

Abstract
The dynamic kinetic resolution of α-keto esters via asymmetric transfer hydrogenation has been developed as a technique for the highly stereoselective construction of structurally diverse β-substituted-α-hydroxy carboxylic acid derivatives. Through the development of a privileged m-terphenylsulfonamide for (arene)RuCl(monosulfonamide) complexes with a high affinity for selective α-keto ester reduction, excellent levels of chemo-, diastereo-, and enantiocontrol can be realized in the reduction of β-aryl- and β-chloro-α-keto esters.
1. INTRODUCTION
The enantioselective construction of α-hydroxy carboxylic acids remains an active area of research due to their prevalence in biologically active molecules1 and use in asymmetric synthesis.2 Reliable methods to prepare these compounds include enantioselective glycolate aldol and alkylation reactions,3,4 reduction or alkylation of α-keto esters,5 Passerini-type reactions,6 asymmetric cyanohydrin synthesis,7 and ester enolate oxygenations.8 Despite advances in these methodologies, the preparation of β-stereogenic glycolic acid derivatives remains much more challenging highlighting the importance of a generalizable strategy to access these substructures.
The reduction of α-keto esters to give β-stereogenic-α-hydroxy esters has largely been limited to the diastereoselective reduction of enantioenriched substrates.9 A more direct, efficient reaction manifold might arise from the asymmetric catalyst-controlled reduction of configurationally labile racemic β-substituted-α-keto esters. Such a reaction could in principle proceed with concomitant formation of two (or more) stereogenic centers in a single step and provide access to a number of functionalized glycolic acid derivatives (Scheme 1).10 This strategy pre-supposes the application of a dynamic kinetic resolution (DKR), a powerful tool for the conversion of racemic materials into enantiomerically enriched products.11 In light of the prominence and utility of DKR reactions of α-stereogenic-β-keto esters, the absence of complementary isomeric variants from racemic α-keto esters was surprising. As part of our laboratory’s continued interest in glycolic acid synthesis,12 we have recently developed a highly stereoselective dynamic kinetic resolution of β-stereogenic-α-keto esters via asymmetric transfer hydrogenation (DKR-ATH), yielding trisubstituted γ-butyrolactones (vide infra).13 It occurred to us that substantial product diversity might arise from a common mechanistic platform simply by varying the identities of the nonhydrogen substituents (X and Y) at the β-carbon. The successful creation of an attractive synthetic protocol would require: (1) simple routes to the needed racemic α-keto ester substrates; (2) reaction conditions that achieve rapid substrate racemization; (3) the identification of a reduction catalyst that is enantiomer-selective, provides strong facial bias during the diastereoselective reduction, and can be applied to functionally diverse substrates. The subject of this paper is the evaluation of this strategy and the presentation of a new (arene)Ru catalyst system for the asymmetric dynamic reduction of a range of racemic β-stereogenic-α-keto esters. The chemistry to be detailed was enabled by a seemingly trivial, yet ultimately crucial deviation from established art in asymmetric Ru-catalyzed transfer hydrogenation.
Scheme 1.

β-Stereogenic glycolic acid derivatives via reduction
2. RESULTS AND DISCUSSION
2.1. Ligand/Catalyst Design
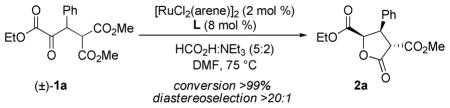
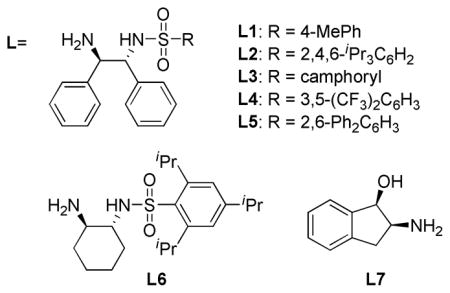
Inspired by the efficacy of Noyori’s (arene)RuCl(monosulfonamide-DPEN)14 in both the asymmetric reduction of simple ketones15 and the dynamic reduction of α-substituted β-keto esters and amides,16 we took this complex as our point of departure in ligand/ catalyst design. Utilizing formic acid:triethylamine (5:2 mixture)17 as the organic reductant and 1a as a test substrate, a screen of ligands and precatalysts was undertaken (Table 1). Initial studies looked at the steric effects of the sulfonamide in Ru(II)-complexes possessing a (1S,2S)-diphenylethylenediamine (DPEN) backbone. Subjecting 1a to 2 mol % of the ruthenium dimer [RuCl2(arene)]2 and Noyori’s ligand L1 (Ru atom: L mole ratio 1:2) in DMF at 75 °C18 provided the desired γ-butyrolactone in high yield (90%) and diastereoselectivity (>20:1 dr), but with low levels of enantiocontrol (57:43 er, entry 1). DPEN-based ligands featuring bulkier sulfonamides (L2–L5) provided only modestly higher levels of selectivity (entries 2–5). Employing L2, a screen of (arene)Ru(II)-precatalysts was conducted to determine the role of the arene in the stereoselectivity of the reduction; however, no improvements were observed moving away from [RuCl2(p-cymene)]2 (entries 6 and 7). In addition to DPEN, 1,2-diaminocyclohexane and 1,2-aminoindanol were also investigated as chiral backbones (L6 and L7), but yielded comparable results (entries 8 and 9).
Table 1.
Evaluation of chiral diamine ligands
| |||
|---|---|---|---|
| entry | L | arene | er |
| 1 | L1 | p-cymene | 57:43 |
| 2 | L2 | p-cymene | 70:30 |
| 3 | L3 | p-cymene | 62:38 |
| 4 | L4 | p-cymene | 70:30 |
| 5 | L5 | p-cymene | 72:28 |
| 6 | L2 | C6Me6 | 62:38 |
| 7 | L2 | benzene | 71:29 |
| 8 | L6 | p-cymene | 73:27 |
| 9 | L7 | p-cymene | 60:40 |
Conditions: 1a (1.0 equiv), [RuCl2(arene)]2 (0.02 equiv), L (0.08 equiv), HCO2H:NEt3 (5.0 equiv), [1a]0 = 0.1 M in DMF, 75 °C, 16 h.
Based on these preliminary findings that asymmetric transfer hydrogenation catalysts from this family present in the literature were found to provide inadequate levels of selectivity, it became clear that new chiral space would need to be explored in order achieve high levels of enantiocontrol. Utilizing the “mother diamine”/diaza-Cope approach to the synthesis of C2-symmetric 1,2-diamines,19 screening of a number of chiral diamine backbones was conducted. The α-naphthyl/triisopropylbenzenesulfonamide ligand L8 considerably increased the selectivity (88:12 er, Table 2). To further optimize the ligand structure, perturbations of the sulfonamide were examined due to its apparent ability to directly impact the chiral environment (Table 1, L1 vs. L2). Since less sterically encumbering sulfonamides (L9–L12) resulted in erosions in selectivity, we sought to synthesize bulkier sulfonamides by exploring new chiral space at the 2,6-positions of the arylsulfonamide.20 A number of diverse sulfonyl chlorides were synthesized through a one-pot double alkylation/sulfonylation of 1,3-dichlorobenzene.21 The simplest m-terphenyl sulfonamide variant L14, distinguished itself as being uniquely effective for providing high levels of enantioselectivity for the title reaction (95:5 er). The use of electron-withdrawing (L15) or releasing substituents (L16) provided no improvement in selectivity. The α-naphthyl backbone and m-terphenylsulfonamide operate synergistically; no improvement in enantiocontrol with the DPEN/m-terphenylsulfonamide ligand L5 was observed (Table 1, entry 5). The α-naphthyl ethylenediamine backbone has been used sporadically in asymmetric synthesis and the use of the m-terphenylsulfonamide for enantioselective catalysis is rarer still.22
Table 2.
Evaluation of sulfonamides on α-naphthyl backbone

|
Conditions: As described for Table 1.
2.2. Synthesis of Trisubstituted γ-Butyrolactones
This DKR-ATH was found to be applicable for a range of β-aryl α-keto esters.13 Electron-rich, electron-poor, and heteroaryl substituents were tolerated at the β-position providing γ-butyrolactone products in high yield and enantioselectivity. 23 Additionally, the reduction of 1g was performed on a 10 g scale employing reduced catalyst loading (1 mol % Ru) yielding enantiopure lactone 2g in 72% yield following a single recrystallization. The absolute stereochemistry of the trisubstituted γ-butyrolactone products was determined by X-ray crystallographic analysis of 2b.24
The obtained functionally rich γ-butyrolactones can be deployed in secondary transformations (Scheme 2). Diastereoselective alkylation of 2a employing allyl bromide and DBU provided tetrasubstituted lactone 3a bearing an all-carbon quaternary carbon center in high yield. Krapcho decarboxylation25 gave access to α-unsubstituted lactone 3b (86% yield) that formally arises from cinnamic acid. When decarboxylation was preceded by alkylation with dibromomethane, dehalodecarboxylation26 resulted and afforded α-alkylidene γ-butyrolactone 3c. This substructure is prominent in natural product chemistry and bioactivities within this subclass are well documented.27
Scheme 2.
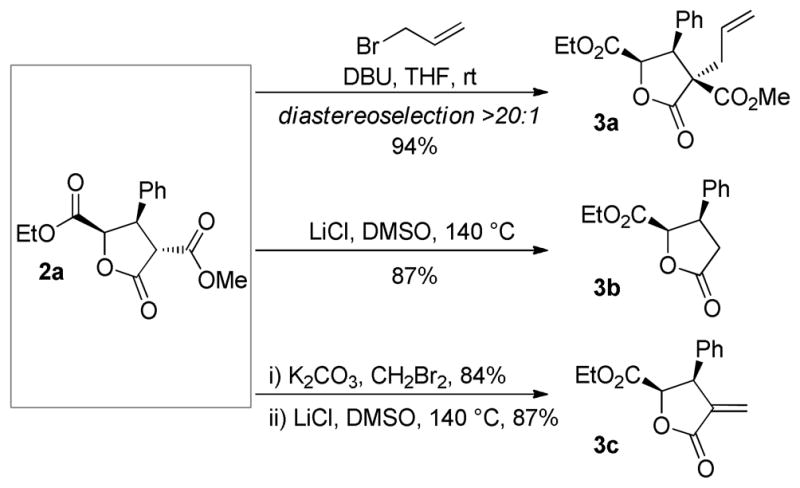
Reactions of lactone reduction product 2a
2.3. “Free” Glycolate Michael Adducts Using Chemoselective DKR-ATH of α,δ-Diketo Esters
2.3.1 DKR-ATH Approach to “Free” Glycolate Michael Adducts
Access to δ-oxygenated glycolic acid derivatives via asymmetric glycolate Michael reactions is limited. The most common approach to this class of compounds is the addition of stoichiometric chiral glycolate enolates to α,β-unsaturated ketones and esters,28 and in each case the protected glycolate is obtained (Scheme 3a). Only recently has a catalytic enantioselective variant been disclosed; that method uses oxazolones as the α-hydroxy acid surrogate (Scheme 3b).29 To the best of our knowledge, no catalytic enantioselective Michael addition of a free glycolate enolate has been reported.30
Scheme 3.
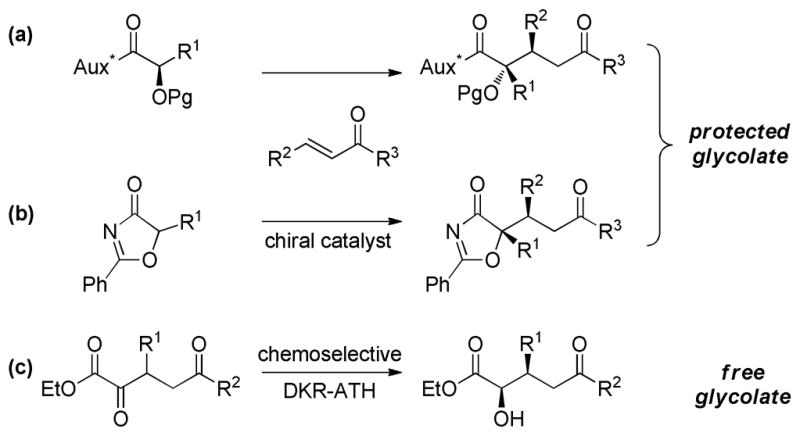
Approaches to δ-oxygenated glycolic acid derivatives
Based on the success of the DKR-ATH of γ,γ-dicarboalkoxy-α-keto esters (1, vide supra), we postulated that β-substituted-δ-keto-α-hydroxy esters might be accessible via a chemoselective dynamic reduction of α,δ-diketo esters directly delivering the formal “free” glycolate Michael adducts. Implicit in this analysis is the need for complete site selectivity in the reduction. Methods for the selective reduction of an aldehyde in the presence of less reactive carbonyls, i.e. ketones and esters, are well-established. 31 While significant progress has been made in achieving the inverse process, the selective reduction of a ketone in the presence of an aldehyde,32 the discrimination between two ketones remains a challenging task. Examples of the latter include the chemoselective reduction of 2,4-diketo acid derivatives using cinchona-modified platinum catalysts33 or baker’s yeast34 and aluminium-mediated selective reductions of diaryl ketones.35 Our tactic takes advantage of the heightened reactivity enjoyed by α-dicarbonyls and establishes a simple catalytic method for achieving the formal asymmetric glycolate Michael construction without recourse to auxiliary control or protection of the hydroxyl group (Scheme 3c).
2.3.2 Glyoxylate Stetter Addition with Enones
Indirect methods for the preparation of the requisite α,δ-diketo esters 5 have been reported by us10b and others,36 but the most direct and atom economical approach to these substrates would be a new Stetter reaction between commercial ethyl glyoxylate and α,β-enones.37 This reaction was previously reported to be unsuccessful with thiazolium carbenes,36c but we have found that effective catalysis can be realized by employing the Rovis triazolium carbene derived from salt 4.38 As outlined in Table 4, this glyoxylate Stetter addition is highly efficient, tolerant of a number of ketonic substrates and substituents at the β-position and can be performed on a multigram scale (entry 1). Notably, with the 1,4-dien-3-one dibenzyli-deneacetone, exclusive monoaddition was observed (entry 5).
Table 4.
Scope of the glyoxylate Stetter addition with enonesa
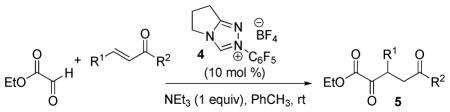
| ||||
|---|---|---|---|---|
| entry | R1 | R2 | 5 | yield (%)b |
| 1c | C6H5 | C6H5 | 5a | 97 |
| 2 | C6H5 | 4-I-C6H4 | 5b | 95 |
| 3 | C6H5 | 4-MeO-C6H5 | 5c | 86 |
| 4 | C6H5 | Me | 5d | 72 |
| 5 | C6H5 | (E)–CH=CHPh | 5e | 91 |
| 6 | C6H5 | piperonyl | 5f | 92 |
| 7 | 4-Cl-C6H4 | C6H5 | 5g | 94 |
| 8 | 4-MeO-C6H4 | C6H5 | 5h | 96 |
| 9 | 2-Me-C6H4 | C6H5 | 5i | 70 |
| 10 | 3-Me-C6H4 | C6H5 | 5j | 93 |
| 11 | 4-Me-C6H4 | C6H5 | 5k | 95 |
| 12 | CO2Et | C6H5 | 5l | 92 |
| 13 | piperonyl | C6H5 | 5m | 94 |
| 14 | N-Boc-Indol-3-yl | C6H5 | 5n | 74 |
Conditions: Unless otherwise noted, all reactions were performed on a 2.0 mmol scale in PhCH3 (4 mL) at ambient temperature.
Isolated yield.
Reaction performed on a 20 mmol scale.
2.3.3 Chemoselective DKR-ATH of α,δ-Diketo Esters
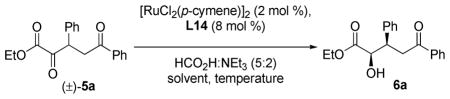
Our investigation into the chemoselective DKR-ATH began with an examination of the reduction of α,δ-diketo ester 5a. As shown in Table 5, our initial studies confirmed the feasibility of selectively reducing the α-keto ester in the presence of an aryl ketone, as we observed exclusive formation of δ-keto-α-hydroxy ester 6a as a single diastereomer. Subjecting 5a to our previously optimized reaction conditions, 2 mol % of [RuCl2(p-cymene)]2 and diamine ligand L14 (Ru atom:L mole ratio 1:2) in DMF at 70 °C using HCO2H/Et3N as the reductant, provided 6a in 96% yield and a 91:9 er (entry 1). Lower levels of selectivity were observed when the reduction was conducted at room temperature (87:13 er, entry 2). Further optimization revealed that high levels of enantioselectivity (97:3 er) could be obtained by performing the reaction in DMSO at room temperature (entry 6). The chemoselectivity observed is remarkable in light of the extensive use of (arene)RuCl(sulfonamide) complexes for asymmetric transfer hydrogenation of aryl ketones.14,15,17
Table 5.
Chemoselective DKR-ATH: reaction optimization a
| ||||
|---|---|---|---|---|
| entry | solvent | T (°C) | yield (%)a | erc |
| 1 | DMF | 70 | 94 | 91:9 |
| 2 | DMF | rt | 93 | 87:13 |
| 3 | 2-MeTHF | 70 | 90 | 65:35 |
| 4 | DCE | 70 | 91 | 78:22 |
| 5 | DMSO | 70 | 96 | 97:3 |
| 6 | DMSO | rt | 98 | 97:3 |
Conditions: 5a (1.0 equiv), [RuCl2(p-cymene)]2 (0.02 equiv), L14 (0.08 equiv), HCO2H:NEt3 (5.0 equiv), [5a]0 = 0.1 M, 16 h.
Isolated yield.
Enantiomeric ratio determined by SFC analysis.
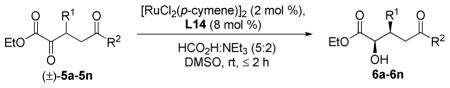
With optimal reaction conditions in hand, we next explored other substrates in the chemoselective dynamic reduction (Table 6). For all substrates examined, exclusive reduction of the α-keto ester was observed, irrespective of the electronic characteristics of the δ-ketone. High yields and enantioselectivities were obtained for substrates incorporating electron-rich, electron-poor, and heteroaryl substituents at the β-position. The level of selectivity for 6l (entry 12, 91:9 er) is noteworthy as this demonstrates that the scope is not limited to β-aryl substrates (vide infra). 39 Additionally, other reducible functional groups remained intact: the retention of the α,β-enone in 6e under the reaction conditions further highlights the catalyst’s strong preference for the α-keto ester (entry 5). Aryl halides were also tolerated (entries 2,7). To evaluate the catalytic efficiency of this system, the reduction of 5a was performed using 0.05 mol % of [RuCl2(p-cymene)]2; no loss in reaction efficiency was observed as 6a was obtained in 98:2 er (entry 1).
Table 6.
Chemoselective dynamic reduction scopea
| |||||
|---|---|---|---|---|---|
| entry | R1 | R2 | 6 | yield (%)b | erc |
| 1d | C6H5 | C6H5 | 6a | 97 | 98:2 |
| 2 | C6H5 | 4-I-C6H4 | 6b | 95 | 96:4 |
| 3 | C6H5 | 4-MeO-C6H5 | 6c | 86 | 96:4 |
| 4 | C6H5 | Me | 6d | 72 | 93:7 |
| 5 | C6H5 | (E)–CH=CHPh | 6e | 91 | 99:1 |
| 6 | C6H5 | piperonyl | 6f | 92 | 99:1 |
| 7 | 4-Cl-C6H4 | C6H5 | 6g | 94 | 98:2 |
| 8 | 4-MeO-C6H4 | C6H5 | 6h | 96 | 96:4 |
| 9 | 2-Me-C6H4 | C6H5 | 6i | 70 | 83:17 |
| 10 | 3-Me-C6H4 | C6H5 | 6j | 93 | 98:2 |
| 11 | 4-Me-C6H4 | C6H5 | 6k | 95 | 98:2 |
| 12 | CO2Et | C6H5 | 6l | 92 | 91:9 |
| 13 | piperonyl | C6H5 | 6m | 94 | 97:3 |
| 14 | N-Boc-Indol-3-yl | C6H5 | 6n | 95 | 98:2 |
Conditions: as described for Table 5.
Isolated yield.
Enantiomeric ratio determined by SFC analysis.
Reaction performed using 0.05 mol % [RuCl2(p-cymene)]2.
The absolute configuration and syn-stereochemical relationship of the α-hydroxy ester products were determined by converting 6c to lactone 3b via a Baeyer-Villiger oxidation followed by in situ lactonization; spectral data and optical rotation were in agreement with those previously obtained by us for 3b.13
To determine if the [RuCl2(p-cymene)]2/L14 catalyst system was uniquely effective for the reduction of α-keto esters, the chemoselectivity of transfer hydrogenation catalysts known to reduce simple ketones was evaluated with 5a (Scheme 5). The use of ligand L2 in the reduction, which is known to reduce acetophenone,40 also afforded 6a as the sole product, albeit with lower levels of enantioselectivity (69:31 er). This result caused us to wonder if the δ-ketone was possibly undergoing in situ “protection” as the lactol 7 following reduction of the α-keto ester and that this intermediate masked the δ-ketone from further reduction. To test this hypothesis, the reduction was monitored by 1H NMR spectroscopy in DMSO-d6, but 7 was not detected: only the diketo ester 5a and hydroxy ester 6a were observed (Figure 1).
Scheme 5.

Investigations into the observed chemoselectivity
Figure 1.

In situ monitoring of the reduction of (±)-5a by 1H NMR spectroscopy in DMSO-d6. Blue panel: α,δ-diketo ester 5a; Red panel: in situ monitoring of reduction of α,δ-diketo ester 5a to 6a (~50% conversion); Green panel: α-hydroxy-δ-keto ester 6a (with added HCO2H/Et3N).
We then examined the transfer hydrogenation of several other ketone substrates using the standard reaction conditions used in Table 6 (Scheme 6). Interestingly, acetophenone and ethyl 2-ethyl-3-oxobutanoate, which are prototypical test substrates for new transfer hydrogenation catalysts, are not reduced with this catalyst system. This lack of reactivity further highlights the unique preference for α-keto esters conferred by the newly developed terphenylsulfonamide/di-α-napthylethylenediamine ligand. Reducing tert-butyl 2-oxo-4-phenylbutanoate under the standard conditions proceeded efficiently, imparting good levels of enantioselection and highlighting the potential applicability of this catalyst for simple α-keto esters.
Scheme 6.

Other keto ester reductions
2.4. Chlorohydrin Synthesis
2.4.1 DKR-ATH Approach to Optically Active Chlorohydrins
The investigations described above provide simple access to useful enantiomerically enriched glycolate building blocks, but Scheme 1 implies a goal of diversification in product structures that had not yet been realized. In considering new substrates that might be useful for DKR-ATH reactions, the potential integration of β-halo substituents was appealing on several levels. Optically active halohydrins are fundamental building blocks in organic chemistry and these functional arrays can be converted to their derived enantioenriched epoxides or engage in nucleophilic substitution to provide a variety of functionalized product classes (Scheme 7). The emergence of halohydrin dehalogenase (HheC), an enzyme produced by Agrobacterium radiobacter AD1, as a biocatalyst for the kinetic resolution of racemic haloalcohols highlights the importance of methods for the preparation of optically pure terminal halohydrins.41 The catalytic asymmetric preparation of halohydrins has been limited principally to desymmetrization reactions of epoxides42 and alkenes43 or kinetic resolution of terminal epoxides.42d,42g,42l,42m,44 Methodology designed to directly access internal halohydrins from unsymmetrical precursors is largely underdeveloped (vide infra). We sought to develop a stereoselective Ru-catalyzed dynamic kinetic resolution of β-chloro-α-keto esters that would provide an efficient route to β-chloro-α-hydroxy esters.
Scheme 7.

Secondary transformations of chlorohydrin products.
Access to chiral β-chloro glycolic acid derivatives is currently limited to enzymatic processes or stereospecific opening of glycidic esters with strong acids. While enzymatic reductions45 and kinetic resolutions46 have been shown to impart good levels of diastereo-and enantioselectivity, these processes are substrate limited and lack generality. Chloride addition to optically pure glycidic esters often necessitates harsh reaction conditions, suffers from non-ideal regio- and stereoselectivity, and lacks significant precedent for aliphatic substrates.47 The dynamic kinetic resolution of α-chloro-β-keto esters has been developed for some time,48 but the dynamic kinetic resolution of β-chloro-α-keto esters that would provide isomeric products is heretofore unknown. Thus, we sought to develop a simple, flexible synthesis of β-chloro glycolic acid derivatives employing a dynamic kinetic resolution asymmetric transfer hydrogenation (DKR-ATH) of β-chloro-α-keto esters (Figure 2).
Figure 2.
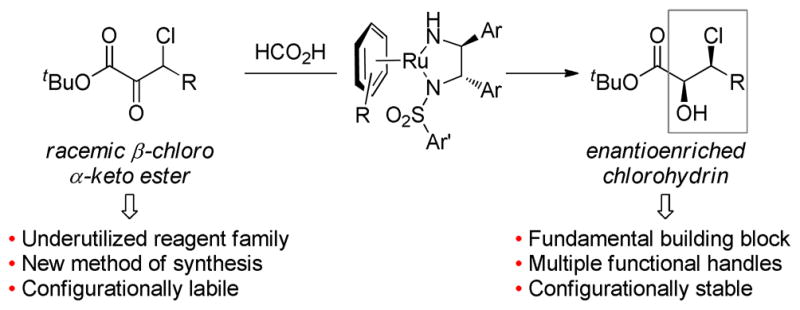
Dynamic kinetic resolution of β-chloro-α-keto esters.
2.4.2 Direct Catalytic β-Chlorination of α-Keto Esters
The relative dearth of direct, catalytic β-functionalizations of α-keto esters presented an obstacle to the implementation of this synthetic plan; in particular, methods for the direct β-halogenation of α-keto esters are scarce.9k,45b,45c,49 Only two examples of the direct β-chlorination of singly activated α-keto esters have been reported and those require either long reaction times or harsh reaction conditions.45b,49a In order to synthesize the requisite β-chloro substrates for the anticipated DKR-ATH, the development of a mild chlorination reaction of α-keto esters was pursued. A screen of various Cu(II)-diamine and Ni(II)-diamine complexes led to the identification of Sodeoka’s Ni(OAc)2-diamine complex 89i as an effective catalyst for the β-chlorination of α-keto esters 9 using N-chlorosuccinimide (NCS) under mild reaction conditions.50 As outlined in Table 7, the chlorination proceeds with good selectivity for the singly halogenated product, is tolerant of a variety of functionalized aliphatic substrates, and can be performed on a multigram scale (entry 1). The elevated acidity of β-aryl substrates favored dichlorination (entry 9); therefore, the requisite β-aryl substrates were prepared via Darzens reaction.51
Table 7.
Ni(II)-catalyzed β-chlorination of α-keto esters.a
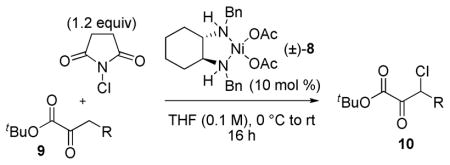
| ||||
|---|---|---|---|---|
| entry | R | 10 | mono:dib | yield (%)c |
| 1d | –CH2Ph | 10a | 9:1 | 81 |
| 2 | –CH2p-ClPh | 10b | 10:1 | 83 |
| 3 | –CH2p-MeOPh | 10c | 12:1 | 84 |
| 4 | –CH2CH2Ph | 10d | 8:1 | 78 |
| 5 | –CH2CH=CH2 | 10e | 13:1 | 86 |
| 6 | –CH2C≡CTMS | 10f | 10:1 | 79 |
| 7 | –(CH2)2CH3 | 10g | 13:1 | 87 |
| 8 | –(CH2)3OBn | 10h | 13:1 | 86 |
| 9 | –Ph | 10i | 1:>20 | trace |
Unless otherwise noted, all reactions were performed on a 1.0 mmol scale.
Determined by 1H NMR analysis of the crude reaction mixture.
Isolated yield.
Performed on 8.0 mmol scale.
2.4.3 DKR-ATH of β-Chloro-α-Keto Esters
The reduction of β-chloro-α-keto esters with NaBH4 proceeds with high levels of diastereoselectivity to afford the syn-diastereomer via Felkin-Ahn control.46a Initial investigations into the DKR-ATH of 10a revealed that ethylenediamine-derived 11a also afforded excellent levels of syn-selectivity (Figure 3); however, the diastereochemical outcome was powerfully influenced by ligand selection. Upon switching to Noyori’s parent Ru(II)-complex possessing a (1S,2S)-diphenylethylenediamine (DPEN) backbone (11b),14 a significant erosion in syn-diastereoselection was observed, albeit with promising levels of enantioselectivity. The bulkier triisopropyl-DPEN ligand (11c) gave modest anti-diastereoselection with improved enantiocontrol. Exploiting the unique properties associated with the terphenylsulfonamide (vide supra), the DPEN-derivative 11d led to appreciable ligand-controlled diastereoselection providing anti-12a with excellent levels of enantioselectivity. The α-naphthyl backbone (11e) employed in the reduction of β-aryl-α-keto esters was found to provide slightly higher levels of diastereoselection albeit with a small loss in enantioselectivity. The diastereoselectivity and enantioselectivity were further improved in DMF at 0 °C employing only 1 mol % of the Ru catalyst (in this case, the conveniently prepared and stored dehydrohalgenated variant of 11d). Considering diastereoselectivity only, the continuum expressed by Figure 3 (>20:1 syn:anti → 1:9 syn:anti at 298 K) represents approximately 2.5 kcal/mol modulation of diastereomeric transition states through simple substituent modifications on a common ligand framework.
Figure 3.

Ligand-controlled switch in diastereoselectivity.a
aReactions were performed on 0.155 mmol scale employing 5 equiv. HCO2H:NEt3 (5:2). bDetermined by 1H NMR analysis of the crude reaction mixture. cDetermined by chiral SFC analysis. dPerformed with 1 mol % catalyst in DMF (0. 1 M) at 0 °C for 10 h. Complex 11da is the dehydrohalogenated variant of 11d; the structure is illustrated in Table 8.
With optimized reaction conditions in hand the relationship between α-keto ester structure and reaction stereoselectivity was assayed (Table 8). A variety of aliphatic substrates were found to be amenable to the reaction conditions providing β-chloro α-hydroxy esters with excellent levels of diastereo- and enantioselection. Alkene, alkyne, and benzyloxy functionality was tolerated under the reaction conditions offering value-added functional handles. The method was also scalable (entry 1). The efficiency of these aliphatic substrates under the DKR-ATH reaction conditions is a marked structural departure from the β-aryl and β-ester requirements in antecedent work from our group and the paradigm that aryl groups are necessary for high levels of enantiocontrol due to ligand/substrate π/C–H interactions.52
Table 8.
β-aliphatic substrates in the DKR-ATH.a
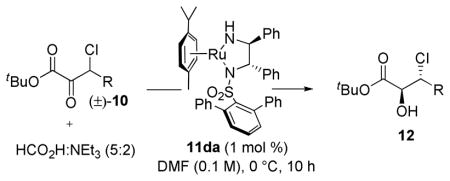
| |||||
|---|---|---|---|---|---|
| entry | R | 12 | yield (%)b | drc | erd |
| 1e | –CH2Ph | 12a | 89 | 12:1 | 99:1 |
| 2 | –CH2p-ClPh | 12b | 90 | 16:1 | 99.5:0.5 |
| 3 | –CH2p-MeOPh | 12c | 93 | 20:1 | 98.5:1.5 |
| 4 | –CH2CH2Ph | 12d | 93 | 16:1 | 98.5:1.5 |
| 5 | –CH2CH=CH2 | 12e | 95 | >20:1 | 98:2f |
| 6g | –CH2C≡CTMS | 12f | 91 | >20:1 | 96.5:3.5f |
| 7 | –(CH2)2CH3 | 12g | 94 | >20:1 | 98.5:1.5f |
| 8 | –(CH2)3OBn | 12h | 91 | 18:1 | 98:2 |
Unless otherwise noted, all reactions were performed on 0.155 mmol scale employing 5 equiv. HCO2H:NEt3 (5:2).
Isolated yield of anti-diastereomer.
Determined by 1H NMR analysis of the crude reaction mixture.
Determined by chiral SFC/HPLC analysis.
Performed on 6.5 mmol scale.
Determined following conversion to the benzoate (see Supporting Information).
Performed at 23 °C for 10 h.
Compatibility with β-aryl substrates was also demonstrated under the optimized reaction conditions, providing adducts with excellent levels of enantioselectivity (Table 9). The electronic character of the aromatic ring was found to significantly impact the diastereoselectivity of the reaction. Electron-releasing groups engendered excellent diastereocontrol (entries 2, 3, and 10) whereas electron-withdrawing groups provided somewhat lower diastereoselection (entries 4, 7, and 8).
Table 9.
Scope of β-aryl substrates in the DKR-ATH.a
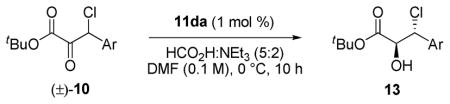
| |||||
|---|---|---|---|---|---|
| entry | Ar | 13 | yield (%)b | drc | erd |
| 1 | Ph | 13a | 93 | 14:1 | 99.5:0.5 |
| 2 | o-MeOPh | 13b | 95 | >20:1 | 99:1 |
| 3 | m-MeOPh | 13c | 94 | 19:1 | 99.5:0.5 |
| 4 | m-NO2Ph | 13d | 74 | 5:1 | 97.5:2.5 |
| 5 | p-ClPh | 13e | 85 | 10:1 | 98.5:1.5 |
| 6 | p-CF3Ph | 13f | 80 | 8:1 | 98.5:1.5 |
| 7 | p-CNPh | 13g | 82 | 6:1 | 98:2 |
| 8 | p-NO2Ph | 13h | 73 | 4:1 | 99:1 |
| 9 | p-MePh | 13i | 91 | 14:1 | 99.5:0.5 |
| 10 | p-MeOPh | 13j | 93 | >20:1 | 99:1 |
Reactions were performed on 0.155 mmol scale employing 5 equiv. HCO2H:NEt3 (5:2).
Isolated yield of anti-diastereomer.
Determined by 1H NMR analysis of the crude reaction mixture.
Determined by chiral SFC/HPLC analysis.
The absolute stereochemistry of the products was determined by comparison to the known epoxide (2R,3S)-14,53 which was synthesized from chlor0hydrin (2S,3R)-13a upon exposure to KOtBu (Scheme 7). To highlight the synthetic utility of the enantioenriched chlorohydrins as synthetic building blocks, illustrative secondary transformations were pursued. Treatment of chlorohydrin 13a with NaN3 afforded the azido alcohol 15 representing a formal synthesis of the paclitaxel C13 side-chain.47b Notably, the syn-product 15 is stereocomplementary to the anti diastereomer obtained from the glycidic esters that one might derive from Darzens or Weitz-Scheffer reactions. Following triflate formation of chloroalcohol 12a, chemoselective displacement with NaN3 affords α-azido-β-chloro ester 16 providing access to β-chloro-α-amino acid derivatives.
The DKR-ATH is also amenable to the reduction of β-fluoro-α-keto esters. Preliminary investigations have revealed that ketone 17 can be reduced under the optimized reaction conditions to afford the derived fluorohydrin 18 in excellent yield as a mixture of diastereomers (Scheme 8). Despite the lack of diastereocontrol in the reaction,54 excellent levels of enantioinduction were observed for both diastereomers. This initial finding is quite encouraging in light of the responsiveness of diastereocontrol to ligand structure in this reaction family (vide supra).
Scheme 8.

DKR-ATH of β-fluoro-α-keto ester 17.
3. CONCLUSIONS
In summary, we have designed new (arene)Ru(monosulfonamide) asymmetric transfer hydrogenation catalysts that have led to the successful development of a highly modular dynamic kinetic resolution of β-substituted-α-keto esters. The productive merger of a common mechanistic framework and a new m-terphenylsulfonamide-based catalyst system allow for rapid, atom-economical construction of highly functionalized glycolic acid derivatives with excellent levels of chemo-, diastereo-, and enantioselectivity.
A formal asymmetric glycolate Michael reaction has been established via a catalytic asymmetric chemoselective dynamic reduction of α,δ-diketo esters. The latter are prepared via a new atom economical carbene-catalyzed Stetter reaction between commercial ethyl glyoxylate and α,β-enones. The enantioselective reduction proceeds with high enantio- and diastereoselectivity for a number of substrates. Initial investigations into the origin of the observed selectivity suggests that the [RuCl2(p-cymene)]2/L14 catalyst system is uniquely effective for the reduction of α-keto esters, as other ketone substrates (even acetophenone) are unreactive under the standard reaction conditions.
Additionally, a highly stereoselective synthesis of β-chloro glycolic acid derivatives via asymmetric transfer hydrogenation was developed. A Ni(II)-catalyzed chlorination of aliphatic α-keto esters was developed to provide the requisite β-chloro-α-keto esters. In the reduction of these ketones, careful catalyst tuning allows for a remarkable ligand-controlled inversion of the preference for syn-selectivity to provide access to anti-chlorohydrins. The DKR-ATH proceeds with high levels of diastereo- and enantioselectivity for a number of aliphatic and aromatic substrates. The obtained chlorohydrins are versatile chemical building blocks for valuable secondary transformations.
These studies collectively provide diverse glycolate-based building blocks for synthesis. This study has highlighted some of the preparative and practical aspects of these reactions, but open questions with respect to mechanism clearly remain (Scheme 9). Additional studies to understand the catalyst-substrate interactions that account for the high levels of selectivity observed are ongoing, with the goal of utilizing of this information in extensions to the dynamic kinetic resolution of other useful frameworks.
Scheme 9.
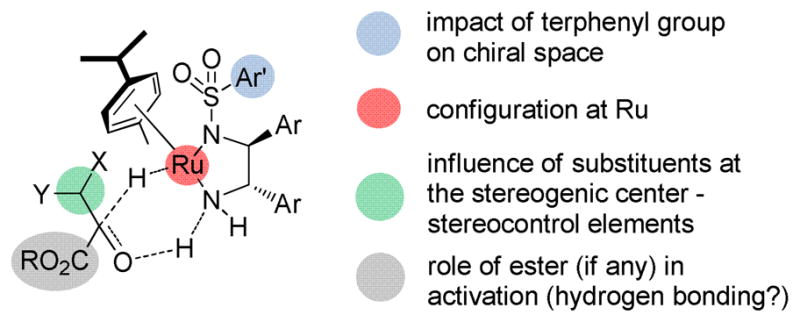
Open mechanistic questions.
Supplementary Material
Scheme 4.

Stereochemical analysis of δ-keto-α-hydroxy esters
Table 3.
DKR-ATH substrate scopea
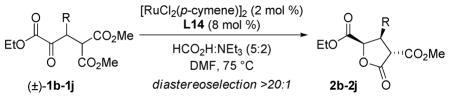
| ||||
|---|---|---|---|---|
| entry | R2 | 2 | yield (%)b | erc |
| 1 | 4-Cl-C6H4 | 2b | 94 | 96:4 |
| 2 | 4-Me-C6H4 | 2c | 84 | 95.5:4.5 |
| 3 | 4-MeO-C6H5 | 2d | 90 | 95:5 |
| 4 | 4-CN-C6H5 | 2e | 88 | 95:5 |
| 5 | 2-Me-C6H5 | 2f | 82 | 89:11 |
| 6d | piperonyl | 2g | 72 | 95:5 |
| 7 | 2-furyl | 2h | 91 | 95:5 |
| 8 | N-Ts-indol-3-yl | 2i | 91 | 96.5:3.5 |
| 9 | N-Boc-indol-3-yl | 2j | 88 | 96:4 |
Conditions: As described for Table 1.
Isolated yield.
Enantiomeric ratio determined by HPLC/SFC analysis.
[RuCl2(p-cymene)]2 (0.5 mol %), L14 (2 mol %), 10 g scale, >99.5:0.5 recrystallized.
Acknowledgments
The project described was supported by Award No. R01 GM084927 from the National Institute of General Medical Sciences. K.M.S. acknowledges an NIH NRSA Fellowship to Promote Diversity in Health-Related Research.
Footnotes
Funding Sources
No competing financial interests have been declared.
Experimental procedures, spectral and HPLC/SFC data and crystallographic data (CIF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Cooper A, Meister GA. Chem Rev. 2002;102:471–490. [Google Scholar]
- 2.Coppola GM, Schuster HF. Hydroxy Acids in Enantioselective Synthesis. VCH; Weinheim: 1997. [Google Scholar]
- 3.For a review see: Ley SV, Sheppard TD, Myers RM, Chorghade MS. Bull Chem Soc Jpn. 2007;80:1451–1472.
- 4.For examples see: Crimmins MT, Long A. Org Lett. 2005;7:4157–4160. doi: 10.1021/ol0515107.Fanjul S, Hulme AN. J Org Chem. 2008;73:9788–9791. doi: 10.1021/jo801720v.Chang JW, Jang DP, Uang BH, Liao FL, Wang SL. Org Lett. 1999;1:2061–2063.Crimmins MT, Stanton MG, Allwein SP. J Am Chem Soc. 2002;124:5958–5959. doi: 10.1021/ja026269v.Andrus MB, Hicken EJ, Stephens JC, Bedke DK. J Org Chem. 2005;70:9470–9479. doi: 10.1021/jo051568z.
- 5.(a) Blaser HU, Jalett HP, Muller M, Studer M. Catal Today. 1997;37:441–463. [Google Scholar]; (b) Li H, Wang B, Deng L. J Am Chem Soc. 2006;128:732–733. doi: 10.1021/ja057237l. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wang F, Xiong Y, Liu X, Feng X. Adv Synth Catal. 2007;349:2665–2668. [Google Scholar]
- 6.(a) Seebach D, Adam G, Gees T, Schiess M, Weigand W. Chem Ber. 1988;121:507–517. [Google Scholar]; (b) Denmark SE, Fan Y. J Am Chem Soc. 2003;125:7826–7827. doi: 10.1021/ja035410c. [DOI] [PubMed] [Google Scholar]; (c) Kusebauch U, Beck B, Messer K, Herdtweck E, Dömling A. Org Lett. 2003;5:4021–4024. doi: 10.1021/ol035010u. [DOI] [PubMed] [Google Scholar]; (d) Andreana PR, Liu CC, Schreiber SL. Org Lett. 2004;6:4231–4233. doi: 10.1021/ol0482893. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Denmark SE, Fan Y. J Org Chem. 2005;70:9667–9676. doi: 10.1021/jo050549m. [DOI] [PubMed] [Google Scholar]; (f) Wang SX, Wang MX, Wang DX, Zhu J. Angew Chem, Int Ed. 2007;47:388–391. doi: 10.1002/anie.200704315. [DOI] [PubMed] [Google Scholar]
- 7.For reviews on asymmetric cyanohydrin synthesis see: Effenberger F. Angew Chem Int Ed Engl. 1994;33:1555–1564.Gregory RJH. Chem Rev. 1999;99:3649–3682. doi: 10.1021/cr9902906.North M. Tetrahedron: Asymmetry. 2003;14:147–176.Brunel JM, Holmes IP. Angew Chem, Int Ed. 2004;43:2752–2778. doi: 10.1002/anie.200300604.North M, Usanov DL, Young C. Chem Rev. 2008;108:5146–5226. doi: 10.1021/cr800255k.Wang W, Liu X, Lin L, Feng X. Eur J Org Chem. 2010:4751–4769.
- 8.(a) Smith AMR, Hii KK. Chem Rev. 2011;111:1637–1656. doi: 10.1021/cr100197z. [DOI] [PubMed] [Google Scholar]; (b) Chen BC, Zhou P, Davis FA, Ciganek E. Org React (NY) 2003;62:1–356. [Google Scholar]; (c) Davis FA, Chen BC. Chem Rev. 1992;92:919–934. [Google Scholar]
- 9.Selected examples: Juhl K, Jørgensen KA. J Am Chem Soc. 2002;124:2420–2421. doi: 10.1021/ja0175486.Lee JM, Lim HS, Seo KC, Chung SK. Tetrahedron: Asymmetry. 2003;14:3639–3641.Yao W, Wang J Org Lett. 2003;5:1527–1530. doi: 10.1021/ol0343257.Zhao Y, Ma Z, Zhang X, Zou Y, Jin X, Wang J. Angew Chem, Int Ed. 2004;43:5977–5980. doi: 10.1002/anie.200460730.Abraham L, Körner M, Schwab P, Hiersemann M. Adv Synth Catal. 2004;346:1281–1294.Uraguchi D, Sorimachi K, Terada M. J Am Chem Soc. 2005;127:9360–9361. doi: 10.1021/ja051922a.Trost BM, Malhotra S, Fried BA. J Am Chem Soc. 2009;131:1674–1675. doi: 10.1021/ja809181m.Cowen BJ, Saunders LB, Miller SJ. J Am Chem Soc. 2009;131:6105–6107. doi: 10.1021/ja901279m.Nakamura A, Lectard S, Hashizume D, Hamashima Y, Sodeoka M. J Am Chem Soc. 2010;132:4036–4037. doi: 10.1021/ja909457b.Terada M, Amagai K, Ando K, Kwon E, Ube H. Chem Asian J. 2011;17:9037–9041. doi: 10.1002/chem.201101076.Suzuki S, Kitamura Y, Lectard S, Hamashima Y, Sodeoka M. Angew Chem, Int Ed. 2012;51:4581–4585. doi: 10.1002/anie.201201303.
- 10.For example: Patel RN, Banerjee A, Howell JM, McNamee DB, Mirfakhrae D, Nanduri V, Thottathil JK, Szarka LJ. Tetrahedron: Asymmetry. 1993;4:2069–2084.Steward KM, Johnson JS. Org Lett. 2010;12:2864–2867. doi: 10.1021/ol100996w.
- 11.Noyori R, Tokunaga M, Kitamura M. Bull Chem Soc Jpn. 1995;68:36–55.Genet JP. Acc Chem Res. 2003;36:908–918. doi: 10.1021/ar020152u.For recent reviews on dynamic kinetic resolutions see: Pellissier H. Tetrahedron. 2008;64:1563–1601.Pellissier H. Tetrahedron. 2011;67:3769–3802.
- 12.Boyce GR, Greszler SN, Johnson JS, Linghu X, Malinowski JT, Nicewicz DA, Satterfield AD, Schmitt DC, Steward KM. J Org Chem. 2012;77:4503–4515. doi: 10.1021/jo300184h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steward KM, Gentry EC, Johnson JS. J Am Chem Soc. 2012;134:7329–7332. doi: 10.1021/ja3027136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hashiguchi S, Fujii A, Ikariya T, Noyori R. J Am Chem Soc. 1995;117:7562–7563. [Google Scholar]
- 15.Noyori R, Hashiguchi S. Acc Chem Res. 1997;30:97–102. [Google Scholar]
- 16.(a) Ros A, Magriz A, Dietrich H, Lassaletta JM, Fernández R. Tetrahedron. 2007;63:6755–6763. [Google Scholar]; (b) Limato J, Krska SW, Dorner BT, Vazquez E, Yoshikawa N, Tan L. Org Lett. 2010;12:512–515. doi: 10.1021/ol902715d. [DOI] [PubMed] [Google Scholar]; (c) Seashore-Ludlow B, Villo P, Häcker C, Somfai P. Org Lett. 2010;12:5274–5277. doi: 10.1021/ol102323k. [DOI] [PubMed] [Google Scholar]
- 17.Fujii A, Hashiguchi S, Uematsu N, Ikariya T, Noyori R. J Am Chem Soc. 1996;118:2521–2522. [Google Scholar]
- 18.Reactions performed at 25 °C gave comparable levels of selectivity but mixtures of acyclic hydroxy triester and lactone diester were obtained.
- 19.Kim H, Nguyen Y, Yen CPH, Chagal L, Lough AJ, Kim BM, Chin J. J Am Chem Soc. 2008;130:12184–12191. doi: 10.1021/ja803951u. [DOI] [PubMed] [Google Scholar]
- 20.Recent examples of o-Ph aryl substituents directly impacting the chiral environment: Ohmatsu K, Kiyokawa M, Ooi T. J Am Chem Soc. 2011;133:1307–1309. doi: 10.1021/ja1102844.Ohmatsu K, Hamajima Y, Ooi T. J Am Chem Soc. 2012;134:8794–8797. doi: 10.1021/ja3028668.Ohmatsu K, Goto A, Ooi T. Chem Commun. 2012;48:7913–7915. doi: 10.1039/c2cc32398b.
- 21.(a) Saednya A, Hart H. Synthesis. 1996:1455–1458. [Google Scholar]; (b) Luning U, Baumgartner H, Manthey C, Meynhardt B. J Org Chem. 1996;61:7922–7926. doi: 10.1021/jo961094r. [DOI] [PubMed] [Google Scholar]
- 22.α-Naphthyl ethylenediamine: Denmark SE, Su X, Nishigaichi Y, Coe DM, Wong KT, Winter SBD, Choi JY. J Org Chem. 1999;64:1958–1967. doi: 10.1021/jo9820723.Denmark SE, Pham SM, Stavenger RA, Su X, Wong KT, Nishigaichi Y. J Org Chem. 2006;71:3904–3922. doi: 10.1021/jo060243v.Basak AK, Shimada N, Bow WF, Vicic DA, Tius MA. J Am Chem Soc. 2010;132:8266–8267. doi: 10.1021/ja103028r.m-Terphenylsulfonamide: Kosugi Y, Akakura M, Ishihara K. Tetrahedron. 2007;63:6191–6203.
- 23.At our current levels of optimization, α-keto ester 1k affords the corresponding γ-butyrolactone in 67:33 er.
- 24.CCDC 871630 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Centre via www.ccdc.cam.ac.uk/data_request/cif.
- 25.Krapcho AP. Synthesis. 1982:893–914. [Google Scholar]
- 26.Bukowsa M, Kolodziejek W, Prejzner J. Pol J Chem. 1990;64:573–576. [Google Scholar]
- 27.Kitson RRA, Millemaggi A, Taylor RJK. Angew Chem, Int Ed. 2009;48:9426–9451. doi: 10.1002/anie.200903108. [DOI] [PubMed] [Google Scholar]
- 28.(a) Calderari G, Seebach D. Helv Chim Acta. 1985;68:1592–1604. [Google Scholar]; (b) Jang DP, Chang JW, Uang BJ. Org Lett. 2001;3:983–985. [PubMed] [Google Scholar]; (c) Blay G, Fernández I, Monje B, Pedro JR, Ruiz R. Tetrahedron Lett. 2002;43:8463–8466. [Google Scholar]
- 29.Huang H, Zhu K, Wu W, Jin Z, Ye J. Chem Commun. 2012;48:461–463. doi: 10.1039/c1cc15928c.Misaki T, Kawano K, Sugimura T. J Am Chem Soc. 2011;133:5695–5697. doi: 10.1021/ja200283n.Alemán J, Milelli A, Cabrera S, Reyes E, Jørgensen KA. Chem Eur J. 2008;14:10958–10966. doi: 10.1002/chem.200802030.For additions to nitrostyrenes see: Trost BM, Hirano K. Angew Chem, Int Ed. 2012;51:6480–6483. doi: 10.1002/anie.201201116.Hynes PS, Stranges D, Stupple PA, Guarna A, Dixon DJ. Org Lett. 2007;9:2107–2110. doi: 10.1021/ol070532l.
- 30.For a direct Michael addition of unmodified hydroxy ketones see: Harada S, Kumagai N, Kinoshita T, Matsunaga S, Shibasaki M. J Am Chem Soc. 2003;125:2582–2590. doi: 10.1021/ja028928+.
- 31.Burke SD, Danheiser RL. Oxidizing and Reducing Agents. Wiley; Chichester: 1999. Handbook of Reagents for Organic Synthesis. [Google Scholar]
- 32.(a) Gemal AL, Luche JL. J Org Chem. 1979;44:4187–4189. [Google Scholar]; (b) Luche JL, Gemal AL. J Am Chem Soc. 1979;101:5848–5849. [Google Scholar]; (c) Bastug G, Dierick S, Lebreux F, Markó IE. Org Lett. 2012;14:1306–1309. doi: 10.1021/ol300188e. [DOI] [PubMed] [Google Scholar]; (d) Paradisi MP, Zecchini GP, Ortar G. Tetrahedron Lett. 1980;21:5085–5088. [Google Scholar]
- 33.Blandin V, Carpentier JF, Mortreux A. Eur J Org Chem. 1999:1787–1793. [Google Scholar]
- 34.Baraldi PG, Manfredini S, Pollini GP, Romagnoli R, Simoni D, Zanirato V. Tetrahedron Lett. 1992;33:2871–2874. [Google Scholar]
- 35.Bhar S, Guha S. Tetrahedron Lett. 2004;45:3775–3777. [Google Scholar]
- 36.(a) Thompson W, Buhr CA. J Org Chem. 1983;48:2769–2772. [Google Scholar]; (b) Xian M, Alaux S, Sagot E, Gefflaut T. J Org Chem. 2007;72:7560–7566. doi: 10.1021/jo070805q. [DOI] [PubMed] [Google Scholar]; (c) Kim SH, Kim KH, Nyoung J. Adv Synth Catal. 2011;353:3335–3339. [Google Scholar]
- 37.For the Stetter reaction between ethyl glyoxylate and benzylidene malonates see ref. 13.
- 38.(a) Stetter H, Skrobel H. Chem Ber. 1987;120:643–645. [Google Scholar]; (b) Liu Q, Perreault S, Rovis T. J Am Chem Soc. 2008;130:14066–14067. doi: 10.1021/ja805680z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Liu Q, Rovis T. Org Lett. 2009;11:2856–2859. doi: 10.1021/ol901081a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Vora HU, Lathrop HP, Reynolds NT, Kerr MS, Read de Alaniz J, Rovis T. Org Synth. 2010;87:350–361. [Google Scholar]
- 39.In contrast, simple alkyl substituents to date have provided poor selectivity (see, e.g., reference 23).
- 40.This reduction was performed during the course of this study.
- 41.(a) Lutje Spelberg JH, van Hylckama Vlieg JET, Bosma T, Kellogg RM, Janssen DB. Tetrahedron: Asymmetry. 1999;10:2863–2870. [Google Scholar]; (b) Pàmies O, Bäckvall JE. J Org Chem. 2002;67:9006–9010. doi: 10.1021/jo026157m. [DOI] [PubMed] [Google Scholar]; (c) Haak RM, Tarabiono C, Janssen DB, Minnaard AJ, De Vries JG, Feringa BL. Org Biomol Chem. 2007;5:318–323. doi: 10.1039/b613937j. [DOI] [PubMed] [Google Scholar]; (d) Janssen DB. Adv Appl Microbiol. 2007;61:233–252. doi: 10.1016/S0065-2164(06)61006-X. [DOI] [PubMed] [Google Scholar]; (e) Haak RM, Berthiol F, Jerphagnon T, Gayet AJA, Tarabiono C, Postema CP, Ritleng V, Pfeffer M, Janssen DB, Minnaard AJ, Feringa BL, De Vries JG. J Am Chem Soc. 2008;130:13508–13509. doi: 10.1021/ja805128x. [DOI] [PubMed] [Google Scholar]
- 42.Selected examples: Naruse Y, Esaki T, Yamamoto H. Tetrahedron. 1988;44:4747–4756.Denmark SE, Barsanti PA, Wong KT, Stavenger RA. J Org Chem. 1998;63:2428–2429. doi: 10.1021/jo9801420.Nugent WA. J Am Chem Soc. 1998;120:7139–7140.Bruns S, Haufe G. J Fluorine Chem. 2000;104:247–254.Tao B, Lo MMC, Fu GC. J Am Chem Soc. 2001;123:353–354. doi: 10.1021/ja003573k.Nakajima M, Saito M, Uemura M, Hashimoto S. Tetrahedron Lett. 2002;43:8827–8829.Haufe G, Bruns S. Adv Synth Catal. 2002;344:165–171.Tokuoka E, Kotani S, Matsunaga H, Ishizuka T, Hashimoto S, Nakajima M. Tetrahedron: Asymmetry. 2005;16:2391–2392.Denmark SE, Barsanti PA, Beutner GL, Wilson TW. Adv Synth Catal. 2007;349:567–582.Takenaka N, Sarangthem RS, Captain B. Angew Chem, Int Ed. 2008;47:9708–9710. doi: 10.1002/anie.200803338.Pu X, Qi X, Ready JM. J Am Chem Soc. 2009;131:10364–10365. doi: 10.1021/ja9041127.Kalow JA, Doyle AG. J Am Chem Soc. 2010;132:3268–3269. doi: 10.1021/ja100161d.Kalow JA, Doyle AG. J Am Chem Soc. 2011;133:16001–16012. doi: 10.1021/ja207256s.
- 43.Sakurada I, Yamasaki S, Göttlich R, Iida T, Kanai M, Shibasaki M. J Am Chem Soc. 2000;122:1245–1246.A number of reports have disclosed enantioselective halocyclizations of alkenes. For recent reviews see: Castellanos A, Fletcher SP. Chem Eur J. 2011;17:5766–5776. doi: 10.1002/chem.201100105.Hennecke U. Chem Asian J. 2012;7:456–465. doi: 10.1002/asia.201100856.
- 44.(a) Thakur SS, Li W, Kim SJ, Kim GJ. Tetrahedron Lett. 2005;46:2263–2266. [Google Scholar]; (b) Thakur SS, Chen SW, Li W, Shin CK, Kim SJ, Koo WM, Kim GJ. J Organomet Chem. 2006;691:1862–1872. [Google Scholar]
- 45.(a) Tsuboi S, Furutani H, Utaka M, Takeda A. Tetrahedron Lett. 1987;28:2709–2712. [Google Scholar]; (b) Tsuboi S, Furutani H, Ansari MH, Sakai T, Utaka M, Takeda A. J Org Chem. 1993;58:486–492. [Google Scholar]; (c) Rodrigues JAR, Milagre HMS, Milagre CDF, Moran PJS. Tetrahedron: Asymmetry. 2005;16:3099–3106. [Google Scholar]
- 46.(a) Tsuboi S, Yamafuji N, Utaka M. Tetrahedron: Asymmetry. 1997;8:375–379. [Google Scholar]; (b) Hamamoto H, Mamedov VA, Kitamoto M, Hayashi N, Tsuboi S. Tetrahedron: Asymmetry. 2000;11:4485–4498. [Google Scholar]
- 47.(a) Akita H, Kawaguchi T, Enoki Y, Oishi T. Chem Pharm Bull. 1990;38:323–328. [Google Scholar]; (b) Adger BM, Barkley JV, Bergeron S, Cappi MW, Flowerdew BE, Jackson MP, McCague R, Nugent TC, Roberts SM. J Chem Soc, Perkin Trans. 1997;1:3501–3508. [Google Scholar]; (c) Zhou Z, Mei X, Chang J, Feng D. Synth Commun. 2001;31:3609–3615. [Google Scholar]
- 48.(a) Genêt J-P, Caño de Andrade MC, Ratovelomanana-Vidal V. Tetrahedron Lett. 1995;36:2063–2066. [Google Scholar]; (b) Ratovelomanana-Vidal V, Genêt JP. J Organomet Chem. 1998;567:163–171. [Google Scholar]; (c) Qiu L, Qi J, Pai CC, Chan S, Zhou Z, Choi MCK, Chan ASC. Org Lett. 2002;4:4599–4602. doi: 10.1021/ol026817+. [DOI] [PubMed] [Google Scholar]; (d) Bai J, Miao S, Wu Y, Zhang Y. Chin J Chem. 2011;29:2476–2480. [Google Scholar]
- 49.(a) Takeda A, Wada S, Fujii M, Nakasima I, Hirata S. Bull Chem Soc Jpn. 1971;44:1342–1345. [Google Scholar]; (b) Okonya JF, Johnson MC, Hoffman RV. J Org Chem. 1998;63:6409–6413. doi: 10.1021/jo9806761. [DOI] [PubMed] [Google Scholar]; (c) Okonya JF, Hoffman RV, Johnson MC. J Org Chem. 2002;67:1102–1108. doi: 10.1021/jo010630z. [DOI] [PubMed] [Google Scholar]; (d) Suzuki S, Kitamura Y, Lectard S, Hamashima Y, Sodeoka M. Angew Chem, Int Ed. 2012;51:4581–4585. doi: 10.1002/anie.201201303. [DOI] [PubMed] [Google Scholar]
- 50.Initial results employing chiral Ni(II)-diamine complex 1 afforded the syn-β-chloro-α-hydroxy ester 5a in >20:1 dr and 61.5:38.5 er following reduction with NaBH4.
- 51.Mamedov VA, Berdnikov EA, Tsuboi S, Hamamoto H, Komiyama T, Gorbunova EA, Gubaidullin AT, Litvinov IA. Russ Chem Bull, Int Ed. 2006;55:1455–1463. [Google Scholar]
- 52.Noyori R, Yamakawa M, Hashiguchi S. J Org Chem. 2001;66:7931–7944. doi: 10.1021/jo010721w. [DOI] [PubMed] [Google Scholar]
- 53.Corey EJ, Choi S. Tetrahedron Lett. 1991;32:2857–2860. [Google Scholar]
- 54.Sodeoka also observed low levels of diastereocontrol in the reduction of β-fluoro-α-keto esters: see reference 9k.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.