlled mainly by protein protein–interactions. Considering its many substrates, regulation by a complex and dynamic interaction network serves as an attractive model because it allows both pathway-specific control of GSK3 activity via signal-dependent formation of protein complexes and precise regulation of GSK activity towards particular substrates. It remains to be determined whether multiprotein complexes, as exemplified in the Wnt signaling, are a widespread mechanism for controlling the specificity of GSK3 actions. A distinguishable characteristic of GSK3 is the requirement of a priming event, which involves activation of another kinase. Thus, in combination with the signaling that directly regulates GSK3 activity, signaling that regulates the phosphorylation status of its substrates allows GSK signaling to achieve further specificity.
With respect to GSK3 regulation, studies so far have focused on the inhibitory mechanism. However, it is becoming increasingly apparent that activation of GSK3 comprises an important aspect of the regulatory scheme, suggesting that a delicate balance between activation and inactivation is required to shape GSK3 signaling. The data so far support the view that GSK3 activity is subjected to multiple layers of sophisticated regulation to ensure that GSK3 encounters its substrates in the right place at the right time. It will be of great interest to investigate how the complexity of GSK3 signaling is orchestrated in vivo to exert exquisite control over a plethora of cellular processes during neural development.
At-a-glance summary.
Recent studies provide strong evidence that GSK3 signaling have important roles in many neurodevelopmental processes, such as neurogenesis, neuronal polarization, and axon outgrowth, etc.
Results from GSK3α-S21A/GSK3β-S9A double knock-in mice suggest that phosphorylation of GSK3s at their N-terminal serine residues is not the major regulatory mechanism to control GSK3 activity in the nervous system. Other players such as DISC1, Par3, and LRP6, may play more important roles in regulation of GSK3 activity.
During neurogenesis, evidence suggests that GSK3 inactivation promotes neural progenitor proliferation, whereas GSK3 activation promotes neuronal differentiation. The role of GSK3 in neurogenesis might be achieved through regulation of protein stability and microtubule assembly.
Several lines of evidence suggest that GSK3 signaling controls neuronal polarization by regulating microtubule stability through several microtubule-binding proteins. GSK3 signaling appears to be the converging point of many pathways that regulate neuronal polarity, such as PI3K-AKT, Par3/Par6/aPKC, LKB1-SAD, and TSC-mTor pathways.
GSK3 plays pleiotropic roles in controlling axon outgrowth depending on the substrates involved. Local inactivation of GSK3 towards primed substrates at distal axon is necessary for efficient axon outgrowth, whereas strong suppression of GSK3 activity prevents axon outgrowth, probably through regulation of unprimed substrates. GSK3 signaling can also regulate axon outgrowth by controlling transcriptional factors in the cell soma.
The important roles GSK3 signaling in neural development is consistent with its involvement in many neurodevelopmental disorders, such as schizophrenia and autism. Further studies may open possibilities of novel therapeutic treatments by targeting GSK3 signaling.
Acknowledgments
The authors were supported by grants from the Whitehall Foundation (to F.Q.Z), the Basil O’Connor Starter Scholar Research Award from the March of Dimes foundation (to F.Q.Z), and the NIH (1R01NS064288 to F.Q.Z). We apologize to authors whose work we were unable to include owing to space constraints.
Glossary terms
- Mitotic spindle reorganization
reorganization of microtubules and associated molecules during cell division to form a bipolar spindle between duplicated centrosome components, which leads to the segregation of chromosomes
- Leading process
a process that extends in the direction of neuronal migration prior to nuclear movement
- Microtubule minus end-anchoring proteins
a family of proteins that specifically bind to the microtubule minus-ends and function to anchor the microtubule to specific cellular structures, such as the centriole
- Microtubule plus end-binding proteins
a family of microtubule-binding proteins that specifically track the growing ends (plus ends) of microtubules and function to stabilize microtubules or to mediate the interactions between microtubules and other cellular structures
- Leading edge
the region of a migrating cell that protrudes forward
- Asymmetrical division
division of cells that leads to the production of two daughter cells with different developmental potentials
- Centrosome
an organelle that controls the organization of microtubules and regulates cell-cycle progression
- Centriole
one of two perpendicular structures in the centrosome that are composed of a ring of nine microtubules
- Ventricular zone
a region in the brain next to the ventricles composed of neuroepithelial cells that generate neuronal and glial cells
- Microtubule minus end
the slow-growing end of purified microtubule polymers in vitro. In animal cells, minus ends are generally anchored at centrosomes and not growing
- Microtubule plus end
the more quickly polymerizing end of microtubule polymers
- Kinetochore
a specialized condensed region on the centromere to which the spindle fibers attach during cell division
- Cell cortex
a cytoplasmic region beneath the plasma membrane that functions as a mechanical support of the plasma membrane
- Myristoylation
an irreversible protein modification that covalently attaches a myristoyl group via an amide bond to the alpha-amino group of an N-terminal amino acid of a nascent polypeptide
Biography
Eun-Mi Hur received her Ph.D. in cell biology from Pohang University of Science and Technology, South Korea. After completing her Ph.D. under the supervision of Dr. Kyong-Tai Kim on receptor-mediated signal transduction and crosstalk, she joined the laboratory of Feng-Quan Zhou at Johns Hopkins University School of Medicine, where she studies the role of GSK3 signaling in neural development and axon regeneration. She is the recipient of the postdoctoral fellowship from the Christopher and Dana Reeve Foundation.
Feng-Quan Zhou is an Assistant Professor in the Department of Orthopaedic Surgery and The Soloman H. Snyder Department of Neuroscience at Johns Hopkins University School of Medicine. He received his Ph.D. in Anatomy and Cell Biology from State University of New York at Buffalo (mentored by Christopher Cohan), and postdoctoral training at University of North Carolina, Chapel Hill (mentored by William Snider). His current research focuses on the role of GSK3 signaling in neural development and the molecular mechanisms regulating axon regeneration. He is the recipient of the Basil O’Connor Starter Scholar Research Award.
References
- 1.Woodgett JR, Cohen P. Multisite phosphorylation of glycogen synthase. Molecular basis for the substrate specificity of glycogen synthase kinase-3 and casein kinase-II (glycogen synthase kinase-5) Biochim Biophys Acta. 1984;788:339–47. doi: 10.1016/0167-4838(84)90047-5. [DOI] [PubMed] [Google Scholar]
- 2.Wang Y, Roach PJ. Inactivation of rabbit muscle glycogen synthase by glycogen synthase kinase-3. Dominant role of the phosphorylation of Ser-640 (site-3a) J Biol Chem. 1993;268:23876–80. [PubMed] [Google Scholar]
- 3.Woodgett JR. Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 1990;9:2431–8. doi: 10.1002/j.1460-2075.1990.tb07419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mukai F, Ishiguro K, Sano Y, Fujita SC. Alternative splicing isoform of tau protein kinase I/glycogen synthase kinase 3beta. J Neurochem. 2002;81:1073–83. doi: 10.1046/j.1471-4159.2002.00918.x. [DOI] [PubMed] [Google Scholar]
- 5.Castano Z, Gordon-Weeks PR, Kypta RM. The neuron-specific isoform of glycogen synthase kinase-3beta is required for axon growth. J Neurochem. 2010;113:117–30. doi: 10.1111/j.1471-4159.2010.06581.x. [DOI] [PubMed] [Google Scholar]
- 6.Wood-Kaczmar A, Kraus M, Ishiguro K, Philpott KL, Gordon-Weeks PR. An alternatively spliced form of glycogen synthase kinase-3beta is targeted to growing neurites and growth cones. Mol Cell Neurosci. 2009;42:184–94. doi: 10.1016/j.mcn.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 7.Woodgett JR. Judging a protein by more than its name: GSK-3. Sci STKE. 2001;2001:RE12. doi: 10.1126/stke.2001.100.re12. [DOI] [PubMed] [Google Scholar]
- 8.Grimes CA, Jope RS. CREB DNA binding activity is inhibited by glycogen synthase kinase-3 beta and facilitated by lithium. J Neurochem. 2001;78:1219–32. doi: 10.1046/j.1471-4159.2001.00495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beals CR, Sheridan CM, Turck CW, Gardner P, Crabtree GR. Nuclear export of NF-ATc enhanced by glycogen synthase kinase-3. Science. 1997;275:1930–4. doi: 10.1126/science.275.5308.1930. [DOI] [PubMed] [Google Scholar]
- 10.Neal JW, Clipstone NA. Glycogen synthase kinase-3 inhibits the DNA binding activity of NFATc. J Biol Chem. 2001;276:3666–73. doi: 10.1074/jbc.M004888200. [DOI] [PubMed] [Google Scholar]
- 11.Ma YC, et al. Regulation of motor neuron specification by phosphorylation of neurogenin 2. Neuron. 2008;58:65–77. doi: 10.1016/j.neuron.2008.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fuentealba LC, et al. Integrating patterning signals: Wnt/GSK3 regulates the duration of the BMP/Smad1 signal. Cell. 2007;131:980–93. doi: 10.1016/j.cell.2007.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Groot RP, Auwerx J, Bourouis M, Sassone-Corsi P. Negative regulation of Jun/AP-1: conserved function of glycogen synthase kinase 3 and the Drosophila kinase shaggy. Oncogene. 1993;8:841–7. [PubMed] [Google Scholar]
- 14.Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997;16:3797–804. doi: 10.1093/emboj/16.13.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou FQ, Snider WD. Cell biology. GSK-3beta and microtubule assembly in axons. Science. 2005;308:211–4. doi: 10.1126/science.1110301. [DOI] [PubMed] [Google Scholar]
- 16.Beaulieu JM. Not only lithium: regulation of glycogen synthase kinase-3 by antipsychotics and serotonergic drugs. Int J Neuropsychopharmacol. 2007;10:3–6. doi: 10.1017/S1461145706006857. [DOI] [PubMed] [Google Scholar]
- 17.Beaulieu JM, Gainetdinov RR, Caron MG. Akt/GSK3 signaling in the action of psychotropic drugs. Annu Rev Pharmacol Toxicol. 2009;49:327–47. doi: 10.1146/annurev.pharmtox.011008.145634. [DOI] [PubMed] [Google Scholar]
- 18.Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci U S A. 1996;93:8455–9. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martinez A. Preclinical efficacy on GSK-3 inhibitors: towards a future generation of powerful drugs. Med Res Rev. 2008;28:773–96. doi: 10.1002/med.20119. [DOI] [PubMed] [Google Scholar]
- 20.Lovestone S, Killick R, Di Forti M, Murray R. Schizophrenia as a GSK-3 dysregulation disorder. Trends Neurosci. 2007;30:142–9. doi: 10.1016/j.tins.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 21.Kwon CH, et al. Pten regulates neuronal arborization and social interaction in mice. Neuron. 2006;50:377–88. doi: 10.1016/j.neuron.2006.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mao Y, et al. Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling. Cell. 2009;136:1017–31. doi: 10.1016/j.cell.2008.12.044. This study shows that DISC1 physically interacts with GSK3β to specifically inhibit its activity towards β-catenin in neural progenitors. It suggests a link between GSK3 signaling and DISC1-related psychiatric disroders. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beaulieu JM, et al. Role of GSK3 beta in behavioral abnormalities induced by serotonin deficiency. Proc Natl Acad Sci U S A. 2008;105:1333–8. doi: 10.1073/pnas.0711496105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang HH, Lipovsky AI, Dibble CC, Sahin M, Manning BD. S6K1 regulates GSK3 under conditions of mTOR-dependent feedback inhibition of Akt. Mol Cell. 2006;24:185–97. doi: 10.1016/j.molcel.2006.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou XL, et al. Association of adenomatous polyposis coli (APC) gene polymorphisms with autism spectrum disorder (ASD) Am J Med Genet B Neuropsychiatr Genet. 2007;144B:351–4. doi: 10.1002/ajmg.b.30415. [DOI] [PubMed] [Google Scholar]
- 26.Mili S, Moissoglu K, Macara IG. Genome-wide screen reveals APC-associated RNAs enriched in cell protrusions. Nature. 2008;453:115–9. doi: 10.1038/nature06888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–9. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 28.Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. Biochem J. 2001;359:1–16. doi: 10.1042/0264-6021:3590001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McManus EJ, et al. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. Embo J. 2005;24:1571–83. doi: 10.1038/sj.emboj.7600633. This study shows that mutant mice harboring mutations in GSK3 at their N-terminal serine residues (replaced by alanine and thus cannot be phosphorylated) survive into adulthood without any obvious phenotype in their nervous system. It provides unequivocal geneitic evidence that phosphorylation of GSK3s at their N-terminal serine residues is not a major mechanism that regulates GSK3 activity in the nervous system. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thornton TM, et al. Phosphorylation by p38 MAPK as an alternative pathway for GSK3beta inactivation. Science. 2008;320:667–70. doi: 10.1126/science.1156037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim WY, et al. Essential roles for GSK-3s and GSK-3-primed substrates in neurotrophin-induced and hippocampal axon growth. Neuron. 2006;52:981–96. doi: 10.1016/j.neuron.2006.10.031. This study provides a plausible explanation for the conflicting results regarding how GSK3 signaling controls axon growth. The results suggest that inactivation of GSK3 towards primed substrates is necessary for efficient axon growth, whereas inhibtion of GSK3 activity toward unprimed substrates prevents axon growth. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Piao S, et al. Direct inhibition of GSK3beta by the phosphorylated cytoplasmic domain of LRP6 in Wnt/beta-catenin signaling. PLoS ONE. 2008;3:e4046. doi: 10.1371/journal.pone.0004046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu G, Huang H, Garcia Abreu J, He X. Inhibition of GSK3 phosphorylation of beta-catenin via phosphorylated PPPSPXS motifs of Wnt coreceptor LRP6. PLoS ONE. 2009;4:e4926. doi: 10.1371/journal.pone.0004926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu X, et al. Cdc42 controls progenitor cell differentiation and beta-catenin turnover in skin. Genes Dev. 2006;20:571–85. doi: 10.1101/gad.361406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hengst U, Deglincerti A, Kim HJ, Jeon NL, Jaffrey SR. Axonal elongation triggered by stimulus-induced local translation of a polarity complex protein. Nat Cell Biol. 2009;11:1024–30. doi: 10.1038/ncb1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Etienne-Manneville S, Hall A. Cdc42 regulates GSK-3beta and adenomatous polyposis coli to control cell polarity. Nature. 2003;421:753–6. doi: 10.1038/nature01423. This study shows that the conserved cell polarity pathway Cdc42-Par3/6 acts upstream of GSK3β to control directed cell migration. It provides the first link between GSK3 signaling and the Par3/6 cell polarity pathway. [DOI] [PubMed] [Google Scholar]
- 37.Schlessinger K, McManus EJ, Hall A. Cdc42 and noncanonical Wnt signal transduction pathways cooperate to promote cell polarity. J Cell Biol. 2007;178:355–61. doi: 10.1083/jcb.200701083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Itoh N, et al. Identification of focal adhesion kinase (FAK) and phosphatidylinositol 3-kinase (PI3-kinase) as Par3 partners by proteomic analysis. Cytoskeleton. 2010 doi: 10.1002/cm.20444. [DOI] [PubMed] [Google Scholar]
- 39.Kim WY, et al. GSK-3 is a master regulator of neural progenitor homeostasis. Nat Neurosci. 2009;12:1390–7. doi: 10.1038/nn.2408. By conditionally knocking out both GSK3 isoforms in neural progenitors, this study demonstrates that GSK3s are essential for cortical neurogenesis. Importantly, it provides evidence that GSK3s coordinate multiple signaling pathways, including Wnts, sonic hedgehog (Shh), fibroblast growth factor (FGF), and Notch signaling, to control neurogenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bultje RS, et al. Mammalian Par3 regulates progenitor cell asymmetric division via notch signaling in the developing neocortex. Neuron. 2009;63:189–202. doi: 10.1016/j.neuron.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chenn A, Walsh CA. Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science. 2002;297:365–9. doi: 10.1126/science.1074192. [DOI] [PubMed] [Google Scholar]
- 42.Machold R, et al. Sonic hedgehog is required for progenitor cell maintenance in telencephalic stem cell niches. Neuron. 2003;39:937–50. doi: 10.1016/s0896-6273(03)00561-0. [DOI] [PubMed] [Google Scholar]
- 43.Iwata T, Hevner RF. Fibroblast growth factor signaling in development of the cerebral cortex. Dev Growth Differ. 2009;51:299–323. doi: 10.1111/j.1440-169X.2009.01104.x. [DOI] [PubMed] [Google Scholar]
- 44.Yoon K, Gaiano N. Notch signaling in the mammalian central nervous system: insights from mouse mutants. Nat Neurosci. 2005;8:709–15. doi: 10.1038/nn1475. [DOI] [PubMed] [Google Scholar]
- 45.Jiang J. Regulation of Hh/Gli signaling by dual ubiquitin pathways. Cell Cycle. 2006;5:2457–63. doi: 10.4161/cc.5.21.3406. [DOI] [PubMed] [Google Scholar]
- 46.Foltz DR, Santiago MC, Berechid BE, Nye JS. Glycogen synthase kinase-3beta modulates notch signaling and stability. Curr Biol. 2002;12:1006–11. doi: 10.1016/s0960-9822(02)00888-6. [DOI] [PubMed] [Google Scholar]
- 47.Espinosa L, Ingles-Esteve J, Aguilera C, Bigas A. Phosphorylation by glycogen synthase kinase-3 beta down-regulates Notch activity, a link for Notch and Wnt pathways. J Biol Chem. 2003;278:32227–35. doi: 10.1074/jbc.M304001200. [DOI] [PubMed] [Google Scholar]
- 48.Gregory MA, Qi Y, Hann SR. Phosphorylation by glycogen synthase kinase-3 controls c-myc proteolysis and subnuclear localization. J Biol Chem. 2003;278:51606–12. doi: 10.1074/jbc.M310722200. [DOI] [PubMed] [Google Scholar]
- 49.Sayas CL, Avila J, Wandosell F. Glycogen synthase kinase-3 is activated in neuronal cells by Galpha12 and Galpha13 by Rho-independent and Rho-dependent mechanisms. J Neurosci. 2002;22:6863–75. doi: 10.1523/JNEUROSCI.22-16-06863.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kingsbury MA, Rehen SK, Contos JJ, Higgins CM, Chun J. Non-proliferative effects of lysophosphatidic acid enhance cortical growth and folding. Nat Neurosci. 2003;6:1292–9. doi: 10.1038/nn1157. [DOI] [PubMed] [Google Scholar]
- 51.Gauthier-Fisher A, et al. Lfc and Tctex-1 regulate the genesis of neurons from cortical precursor cells. Nat Neurosci. 2009;12:735–44. doi: 10.1038/nn.2339. [DOI] [PubMed] [Google Scholar]
- 52.Siegenthaler JA, et al. Retinoic acid from the meninges regulates cortical neuron generation. Cell. 2009;139:597–609. doi: 10.1016/j.cell.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Benkoussa M, Brand C, Delmotte MH, Formstecher P, Lefebvre P. Retinoic acid receptors inhibit AP1 activation by regulating extracellular signal-regulated kinase and CBP recruitment to an AP1-responsive promoter. Mol Cell Biol. 2002;22:4522–34. doi: 10.1128/MCB.22.13.4522-4534.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Spittaels K, et al. Neonatal neuronal overexpression of glycogen synthase kinase-3 beta reduces brain size in transgenic mice. Neuroscience. 2002;113:797–808. doi: 10.1016/s0306-4522(02)00236-1. [DOI] [PubMed] [Google Scholar]
- 55.Kim NG, Xu C, Gumbiner BM. Identification of targets of the Wnt pathway destruction complex in addition to beta-catenin. Proc Natl Acad Sci U S A. 2009;106:5165–70. doi: 10.1073/pnas.0810185106. This study, to our knowledge, is the first attempt to discover additional GSK3 substrates by unbiased screening approach. Identification of new GSK3 substrates will help reveal novel functions of GSK3 signaling in the nervous system. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu C, Kim NG, Gumbiner BM. Regulation of protein stability by GSK3 mediated phosphorylation. Cell Cycle. 2009;8:4032–9. doi: 10.4161/cc.8.24.10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fuentealba LC, Eivers E, Geissert D, Taelman V, De Robertis EM. Asymmetric mitosis: Unequal segregation of proteins destined for degradation. Proc Natl Acad Sci U S A. 2008;105:7732–7. doi: 10.1073/pnas.0803027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang X, et al. Asymmetric centrosome inheritance maintains neural progenitors in the neocortex. Nature. 2009;461:947–55. doi: 10.1038/nature08435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hong YR, et al. Cloning and characterization of a novel human ninein protein that interacts with the glycogen synthase kinase 3beta. Biochim Biophys Acta. 2000;1492:513–6. doi: 10.1016/s0167-4781(00)00127-5. [DOI] [PubMed] [Google Scholar]
- 60.Hong YR, Chen CH, Chuo MH, Liou SY, Howng SL. Genomic organization and molecular characterization of the human ninein gene. Biochem Biophys Res Commun. 2000;279:989–95. doi: 10.1006/bbrc.2000.4050. [DOI] [PubMed] [Google Scholar]
- 61.Howng SL, et al. A novel ninein-interaction protein, CGI-99, blocks ninein phosphorylation by GSK3beta and is highly expressed in brain tumors. FEBS Lett. 2004;566:162–8. doi: 10.1016/j.febslet.2004.04.024. [DOI] [PubMed] [Google Scholar]
- 62.Didier C, Merdes A, Gairin JE, Jabrane-Ferrat N. Inhibition of proteasome activity impairs centrosome-dependent microtubule nucleation and organization. Mol Biol Cell. 2008;19:1220–9. doi: 10.1091/mbc.E06-12-1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mimori-Kiyosue Y, Tsukita S. Where is APC going? J Cell Biol. 2001;154:1105–9. doi: 10.1083/jcb.200106113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yamashita YM, Jones DL, Fuller MT. Orientation of asymmetric stem cell division by the APC tumor suppressor and centrosome. Science. 2003;301:1547–50. doi: 10.1126/science.1087795. [DOI] [PubMed] [Google Scholar]
- 65.Yokota Y, et al. The adenomatous polyposis coli protein is an essential regulator of radial glial polarity and construction of the cerebral cortex. Neuron. 2009;61:42–56. doi: 10.1016/j.neuron.2008.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Asada N, Sanada K, Fukada Y. LKB1 regulates neuronal migration and neuronal differentiation in the developing neocortex through centrosomal positioning. J Neurosci. 2007;27:11769–75. doi: 10.1523/JNEUROSCI.1938-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Barnes AP, et al. LKB1 and SAD kinases define a pathway required for the polarization of cortical neurons. Cell. 2007;129:549–63. doi: 10.1016/j.cell.2007.03.025. [DOI] [PubMed] [Google Scholar]
- 68.Chen YM, et al. Microtubule affinity-regulating kinase 2 functions downstream of the PAR-3/PAR-6/atypical PKC complex in regulating hippocampal neuronal polarity. Proc Natl Acad Sci U S A. 2006;103:8534–9. doi: 10.1073/pnas.0509955103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jiang H, Guo W, Liang X, Rao Y. Both the establishment and the maintenance of neuronal polarity require active mechanisms: critical roles of GSK-3beta and its upstream regulators. Cell. 2005;120:123–35. doi: 10.1016/j.cell.2004.12.033. This study, together with Yoshimura et. al (Ref. 76), show that asymmetrical inactivation of GSK3 is necessary and sufficient for axon formation during neuronal polarization. These results place GSK3 as a major determinant of neuronal polarization. [DOI] [PubMed] [Google Scholar]
- 70.Kishi M, Pan YA, Crump JG, Sanes JR. Mammalian SAD kinases are required for neuronal polarization. Science. 2005;307:929–32. doi: 10.1126/science.1107403. [DOI] [PubMed] [Google Scholar]
- 71.Schwamborn JC, Puschel AW. The sequential activity of the GTPases Rap1B and Cdc42 determines neuronal polarity. Nat Neurosci. 2004;7:923–9. doi: 10.1038/nn1295. [DOI] [PubMed] [Google Scholar]
- 72.Shelly M, Cancedda L, Heilshorn S, Sumbre G, Poo MM. LKB1/STRAD promotes axon initiation during neuronal polarization. Cell. 2007;129:565–77. doi: 10.1016/j.cell.2007.04.012. [DOI] [PubMed] [Google Scholar]
- 73.Shi SH, Cheng T, Jan LY, Jan YN. APC and GSK-3beta are involved in mPar3 targeting to the nascent axon and establishment of neuronal polarity. Curr Biol. 2004;14:2025–32. doi: 10.1016/j.cub.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 74.Shi SH, Jan LY, Jan YN. Hippocampal neuronal polarity specified by spatially localized mPar3/mPar6 and PI 3-kinase activity. Cell. 2003;112:63–75. doi: 10.1016/s0092-8674(02)01249-7. [DOI] [PubMed] [Google Scholar]
- 75.Yoshimura T, et al. Ras regulates neuronal polarity via the PI3-kinase/Akt/GSK-3beta/CRMP-2 pathway. Biochem Biophys Res Commun. 2006;340:62–8. doi: 10.1016/j.bbrc.2005.11.147. [DOI] [PubMed] [Google Scholar]
- 76.Yoshimura T, et al. GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell. 2005;120:137–49. doi: 10.1016/j.cell.2004.11.012. See Ref. 69. [DOI] [PubMed] [Google Scholar]
- 77.Linding R, et al. Systematic discovery of in vivo phosphorylation networks. Cell. 2007;129:1415–26. doi: 10.1016/j.cell.2007.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Challacombe JF, Snow DM, Letourneau PC. Dynamic microtubule ends are required for growth cone turning to avoid an inhibitory guidance cue. J Neurosci. 1997;17:3085–95. doi: 10.1523/JNEUROSCI.17-09-03085.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Geraldo S, Khanzada UK, Parsons M, Chilton JK, Gordon-Weeks PR. Targeting of the F-actin-binding protein drebrin by the microtubule plus-tip protein EB3 is required for neuritogenesis. Nat Cell Biol. 2008;10:1181–9. doi: 10.1038/ncb1778. [DOI] [PubMed] [Google Scholar]
- 80.Tanaka E, Ho T, Kirschner MW. The role of microtubule dynamics in growth cone motility and axonal growth. J Cell Biol. 1995;128:139–55. doi: 10.1083/jcb.128.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bradke F, Dotti CG. The role of local actin instability in axon formation. Science. 1999;283:1931–4. doi: 10.1126/science.283.5409.1931. [DOI] [PubMed] [Google Scholar]
- 82.Da Silva JS, Hasegawa T, Miyagi T, Dotti CG, Abad-Rodriguez J. Asymmetric membrane ganglioside sialidase activity specifies axonal fate. Nat Neurosci. 2005;8:606–15. doi: 10.1038/nn1442. [DOI] [PubMed] [Google Scholar]
- 83.Witte H, Neukirchen D, Bradke F. Microtubule stabilization specifies initial neuronal polarization. J Cell Biol. 2008;180:619–32. doi: 10.1083/jcb.200707042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Conde C, Caceres A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat Rev Neurosci. 2009;10:319–32. doi: 10.1038/nrn2631. [DOI] [PubMed] [Google Scholar]
- 85.Fukata Y, et al. CRMP-2 binds to tubulin heterodimers to promote microtubule assembly. Nat Cell Biol. 2002;4:583–91. doi: 10.1038/ncb825. [DOI] [PubMed] [Google Scholar]
- 86.Inagaki N, et al. CRMP-2 induces axons in cultured hippocampal neurons. Nat Neurosci. 2001;4:781–2. doi: 10.1038/90476. [DOI] [PubMed] [Google Scholar]
- 87.Votin V, Nelson WJ, Barth AI. Neurite outgrowth involves adenomatous polyposis coli protein and beta-catenin. J Cell Sci. 2005;118:5699–708. doi: 10.1242/jcs.02679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zumbrunn J, Kinoshita K, Hyman AA, Nathke IS. Binding of the adenomatous polyposis coli protein to microtubules increases microtubule stability and is regulated by GSK3 beta phosphorylation. Curr Biol. 2001;11:44–9. doi: 10.1016/s0960-9822(01)00002-1. [DOI] [PubMed] [Google Scholar]
- 89.Stoothoff WH, Johnson GV. Tau phosphorylation: physiological and pathological consequences. Biochim Biophys Acta. 2005;1739:280–97. doi: 10.1016/j.bbadis.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 90.Trivedi N, Marsh P, Goold RG, Wood-Kaczmar A, Gordon-Weeks PR. Glycogen synthase kinase-3beta phosphorylation of MAP1B at Ser1260 and Thr1265 is spatially restricted to growing axons. J Cell Sci. 2005;118:993–1005. doi: 10.1242/jcs.01697. [DOI] [PubMed] [Google Scholar]
- 91.Takei Y, Teng J, Harada A, Hirokawa N. Defects in axonal elongation and neuronal migration in mice with disrupted tau and map1b genes. J Cell Biol. 2000;150:989–1000. doi: 10.1083/jcb.150.5.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gartner A, Huang X, Hall A. Neuronal polarity is regulated by glycogen synthase kinase-3 (GSK-3beta) independently of Akt/PKB serine phosphorylation. J Cell Sci. 2006;119:3927–34. doi: 10.1242/jcs.03159. By using neurons from the GSK3α-S21A/GSK3β-S9A double knock-in mice, this study shows clearly that phosphorylation of GSK3s at their N-terminal serine residues is not necessary for neuronal polarization, although inactivation of GSK3s is still required. [DOI] [PubMed] [Google Scholar]
- 93.Nishimura T, et al. PAR-6-PAR-3 mediates Cdc42-induced Rac activation through the Rac GEFs STEF/Tiam1. Nat Cell Biol. 2005;7:270–7. doi: 10.1038/ncb1227. [DOI] [PubMed] [Google Scholar]
- 94.Zhang X, et al. Dishevelled promotes axon differentiation by regulating atypical protein kinase C. Nat Cell Biol. 2007;9:743–54. doi: 10.1038/ncb1603. [DOI] [PubMed] [Google Scholar]
- 95.Nishimura I, Yang Y, Lu B. PAR-1 kinase plays an initiator role in a temporally ordered phosphorylation process that confers tau toxicity in Drosophila. Cell. 2004;116:671–82. doi: 10.1016/s0092-8674(04)00170-9. [DOI] [PubMed] [Google Scholar]
- 96.Kosuga S, et al. GSK-3beta directly phosphorylates and activates MARK2/PAR-1. J Biol Chem. 2005;280:42715–22. doi: 10.1074/jbc.M507941200. [DOI] [PubMed] [Google Scholar]
- 97.Timm T, et al. Glycogen synthase kinase (GSK) 3beta directly phosphorylates Serine 212 in the regulatory loop and inhibits microtubule affinity-regulating kinase (MARK) 2. J Biol Chem. 2008;283:18873–82. doi: 10.1074/jbc.M706596200. [DOI] [PubMed] [Google Scholar]
- 98.Wildonger J, Jan LY, Jan YN. The Tsc1-Tsc2 complex influences neuronal polarity by modulating TORC1 activity and SAD levels. Genes Dev. 2008;22:2447–53. doi: 10.1101/gad.1724108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ossipova O, Bardeesy N, DePinho RA, Green JB. LKB1 (XEEK1) regulates Wnt signalling in vertebrate development. Nat Cell Biol. 2003;5:889–94. doi: 10.1038/ncb1048. [DOI] [PubMed] [Google Scholar]
- 100.Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355:1345–56. doi: 10.1056/NEJMra055323. [DOI] [PubMed] [Google Scholar]
- 101.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–84. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 102.Polak P, Hall MN. mTOR and the control of whole body metabolism. Curr Opin Cell Biol. 2009;21:209–18. doi: 10.1016/j.ceb.2009.01.024. [DOI] [PubMed] [Google Scholar]
- 103.Plas DR, Thomas G. Tubers and tumors: rapamycin therapy for benign and malignant tumors. Curr Opin Cell Biol. 2009;21:230–6. doi: 10.1016/j.ceb.2008.12.013. [DOI] [PubMed] [Google Scholar]
- 104.Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 105.Choi YJ, et al. Tuberous sclerosis complex proteins control axon formation. Genes Dev. 2008;22:2485–95. doi: 10.1101/gad.1685008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Morita T, Sobue K. Specification of neuronal polarity regulated by local translation of CRMP2 and Tau via the mTOR-p70S6K pathway. J Biol Chem. 2009;284:27734–45. doi: 10.1074/jbc.M109.008177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Inoki K, et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006;126:955–68. doi: 10.1016/j.cell.2006.06.055. [DOI] [PubMed] [Google Scholar]
- 108.Yan D, Guo L, Wang Y. Requirement of dendritic Akt degradation by the ubiquitin-proteasome system for neuronal polarity. J Cell Biol. 2006;174:415–24. doi: 10.1083/jcb.200511028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhou FQ, Snider WD. Intracellular control of developmental and regenerative axon growth. Philos Trans R Soc Lond B Biol Sci. 2006;361:1575–92. doi: 10.1098/rstb.2006.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhou FQ, Zhou J, Dedhar S, Wu YH, Snider WD. NGF-induced axon growth is mediated by localized inactivation of GSK-3beta and functions of the microtubule plus end binding protein APC. Neuron. 2004;42:897–912. doi: 10.1016/j.neuron.2004.05.011. This paper identified a novel pathway that links NGF to axonal microtubules via GSK3 and a microtubule plus end tracking protein. It is the first study that illustrates how GSK3 signaling is involved in regulation of axon growth during neural development. [DOI] [PubMed] [Google Scholar]
- 111.Frame S, Cohen P, Biondi RM. A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Mol Cell. 2001;7:1321–7. doi: 10.1016/s1097-2765(01)00253-2. [DOI] [PubMed] [Google Scholar]
- 112.Dent EW, Gertler FB. Cytoskeletal dynamics and transport in growth cone motility and axon guidance. Neuron. 2003;40:209–27. doi: 10.1016/s0896-6273(03)00633-0. [DOI] [PubMed] [Google Scholar]
- 113.Lu W, Yamamoto V, Ortega B, Baltimore D. Mammalian Ryk is a Wnt coreceptor required for stimulation of neurite outgrowth. Cell. 2004;119:97–108. doi: 10.1016/j.cell.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 114.Lonze BE, Riccio A, Cohen S, Ginty DD. Apoptosis, axonal growth defects, and degeneration of peripheral neurons in mice lacking CREB. Neuron. 2002;34:371–85. doi: 10.1016/s0896-6273(02)00686-4. [DOI] [PubMed] [Google Scholar]
- 115.Vo N, Goodman RH. CREB-binding protein and p300 in transcriptional regulation. J Biol Chem. 2001;276:13505–8. doi: 10.1074/jbc.R000025200. [DOI] [PubMed] [Google Scholar]
- 116.Riccio A, et al. A nitric oxide signaling pathway controls CREB-mediated gene expression in neurons. Mol Cell. 2006;21:283–94. doi: 10.1016/j.molcel.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 117.Graef IA, et al. Neurotrophins and netrins require calcineurin/NFAT signaling to stimulate outgrowth of embryonic axons. Cell. 2003;113:657–70. doi: 10.1016/s0092-8674(03)00390-8. [DOI] [PubMed] [Google Scholar]
- 118.Mi K, Dolan PJ, Johnson GV. The low density lipoprotein receptor-related protein 6 interacts with glycogen synthase kinase 3 and attenuates activity. J Biol Chem. 2006;281:4787–94. doi: 10.1074/jbc.M508657200. [DOI] [PubMed] [Google Scholar]
- 119.Dajani R, et al. Crystal structure of glycogen synthase kinase 3 beta: structural basis for phosphate-primed substrate specificity and autoinhibition. Cell. 2001;105:721–32. doi: 10.1016/s0092-8674(01)00374-9. [DOI] [PubMed] [Google Scholar]
- 120.Hartigan JA, Xiong WC, Johnson GV. Glycogen synthase kinase 3beta is tyrosine phosphorylated by PYK2. Biochem Biophys Res Commun. 2001;284:485–9. doi: 10.1006/bbrc.2001.4986. [DOI] [PubMed] [Google Scholar]
- 121.Cole A, Frame S, Cohen P. Further evidence that the tyrosine phosphorylation of glycogen synthase kinase-3 (GSK3) in mammalian cells is an autophosphorylation event. Biochem J. 2004;377:249–55. doi: 10.1042/BJ20031259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Liu C, et al. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell. 2002;108:837–47. doi: 10.1016/s0092-8674(02)00685-2. [DOI] [PubMed] [Google Scholar]
- 123.Wei W, Jin J, Schlisio S, Harper JW, Kaelin WG., Jr The v-Jun point mutation allows c-Jun to escape GSK3-dependent recognition and destruction by the Fbw7 ubiquitin ligase. Cancer Cell. 2005;8:25–33. doi: 10.1016/j.ccr.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 124.Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499–511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Welcker M, et al. Multisite phosphorylation by Cdk2 and GSK3 controls cyclin E degradation. Mol Cell. 2003;12:381–92. doi: 10.1016/s1097-2765(03)00287-9. [DOI] [PubMed] [Google Scholar]
- 126.Kumar P, et al. GSK3beta phosphorylation modulates CLASP-microtubule association and lamella microtubule attachment. J Cell Biol. 2009;184:895–908. doi: 10.1083/jcb.200901042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Goold RG, Owen R, Gordon-Weeks PR. Glycogen synthase kinase 3beta phosphorylation of microtubule-associated protein 1B regulates the stability of microtubules in growth cones. J Cell Sci. 1999;112(Pt 19):3373–84. doi: 10.1242/jcs.112.19.3373. [DOI] [PubMed] [Google Scholar]
- 128.Lee H, et al. The microtubule plus end tracking protein Orbit/MAST/CLASP acts downstream of the tyrosine kinase Abl in mediating axon guidance. Neuron. 2004;42:913–26. doi: 10.1016/j.neuron.2004.05.020. [DOI] [PubMed] [Google Scholar]
- 129.van Diepen MT, et al. MyosinV controls PTEN function and neuronal cell size. Nat Cell Biol. 2009;11:1191–6. doi: 10.1038/ncb1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Morfini G, Szebenyi G, Elluru R, Ratner N, Brady ST. Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. EMBO J. 2002;21:281–93. doi: 10.1093/emboj/21.3.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Welsh GI, Miller CM, Loughlin AJ, Price NT, Proud CG. Regulation of eukaryotic initiation factor eIF2B: glycogen synthase kinase-3 phosphorylates a conserved serine which undergoes dephosphorylation in response to insulin. FEBS Lett. 1998;421:125–30. doi: 10.1016/s0014-5793(97)01548-2. [DOI] [PubMed] [Google Scholar]