

Abstract
A novel family of functionalized peptide toxins, aculeines (ACUs), was isolated from the marine sponge Axinyssa aculeate. ACUs are polypeptides with N-terminal residues that are modified by the addition of long-chain polyamines (LCPA). Aculeines were present in the sponge extract as a complex mixture with differing polyamine chain lengths and peptide structures. ACU-A and B, which were purified in this study, share a common polypeptide chain but differ in their N-terminal residue modifications. The amino acid sequence of the polypeptide portion of ACU-A and B was deduced from 3′ and 5′ RACE, and supported by Edman degradation and mass spectral analysis of peptide fragments. ACU induced convulsions upon intracerebroventricular (i.c.v.) injection in mice, and disrupted neuronal membrane integrity in electrophysiological assays. ACU also lysed erythrocytes with a potency that differed between animal species. Here we describe the isolation, amino acid sequence, and biological activity of this new group of cytotoxic sponge peptides.
Keywords: long-chain polyamines, marine sponges, peptides, protein modifications, toxicology
Introduction
Diverse peptide toxins occur in a wide range of organisms, including venomous animals such as snakes,[1] spiders,[2, 3] scorpions,[4] sea anemones,[5] and cone snails.[6] Macrocyclic peptide toxins that likely serve defensive roles also occur in higher plants.[7] An impressive array of biological activities has been documented for many of these peptides. Cellular targets of these peptides often underlie important cellular functions, and include ion channels, receptors, enzymes, and lipid membranes.[8-12] Often peptide toxins acquire stability through the formation of intramolecular disulfide bonds, N- or C-terminal modification, or by associating with nonproteinous amino acids; they thereby become resistant to enzymatic degradation in vivo. Some peptide toxins have received considerable attention as potential drugs because of their keen target specificity and biochemical stability;[13] indeed, ω-conotoxin MVII-A, a 32 amino acid peptide that selectively blocks N-type calcium channels at mammalian synapses[14] was recently approved for clinical use in the management of severe chronic pain.
Marine sponges are also known to be a rich source of bioactive peptides. However, most molecules found in this class of organism are biosynthesized through nonribosomal peptide synthesis (NRPS) pathways, and are therefore presumed to originate in symbiotic bacteria.[15-17] Although there are precedents for sponge-derived bioactive proteins, asteropine, a sialidase inhibitor found in Asteropus simplex, was the first small ribosome-derived sponge peptide.[18] Asteropine is a 38-residue peptide with six cysteine residues that forms a conotoxin-like inhibitory cystine-knot motif, thus suggesting that this structural motif is found in species as diverse as nonvenomous animals and sponges.
In our continuing effort to discover novel marine-derived neuroactive molecules, we found ribosomal peptide toxins, referred to as aculeines (ACUs), in an aqueous extract of the marine sponge Axinyssa aculeata collected in Iriomote, Japan. ACUs are a new family of small peptides that are post-translationally modified by the attachement of long-chain polyamines (LCPAs)[19] to the N-termini of the peptides. Here we describe the isolation and structural identification of ACUs and report that they cause loss of plasma membrane integrity, thereby leading to cytotoxicity, which is likely responsible for the observed convulsant activity.
Results and Discussion
Isolation and structural identification of aculeines
We first observed that intracerebroventricular (i.c.v.) administration of an aqueous extract (20 μg) of the marine sponge Axinyssa aculeata induced whole body convulsions in mice, thus suggesting that it contained neurotoxic molecules. We previously reported that novel LCPAs from the same sponge were putative chemical factors involved in silica biomineralization.[19] LCPAs alone (20 μg), however, did not show acute toxicity in mice (data not shown). Thin-layer chromatography (TLC) of the active fraction obtained from a Sephadex LH20 column yielded ninhydrin-reactive spots in both in normal and reversed-phase plates (Figure 1A, B), and SDS-PAGE in tricine buffer successfully resolved components with apparent molecular weights between 1 and 7 kDa (Figure 1C). Among the several chromatographic supports tested, only large pore reversed-phase gels (300 Å) resulted in reasonable separations of the extracted components. Those fractions eluting after the LCPA in a reversed-phase chromatography were immediately toxic in mice.
Figure 1.
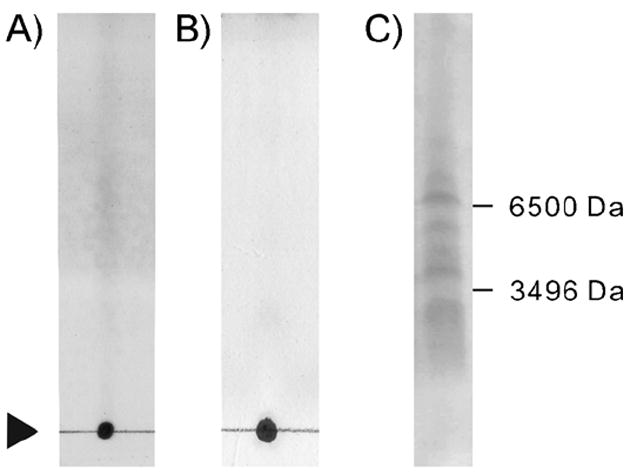
TLCs of the toxic fraction from Sephadex-LH20 separation visualized by A) ninhydrin on a SiO2 developed with pyridine/ethyl acetate/acetic acid/water, (75:35:15:30), or B) on a reversed-phase C18 developed with 50 % MeCN. C) An electrophoresis (SDS-PAGE) of the same fraction.
MALDI-TOF MS for the toxic fractions was highly complex and had multiple ion clusters. These clusters were largely divided into three molecular ranges centered at about 6500 (cluster A), 5700 (cluster B), and 2700 (cluster C; Figure 2A–C). Medium-pressure, reversed-phase liquid chromatography (MPLC) followed by reversed-phase protein HPLC afforded ACU-A and B as major toxic components (Figure 2D and E). ACU-A and B gave ion clusters centered at m/z of 6574 and 5706, respectively, with either quintet- or triplet-like peaks at 57 mu separation; this suggests that they are molecules containing different lengths of LCPA (Figure 2A and B). The MALDI-TOF mass spectrum for cluster C, however, was more complex, with more than eleven peaks between 2612 and 2786; this suggests that ACU-C is a complex heterogeneous mixture (Figure 2C). Separation and structure elucidation of ACU-C will be reported elsewhere.
Figure 2.
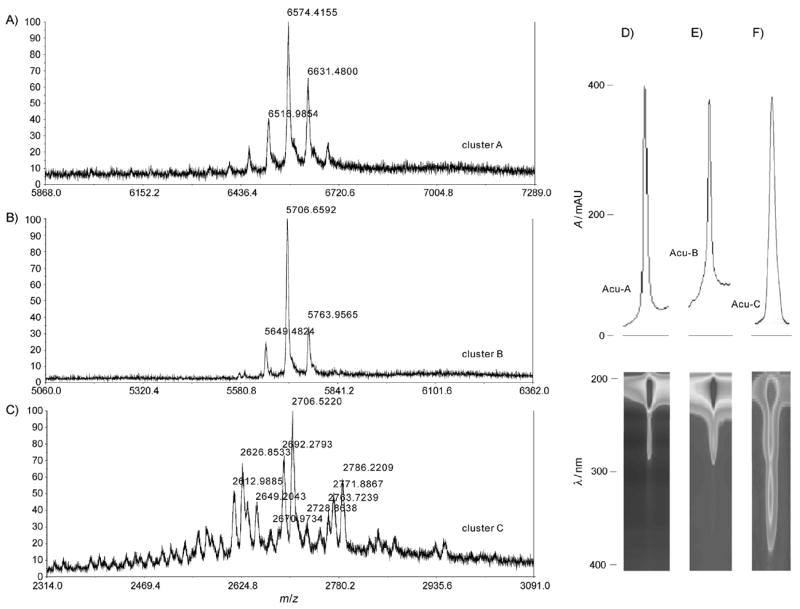
MALDI-TOF MS spectra for A) ACU-A, B) ACU-B, and C) ACU-C. HPLC chromatograms (top) and 2D UV absorption images (bottom) of D) ACU-A, E) ACU-B, and F) ACU-C.
In previous observations, the molecular mass of free LCPAs obtained from this sponge ranged from 303 to 873, consistent with penta- to 15-mer propaneamine units.[19] The mass spectra data thus suggested that both ACU-A and B were polypeptides modified by LCPAs of various chain lengths. Of note, a large ion observed at m/z 96.9 in the negative ion MALDI-TOFMS data for ACU-A and B (Figure S11 in the Supporting Information) suggested that both ACU-A and B present as sulfate as was documented previously in the case of LCPA isolated from the same sponge.[19]
1H NMR spectra of both ACU-A and B in D2O gave complex sets of signals between δ = 0.1 and 5.3 and between δ = 6.2 and 9.6 (typical of polypeptides), but two large broad singlets were also observed at δ = 3.15 and 2.10 (integration ratio of 2:1); this further supports the presence of oligomeric propaneamines (Figures S2 and S3). It should be noted that many amide protons were observed between δ = 6.2 and 9.6 despite the use of D2O, thus indicating that the exchange rates of those amide protons were slow in the conditions employed in the NMR measurement, and suggesting that the peptides might have highly folded structures.[20]
To further establish that ACUs were modified by LPCA addition, we subjected ACU-A to hydrolysis with hydrochloric acid. MALDI-TOF MS data showed a series of ions with 57 mu separation between 417 and 873 (Figure 3A). The 1H NMR spectra of the LCPA pool released by hydrolysis showed two signals at δ = 3.20 and 2.15 (with 2:1 ratio), thus confirming the general structure of the amine to be a propaneamine oligomer (Figure 3 B). A similar result was obtained upon hydrolysis of ACU-B (Figure 3 C). These data confirmed that ACU-A and B were obtained as mixtures containing different lengths of polyamine residues attached to the peptidyl molecules. Further purification of the LPCA homologues, however, was not successful because the mixture of homologues eluted as a single peak in the HPLC. In subsequent experiments, we focused on structural elucidation of the ACU peptide component and on biological exploration of its convulsant activity.
Figure 3.
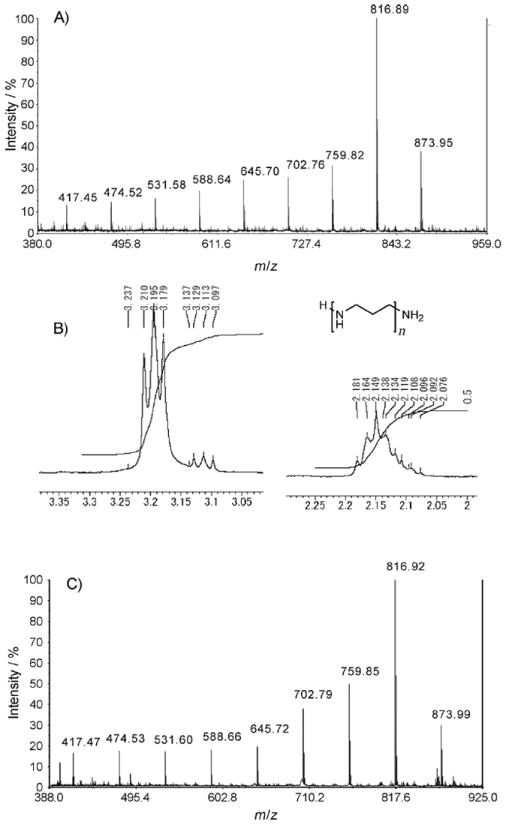
A) MALDI-TOF MS spectrum of the acid hydrolyzate of ACU-A. B) 1H NMR spectrum of LCPA isolated from the acid hydrolyzate of ACU-A. C) MALDI-TOF MS spectrum of the acid hydrolyzate of ACU-B.
Amino acid sequence of ACU-A and -B
Because the 1H NMR strongly suggested that the ACUs were modified peptides, the N-terminal amino acid sequence of ACU-A was first determined by Edman degradation. The 19-residue peptide sequence derived from this experiment was XYDTVAXFREDEIXSGVPI (X denotes an unidentified amino acid). The unidentified residues were hypothesized to be either cysteines or modified amino acids that were unrecognized by the automated peptide sequencer. Edman degradation of ACU-B showed the same amino acid sequence of 19 residues.
Cloning of acu-A and gene sequence analysis
The complete amino acid sequences for the ACUs were therefore deduced from the nucleotide sequence of the gene encoding ACU-A (acu-A), which was obtained by a combination of 3′ and 5′ RACE of cDNA prepared from the sponge. Freshly collected sponge was preserved in RNA stabilization buffer, and mRNA was extracted. Two degenerate primers were prepared (Acu1st and Acu2nd); these corresponded to the internal amino acid sequences (YDTVAXFR and FREDEI, respectively) obtained from the Edman degradation, and were used for nested 3′ RACE (see the Experimental Section). A PCR product of the 400 base pair size was cloned and sequenced. Twenty-five independent clones contained cDNA encoding a predicted amino acid sequence that matched well with the N-terminal amino acid sequence determined by Edman degradation. The upstream region of acu-A was amplified by nested 5′ RACE with primers based on the nucleotide sequences determined by 3′ RACE. 5′ RACE with primer 5Acu yielded six additional bases beyond the open reading frame (ORF) of acu-A. Of note, two out of the eight clones encoded an Arg to His variation at the ninth residue. PCR products obtained when using the 5Acu_4th primer revealed the same sequence for 70 bases upstream of the first 5′ RACE product. The nucleic acid sequence of both of these cDNA clones predicted tryptophan as the N-terminal amino acid, and Cys7 and Cys14 (unresolved in the Edman degradation). The remaining 26 amino acid sequence of the C-terminal region was deduced from the cDNA sequence as shown in Figure 4. Although the protein predicted by the nucleotide sequence of acu-A corresponded well with the amino acid sequence of ACU-A, the cDNA lacked a conventional start codon and contained an amber stop codon, TAG, 25–27 bases upstream of the ORF. Resolution of this remaining uncertainty and cloning of the complete genomic sequence for acu-A therefore have yet to be determined. In summary, the cloning and sequence analysis of acu-A identified the 45 amino acids of the ACU-A precursor peptide.
Figure 4.

A) Amino acid sequence the ACU-A precursor. B) Nucleotide sequence of acu-A.
Peptide-mass mapping
The amino acid sequence of ACU-A was further confirmed by peptide-mass mapping. ACU-A was treated with dithiothreitol (DTT) in denaturing buffer before alkylation with iodoacetoamide (IAA). A portion of this preparation was digested with either trypsin or V8 protease. Reversed-phase HPLC separation of the V8 digests afforded three internal fragments (A–C), whereas the tryptic digest yielded one C-terminal peptide (fragment D). Each of these fragments was subjected to MALDI-TOF MS and MS/MS analyses (Figures 5 and 6). Fragments A–C gave molecular ions at m/z 1126, 1368, and 2102 [M+H]+, respectively. These molecular weights corresponded well with those predicted for fragments Thr4–Glu12, Ile13–Asp25, and Val26–Glu44 in the putative ACU-A peptide, assuming that all five cysteine residues were labeled by IAA. MS/MS analyses of each of those fragments gave mostly y- and b-type fragments[21] (Figure 5), consistent with the sequence of ACU-A determined by other methods (Figure 4). The trypsin-derived C-terminal fragment D gave a molecular ion at m/z 1021. High resolution mass analysis of this fragment refined this to m/z 1021.418, which matches the predicted molecular weight of a C-terminal fragment containing a free acid (m/z 1021.408) rather than an amide (m/z 1020.424; Figure 6A). These data together further confirmed the primary amino acid sequence of ACU-A from the second to 45th residue. We also identified fragments A–C for the V8 digest of ACU-B (Figures S4–S6).
Figure 5.
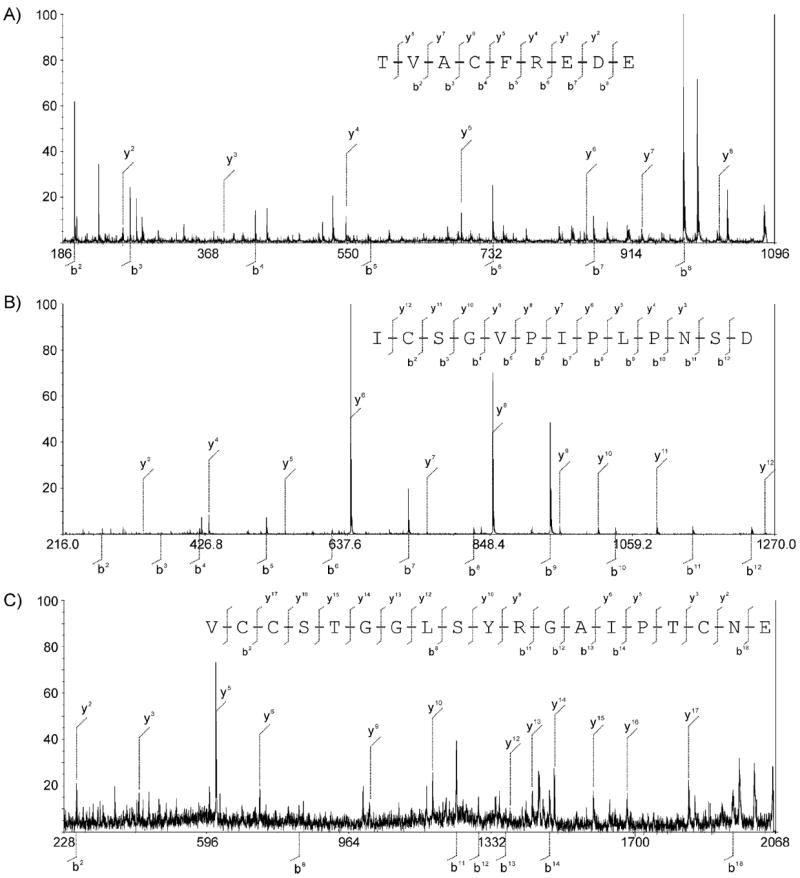
V8 protease digest fragments of ACU-A. MS/MS spectrum for fragments A, B, and C. All cysteines were carbamoyl methylated with iodoacetoamide prior to the analysis.
Figure 6.
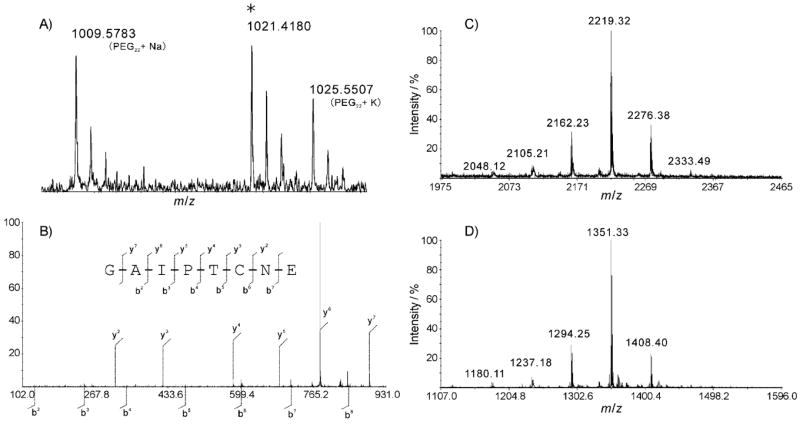
A) HRMS and B) MALDI-TOF MS/MS analysis of fragment D. A cysteine was carbamoyl methylated. PEG22 [M+Na]+ and PEG23 [M+K]+ were used as internal standards. MALDI-TOF mass spectra of N-terminal fragment of C) ACU-A and D) ACU-B obtained from the V8 protease digest.
To determine how Trp1 might have been post-translationally modified, likely by LPCAs, ACU-A was digested with V8 protease without pre-alkylation by IAA, to obtain an intact N-terminal fragment, E. The resulting peptide was separated on a reversed-phase HPLC. A fraction containing LCPA was readily identified from its characteristic mass spectral data, that is, a cluster with 57 mu separation centered at m/z 2219 (Figure 6 C). Edman degradation of fragment E afforded a Tyr2 residue. Amino acid analysis of fragment E using Marfey’s method[22] resulted in identification of l-Tyr and l-Asp. Thus, fragment E was determined to be the three N-terminal residues, X-Tyr-Asp, of ACU-A. In the 1H NMR spectrum, including a two-dimensional COSY spectrum for fragment E, absorptions at δ = 7.56, 7.39, 7.17, 7.10, and 7.09 and at 6.72 indicated a spin system for 1,2- and 1,4-disubstituted benzene rings, respectively. These results are indicative of the presence of a substituted indole and a tyrosine residue. UV absorption at λ = 227 and 279 nm for fragment E also supported these assignments. Furthermore, 1H NMR for the enzyme digest showed large broad singlets at δ = 3.02 and 1.97, a characteristic peak set for LCPA unit. Although further NMR analysis to elucidate detailed structure was difficult because of the limited amount of available sample and severe broadening in the upperfield region of the spectrum, these data illustrated structural feature of the N-terminal unit of ACU-A. Similarly treatment of the reduced ACU-B with V8 protease afforded an N-terminal unit (fragment E′), as in the case of ACU-A; however, the molecular weight of the product, m/z 1351 [M+H]+ by MALDI-TOF MS, was 868 mass units smaller than that of the ACU-A fragment E (Figure 6D). This corresponds to the molecular weight difference between ACU-A and B. These results suggested that ACU-A and B share the same precursor peptide, and are produced from the same gene; however, the N-terminal Trp residues of ACU-A and B are differently modified post-translationally (Figure 7).
Figure 7.
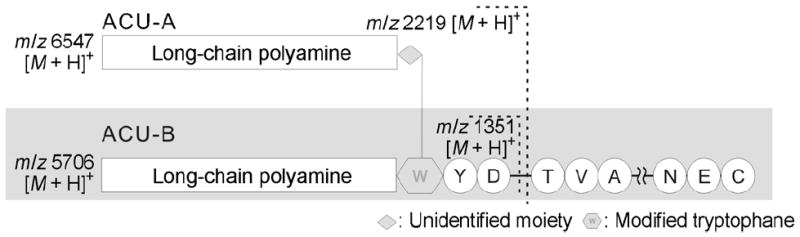
Schematic structures of ACU-A and B.
Detailed structural determination of the N-terminal residues requires additional spectral studies, including detailed NMR analysis of the intact molecule or degraded N-terminal product. In the present study the N-terminal fragment form ACU-A was subjected to preliminary 1H NMR analysis, but that for ACU-B was too small to obtain data. A larger amount of the peptides is in preparation, and the complete structures of aculeines will be reported in due course.
To the best of our knowledge, aculeine is only the second example of a bioactive small ribosome-derived peptide reported from porifera. The structure of ACU is distinguished by its unique post-translational modification at the N-terminus with LCPA. Interestingly, another example of an LCPA-containing peptide, silaffin, has a structure distinct from that of the ACUs.[23] Silaffins were found exclusively in the silica wall of diatoms and contain several modified Lys residues that have N-methylated LCPAs extending from the nitrogen atom of their amino acid side chains. Silaffins and ACUs do not have any primary sequence similarity. Both LCPAs and silaffins are involved in silica wall formation of diatoms,[24, 25] and LCPAs from A. aculeata might similarly contribute to silica spicule formation.[19] Interestingly, ACUs have not been detected in spicules (data not shown); this suggests that the functional roles of ACUs and silaffins (or free LPCA) are different.
The structure of an ACU can be divided into three modules: the LCPA, the modified N-terminal unit, and the polypeptide. We determined that the amino acid sequence of the 44-residue peptide contained six cysteine residues and noted that ACUs might belong to the knottin class of bioactive peptides, which contain inhibitory cystine knot motifs that fold peptides into rigid small molecule-like structures.[26] Bioactive knottins are often found as the toxic components of venom in animals such as cone snails and spiders.[27] A sponge-derived knottin, asteropine, acts as a bacterial sialidase inhibitor.[18] Although the cysteine arrangements in ACUs are similar to those seen in the conotoxins,[28] asteropine, and huwentoxins,[29] the numbers of intervening amino acids between the cysteine residues differ largely between knottin family members (Table 1). ACUs are highly stable in the unreduced form; this could be a result of the cystine knot formation, but additional structural analysis will be necessary to test this theory.
Table 1.
Sequences of representative inhibitory cysteine knots and the precursor peptide of ACU-A.
| Compounds | Amino acid sequences |
|---|---|
| aculeine | WYDTVACFREDEICSGVPIPLPNSDVCCSTGGLSYRGAIPTCNEC |
| asteropine | YCGLFGDLCTLDGTLACCIALELECIPLNDFVGICL |
| huwentoxin I | ACKGVFDACTPGKNE-CC–PNRVCSDKHKW—CKWKL |
| ω-conotoxin GVIA | CKSXGSSCSXTSYN-CC—RSCNXYTKR—CY-NH2 |
| ω-conotoxin MVIIA | CKGKGAKCSRLMYD-CC—TGSC-RSGK—C-NH2 |
Biological activity
Neuronal assays
Aculeines were classified initially as neurotoxic substances because aqueous extracts of sponge induced convulsant behavior in mice upon i.c.v. injection; ACU-A and B were isolated as the principle toxins in the extract, while the ACU-C mixture was also toxic to mice. The peptides induced whole body convulsions with ED50 values of 1.0 (95% confidence intervals; 0.6 to 1.5), 0.6 (0.4 to 0.8), and 0.7 (0.6 to 0.9) μg per mouse, respectively (n = 18 for each peptide).
Although a subset of previously isolated marine sponge-derived convulsants proved to act on mammalian excitatory amino acid receptors, ACUs did not exhibit affinity for rat brain ionotropic glutamate receptors. That is, the peptides did not displace the receptor ligands [3H]CGC19755, [3H]AMPA, or [3H]kainic acid in rat cortical membrane preparations. This is not surprising given the peptidyl nature of the toxins.[30-32]
To explore the biological activity that could underlie the seizurogenic action in mice further, we next carried out electrophysiological recordings from cultured rat hippocampal neurons and human immortalized HEK293 cells. Cells were either maintained at a resting membrane potential of about −60 mV in current clamp, or were held −70 mV in voltage clamp recordings. Application of ACU-A (10 μg mL−1) to neurons in the current clamp caused depolarization, a transient increase in action potential firing, and a loss of membrane integrity within minutes of toxin bath application, as shown in the representative trace in Figure 8A. In the voltage clamp, a large irreversible inward current developed rapidly. Similar effects on membrane integrity were observed with voltage clamp recordings from HEK293 cells. The mean time to loss of membrane integrity in these recordings was approximately 5 min: 5.7 ± 0.3 (n = 3) and 4.7±0.9 mins (n = 4) for neurons and HEK293 cells, respectively (Figure 8A, bottom). These data strongly suggest that ACU-A irreversibly disrupts cell membranes directly by action as an ionophore, or indirectly through actions on integral proteins responsible for maintaining membrane potential.
Figure 8.
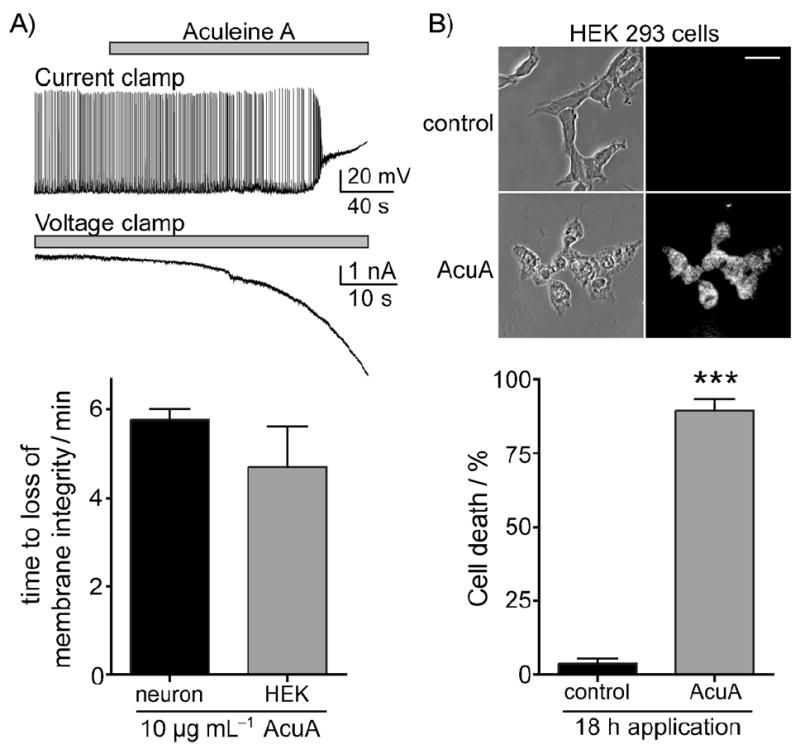
Application of ACU-A causes a profound depolarization and loss of membrane integrity in both rat hippocampal neurons and HEK293-T/17 cells. A) Representative voltage and current recordings from hippocampal neurons held in either current-clamp or voltage-clamp, respectively. The time of onset of depolarizing current was equivalent for both cell types (graph below). B) Cytotoxicity of ACU-A on HEK293-T/17 cells was determined using propidium iodide staining after a 24 h exposure to the purified toxin. Representative phase-contrast and fluorescent images are shown with quantitation (graph below). ***: p < 0.001 in an unpaired t-test.
The loss of membrane integrity observed in the physiological assays was consistent with the hypothesis that ACU-A was acting as a cytotoxic molecule. To more directly assess cytotoxicity, HEK293 cells were exposed to saline (control) or ACU-A (10 μg mL−1) before incubation with propidium iodide, a DNA chelator that stains only dead or dying cells. Phase contrast and ultraviolet images of representative cells acquired on a confocal microscope are shown in Figure 8B, where it is apparent that most cells are positive for propidium iodide after exposure to ACU-A for 18 h, but not to saline. Quantitation of this data revealed that 89 ± 4 % of HEK293 cells were positive for propidium iodide (Figure 8B, bottom). ACU-A induced a robust Ca2+ influx when bath-applied to cultured hippocampal neurons (Figure 9). Application of the toxin in normal extracellular solution (containing 2 mm Ca2+) caused a large increase in intracellular calcium as measured by ratiometric imaging following loading of the cells with the reporter dye Fura2-AM (Figure 9B, triangles, n = 7 cells). In contrast, neurons were unresponsive to ACU-A if the divalent cation was omitted from the extracellular solution. A moderate Ca2+ influx was observed in the neurons upon subsequent addition of calcium (Figure 9B, squares). We conclude from this data that one of the primary actions of ACU-A on neurons is to facilitate massive calcium entry through the plasma membrane. This is consistent with the electrophysiological results and likely, in part, to underlie the cytotoxicity of the peptide. ACU-A to C also showed moderate cytotoxicity to cultured human tumor cell-lines (MDA-MB-231, A 549, and HT-29) with GI50 values of approximately 0.5 μm (Table 2). No significant difference in potency was observed between the cell lines tested.
Figure 9.
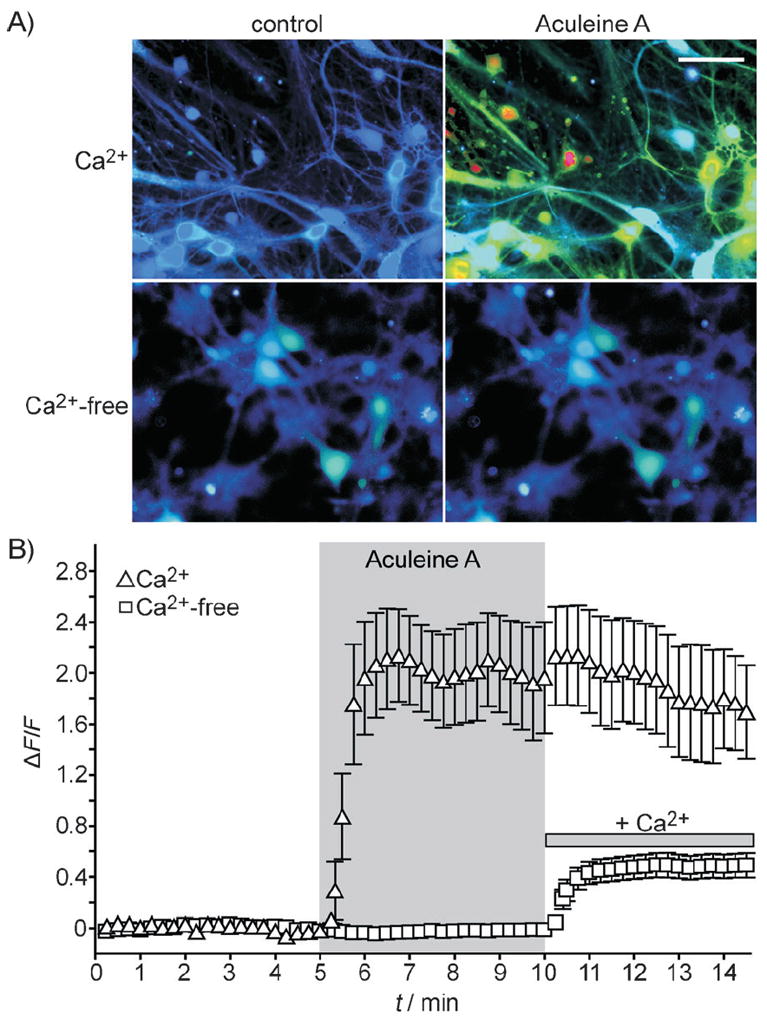
ACU-A (10 μg mL−1) evokes an intracellular calcium transient from cultured hippocampal neurons only the presence of extracellular calcium. A) Ratiometric calcium imaging was performed on Fura-2 AM-loaded neurons. B) Quantitation of intracellular Ca2+ in the presence of ACU-A (ΔF/F = ratiometric change in fluorescence). Upon application of ACU-A in the presence of extracellular calcium (2 mm), a robust intracellular rise in calcium was observed. In contrast, no intracellular fluorescence above baseline was observed if the extracellular solution was nominally calcium-free; replacement of the calcium (2 mm) led to enhanced intracellular signals, thus demonstrating the continued presence of ACU-A.
Table 2.
Cytotoxicity to tumor cell lines.
| GI50 values [μm][a] | |||
|---|---|---|---|
| MDA-MB-231 | A549 | HT-29 | |
| ACU-A | 0.58 | 0.41 | 0.56 |
| ACU-B | 0.68 | 0.51 | 0.68 |
| ACU-C | 2.92 | 1.33 | 1.55 |
GI = Growth inhibition.
Hemolytic activity
Potent hemolytic activity observed with ACU-A to C further confirmed the membrane-targeting activity of the peptides. ACU-A was lytic for mammalian erythrocytes in a concentration-dependent manner. An EC50 value of 7.9 μm was determined from the concentration–activity relationship curves for rabbit erythrocytes (Figure 10 A). In a similar assay, the ED50 value of SDS was determined to be 8.2 mm. The concentration–response curve reached a plateau at OD405 ≈ 0.33, approximately half the maximum of that for SDS. A comparison of the lytic potency for erythrocytes from different animal species indicated that the peptide potency varied from species to species (Figure 10B). These differences could be explained if ACU-A differentiates between membrane structures that arise from the variability (between species) of lipid composition,[33] as has been observed for some antimicrobial peptides.[34, 35]
Figure 10.
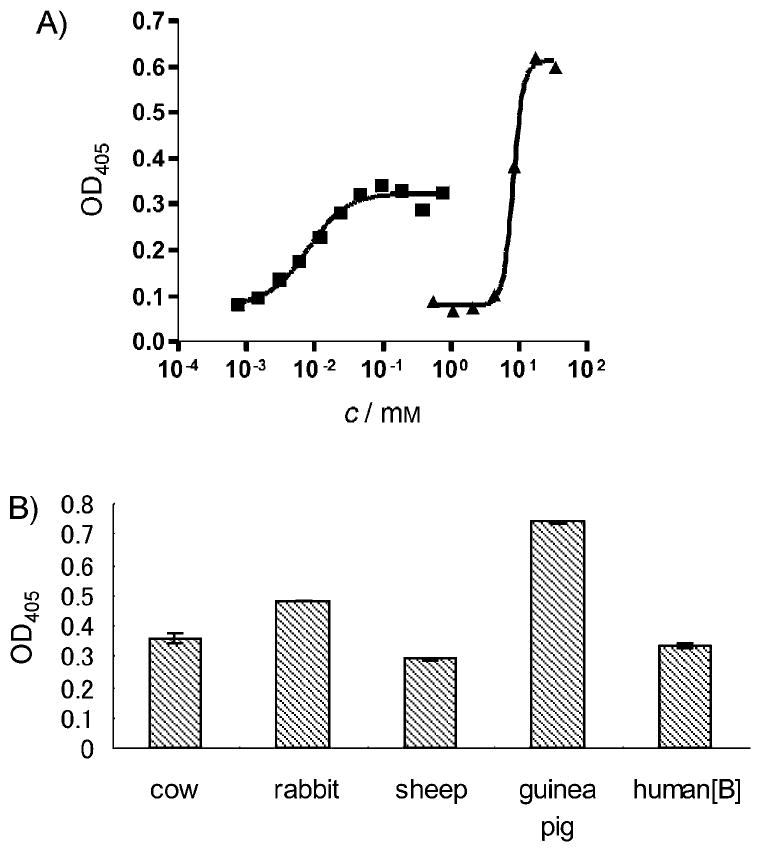
Effect of ACU-A on rabbit erythrocytes. A) ACU-A lysed the cells in a concentration dependent manner (■). SDS was used as positive control (▲). The ED50 value was calculated from sigmoidal curve fitting. B) Comparison of hemolytic potency of ACU-A (5 μm) for different animal species: cow, rabbit, sheep, guinea pig, and human type B. Bars indicate SD (n = 3).
Membrane-permeabilizing activity
We next measured the membrane permeabilizing activity of ACU-A by using 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) liposome with 10% cholesterol, in which the pH-sensitive fluorescent dye BCECF was entrapped. An increase in the membrane permeability produced by the peptide would result in pH change within the vesicle as a result of exchange of H+ (inner) and K+ (outer).[36] ACU-A, however, did not induce such a change at a concentration range comparable to that for its hemolytic activity (1.5–15 μm; Figure S10). This result suggests to us that specific lipid component(s) in addition to POPC/cholesterol or some other factors were required for ACU-A to exhibit full membrane disrupting activity.
Conclusions
In summary, the present study demonstrated that ACUs constitute a novel class of highly modified ribosomal peptides found in marine sponge. Although complete structures are not resolved in this study, we have demonstrated here that ACU-A and B share a common precursor peptide with an intriguing interval of Cys residues, thus suggesting that ACUs are new type of inhibitory cystine knot peptide. Moreover, the N-terminal residues for ACUs were modified by the addition of LCPA; this is a novel structural feature among naturally occurring peptides. These results demonstrate that ACUs are highly unusual, both structurally and biosynthetically. The biological activity of ACUs also seems to involve unique interactions between the peptide and cell membrane. Further structural and functional studies of ACUs might reveal unexpected interactions between these naturally occurring peptide toxins and cells.
Experimental Section
General
1H NMR data were measured on an AMX500 (Bruker), or a UNITY 750 or 600 (Varian), with D2O as the solvent. Chemical shifts are reported in ppm with HDO peak at δH = 4.65 (at 303 K). MALDI-TOF MS and MS/MS were obtained on an AB 4700 spectrometer (Applied Biosystems) with α-cyano-4-hydroxycinnamic acid as a matrix. High-resolution mass spectra were measured on this instrument with polyethylene glycerol as the internal standard.
Extraction and isolation
The sponge specimen was collected from a depth of 10–17 m in May 2002 at Okinawa, Japan. The sample was frozen immediately after the collection and stored at −20°C until use. A portion of the specimen was preserved in 70% ethanol, and sent to Dr. J. Hooper at the Queensland Museum (Australia) for identification. The sponge specimen (200 g wet weight) was homogenized in water and centrifuged, and then the supernatant was lyophilized to afford an aqueous crude extract (15 g), of which 5.0 g was separated on a Sephadex LH-20 column (5×60 cm) by using water as the eluent. Fractions between 350 and 520 mL afforded a mixture of LCPAs and peptides (1.2 g). Fractions containing this mixture were combined (87.4 mg) and separated by reversed-phase MPLC (C18, 300 Å, 2.5 × 30 cm; GL Sciences, Tokyo, Japan) by using a step gradient from water to CH3CN containing 0.1% trifluoroacetic acid (TFA). ACU-A and ACU-B, eluted at 30–40% CH3CN, were concentrated (5.2 mg), and then a portion of the fraction (2.4 mg) was separated by a reversed-phase HPLC with YMC-Pack PROTEIN-RP (YMC, Kyoto, Japan). HPLC was performed at a flow rate of 0.5 mL min−1 with a linear gradient from 0.1% TFA/water to 0.1% TFA/CH3CN, to afford ACU-A (1.1 mg) and ACU-B (0.4 mg). A fraction containing ACU-C eluted immediately before ACU-A on MPLC. This fraction was further separated by HPLC to give 0.7 mg of ACU-C. Each of the separated samples was subjected to further chromatography with a photodiode array detector (Figure 2 D–F), and by tris-tricine SDS-PAGE[37] (Figure 1 C).
N-terminal amino acid analysis of ACU-A and B
ACU-A and ACU-B were separated in a tris-tricine SDS-PAGE, transferred to polyvinylidene fluoride membranes by a semidry electroblotting method, and then submitted to a PPSQ-21A automated protein sequencer (Shimadzu, Kyoto, Japan). Although the N-terminal amino acid was not detected, the second and subsequent residues were assigned: YDTVAXFREDEIXSGVPI.
Cloning of acu-A and gene sequence analysis
The sponge specimen was diced (1 mm cubes), immersed in RNAlater (Qiagen), and stored at −80°C until use. Total RNA was extracted from the sample (60 mg wet wt.) by using the Purescript RNA isolation kit (Qiagen), and mRNA was subsequently purified by using the MPG mRNA purification kit (PureBiotech LCC, Middlesex, NJ). The cDNA libraries (obtained by 3′ and 5′ RACE) were produced by using SMART RACE cDNA Amplification kit (Clontech, Mountain View, CA). Degenerate primers for 3′ RACE (Acu1st and Acu2nd) were synthesized according to N-terminal amino acid sequence (YDTVAXFREDEII; Table 3), and 3′ RACE was carried out by nested PCR to generated a ~400 base pair PCR fragment. PCR products were ligated into pCR4-TOPO vector (Invitrogen), transformed into the competent E. coli DH5α (Takara Bio, Shiga, Japan), and then cultivated on TB agar medium at 37 °C for 16 h. The insert region of the vector was amplified by colony-directed PCR with KOD Dash DNA polymerase (Toyobo, Osaka, Japan) by following the manufacturer’s instruction. Products purified by Sephacryl S-300 (GE Healthcare) were run on a BigDye Terminator v3.1 Cycle Sequencing kit (Applied Biosystems) to reveal 25 clones containing the 3′ poly-A terminal. The 5′ RACE was also carried out as above. A primer, 5Acu, was prepared according to the base sequence of the 3′-terminal region (Table 3). Twelve clones that contained relevant sequence for ACU to six bases upstream of the N-terminal were obtained. Additional 5′ RACE was performed with a primer 5Acu_4th as above. The nucleic acid sequences for six identical clones corresponded to that for the ACU N terminus plus 70 upstream bases.
Table 3.
Primers used for 3′ and 5′ RACE.
| Primer | Nucleotide sequence |
|---|---|
| Acu1st | TAYGAYACNGTNGCNTGYTTYMG[a] |
| Acu2nd | TTYMGNGARGAYGARATHAT[a] |
| 5Acu | TGTAGGTATCGCTCCTCGATATGAGAGC |
| 5Acu_4th | CGTCTTCAYGGAAGCATGCAACTGTGT |
Abbreviations according to IUPAC nucleotide code.
Peptide-mass mapping
ACU-A (0.2 mg) was reduced with DTT (6 μL, 40 mg mL−1) in buffer A (39 μL, urea (8 m), EDTA (1 mm), and Tris-HCl (0.5 m, pH 8.0)) for 90 min at 37 °C. Subsequently, iodoacetamide (45 μL, 40 mg mL−1 in buffer A) was added, and the mixture was incubated at 37°C for 30 min. Carbamoylmethylated ACU-A was purified by RP-HPLC with YMC-Pack Protein-RP (0.1% TFA/acetonitrile–water 10–50%). After lyophilization, the specimen was treated with V8 protease (10 μg in 60 μL Tris-HCl (50 mm, pH 8.0)) at 37°C for 3 h. The product was separated by RP-HPLC as above to afford fragments A–C. MALDI-TOF MS for each fraction showed molecular ions [M+H]+ at m/z 1126, 1368, and 2102, respectively.
A second portion of ACU-A (0.2 mg) was also alkylated as above, then digested by TPCK trypsin (4 μg in 200 μL urea (2 m) with EDTA (0.25 mm) and Tris-HCl (125 mm, pH 8.0)) at 37 °C for 20 h. The specimen afforded C-terminal fragment D after RP-HPLC purification as above.
Enzyme digest and isolation of N-terminal fragment E of ACU
A sample of ACU-A (7.0 mg) was dissolved with excess DTT in buffer A, incubated at 37 °C for 4 h, and the product was purified by RP-HPLC to give reduced ACU-A. The lyophilized sample was dissolved in Tris·HCl (4.5 mL, 50 mm, pH 8.0), mixed with 100 μL V8 protease (2 mg mL−1), and incubated at 37 °C for 18 h. The digested specimen was run on RP-HPLC by 0.1% TFA/10 % acetonitrile-water to give the pure N-terminal fragment of ACU-A (1.1 mg). 1H NMR (500 MHz, D2O): δ = 7.56 (d, J = 7.8 Hz, 1 H), 7.39 (d, J = 8.4 Hz, 1 H), 7.17 (t, J =7.8 Hz, 1 H), 7.10 (m, 3H) and 6.72 (br s, 2 H) (Figures S7 and S8). A sample of ACU-B (0.2 mg) was treated similarly and purified to give the N-terminal fragment of ACU-B (weight not measurable). Amino acid analysis of fragment E was carried out using Marfey’s method. The fragment (50 μg) was hydrolyzed by HCl (6 n) for 12 h at 110°C. The hydrolyzate was treated with 1-fluoro-2,4-dinitrophenyl-5-l-alaninamide (Marfey’s reagent, l-FDAA). The product was analyzed by HPLC (Cosmosil C18-AR, 4.6×250 mm) with 0.05% TFA/acetonitrile-water 10–40%. Peaks for l-Asp and l-Tyr were detected at 50.520 and 51.773 min, respectively.
Toxicity of aculeines in mice
Toxicity was estimated by i.c.v. injection of the aqueous solution (20 μL). The behavioral profiles were scored as reported previously.[32] Dose–response curves were estimated by sigmoidal curve fitting of mean behavioral scores obtained over six doses (Figure S1). Three mice were used at each dose. GraphPad Prism (Graphpad Software, La Jolla, CA) was used to generate a curve (constraints: high score = 7, low score = 0).
Electrophysiology
Electrophysiological recordings from human embryonic kidney cells expressing T-antigen, clone 17 (HEK293-T/17) and cultured rat hippocampal neurons were performed as described previously.[38] HEK293-T/17 cells were cultured at 37°C with 5% CO2 in DMEM, with penicillin (100 μg mL−1), streptomycin (100 μg mL−1), and 10% heat-inactivated fetal bovine serum. Isolated neurons were prepared from E18 (prenatal) rat hippocampi using standard protocols[39] and were used for analysis from between 17 and 28 days in vitro. For voltage clamp recordings from neurons and HEK293-T/17 cells, cells were clamped in whole-cell configuration using an Axopatch 200B amplifier (Molecular Dynamics). The extracellular solution contained NaCl (140 mm), glucose (10 mm), Cs-HEPES (10 mm, pH 7.3), KCl (3 mm), CaCl2 (2 mm), and MgCl2 (1 mm). The internal solution for HEK293-T/17 recordings contained CsF (95 mm), CsCl (25 mm), Cs-HEPES (10 mm, pH 7.3), NaCl (2 mm), CaCl2 (0.5 mm), and EGTA (10 mm), and for neurons additionally contained Mg-ATP (2 mm), QX-314 (10 mm), TEA (5 mm), and 4-AP (5 mm). The internal solution for current clamp recordings contained KMeSO4 (120 mm), KCl (5 mm), NaCl (5 mm), MgCl2 (1 mm), Na-HEPES (11 mm, pH 7.0), phosphocreatine (10 mm), Na-ATP (4 mm), and Na-GTP (0.3 mm). ACUs were bath-applied. Gigaohm seals were established in voltage clamp mode and, after membrane rupture, the recordings were carried out either in voltage clamp mode (clamped at −70 mV) with a 5–15 mΩ series resistance, which was compensated to ~60%, or in current clamp mode (Vm = 60–65 mV) with the bridge balance corrected and pipette resistance neutralized.
Cytotoxicity
Cytotoxicity assays were performed on cultured HEK293-T/17 cells and cultured rat hippocampal neurons maintained on coverslips. Cells were treated with ACU-A (10 μg mL−1) or saline (control), washed twice with phosphate-buffered saline (PBS), stained with propidium iodide (PI; 0.5 μg mL−1) for 15 min at 37°C, and gently washed twice with PBS. Cultures were then fixed in 4% paraformaldehyde, washed and mounted for imaging on an LSM 510 META confocal microscope (Carl Zeiss, Inc.) in the Cell Imaging Core Facility at Northwestern University.
Ca2+ imaging
Cultured hippocampal neurons were incubated with Fura-2 AM (1 μg mL−1, Invitrogen) for 30 min at 37 °C. Coverslips were then transferred to an imaging chamber and perfused with an external solution containing NaCl (140 mm), glucose (10 mm), HEPES (10 mm, pH 7.4), KCl (3 mm), CaCl2 (2 mm), MgCl2 (1 mm). Cells were imaged on an Axiovert 40 CFL inverted microscope (Carl Zeiss, Inc.) through a 40× oil immersion lens. Signals were acquired on a CoolSNAP EZ camera (Photometrics) with Met-aFluor imaging software (Molecular Devices). Regions of interest were manually drawn around the soma of neurons and Fura-2 was sequentially excited at 340 and 380 nm (200 ms exposure time) by using a Xenon arc lamp, and fluorescence emission at 510 nm was recorded. Images were acquired every 2 s throughout the experiment. The ratio of fluorescence emission for each excitation wavelength (340 nm/380 nm) was calculated using Metafluor software.
Hemolytic activity
Rabbit erythrocytes were suspended (4% v/v) in buffer (KCl (2.68 mm), NaCl (137 mm), sodium phosphate (66.6 mm, pH 7.4)). The mixture was centrifuged and a portion (20 μL) was diluted to 120 μL in water, and then the absorption at 405 nm was measured. The ED50 value for ACU-A was estimated from a sigmoidal curve obtained between 0.76 mm and lowest 0.74 μm by using GraphPad Prism. To compare lytic potency of the peptide among different species of animals (cow, rabbit, sheep, guinea pig, and human type B), each erythrocyte was prepared as above and sample of ACU-A (0.01 mm, 50 μL) was mixed with 50 μL of the cell suspension and incubated for 210 min, and then absorptions at 405 nm were measured.
Cytotoxicity to tumor cell lines
Assays were performed at PharmaMar (Madrid, Spain) with human tumor cell lines MDA-MB-231, A 549, and HT-29.
Supplementary Material
Acknowledgments
This work was financially supported by the Naito Foundation and a Grant-in-Aid for Scientific Research from Ministry of Education, Culture, Sports, Science and Technology, Japan (15580183 and 17380125 to R.S.) and ROI NS44322 from NINDS (G.T.S.). Imaging of fixed neurons was performed at the Northwestern University Cell Imaging Facility and was partially subsidized by NCI CCSG P30 CA060553 awarded to the Robert H. Lurie Comprehensive Cancer Center. We thank Dr. Keiko Shimamoto at Suntory Institute for Bioorganic Research for the radioligand binding assay, Dr. John Hooper at Queensland Museum the identification of the sponge specimens, Dr. Kyoko Adachi at the Marine Biotechnology Institute and Katsuhiko Kushida at Agilent technologies for assistance in the NMR measurements, Dr. William Marszalec at Northwestern University for preparation of the rat hippocampal neurons, and PharmaMar for the cytotoxicity assay against tumor cell lines.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/cbic.201100329.
References
- 1.Fry BG. Toxicon. 1999;37:11. doi: 10.1016/s0041-0101(98)00125-1. [DOI] [PubMed] [Google Scholar]
- 2.Corzo G, Escoubas P. Cell Mol Life Sci. 2003;60:2409. doi: 10.1007/s00018-003-3108-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Estrada G, Villegas E, Corzo G. Nat Prod Rep. 2007;24:145. doi: 10.1039/b603083c. [DOI] [PubMed] [Google Scholar]
- 4.Possani LD, Merino E, Corona M, Bolivar F, Becerril B. Biochimie. 2000;82:861. doi: 10.1016/s0300-9084(00)01167-6. [DOI] [PubMed] [Google Scholar]
- 5.Shiomi K. Toxicon. 2009;54:1112. doi: 10.1016/j.toxicon.2009.02.031. [DOI] [PubMed] [Google Scholar]
- 6.Olivera BM, Teichert RW. Mol Interventions. 2007;7:251. doi: 10.1124/mi.7.5.7. [DOI] [PubMed] [Google Scholar]
- 7.Craik DJ, Daly NL, Bond T, Waine C. J Mol Biol. 1999;294:1327. doi: 10.1006/jmbi.1999.3383. [DOI] [PubMed] [Google Scholar]
- 8.Harding L, Scott RH, Kellenberger C, Hietter H, Luu B, Beadle DJ, Bermudez I. J Recept Signal Transduction Res. 1995;15:355. doi: 10.3109/10799899509045226. [DOI] [PubMed] [Google Scholar]
- 9.Gruber CW, Muttenthaler M, Freissmuth M. Curr Pharm Des. 2010;16:3071. doi: 10.2174/138161210793292474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thongyoo P, Bonomelli C, Leatherbarrow RJ, Tate EW. J Med Chem. 2009;52:6197. doi: 10.1021/jm901233u. [DOI] [PubMed] [Google Scholar]
- 11.Huang YH, Colgrave ML, Daly NL, Keleshian A, Martinac B, Craik DJ. J Biol Chem. 2009;284:20699. doi: 10.1074/jbc.M109.003384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pallaghy PK, Nielsen KJ, Craik DJ, Norton RS. Protein Sci. 1994;3:1833. doi: 10.1002/pro.5560031022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Henriques ST, Craik DJ. Drug Discovery Today. 2010;15:57. doi: 10.1016/j.drudis.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 14.Olivera BM, Cruz LJ, De Santos V, LeCheminant G, Griffin D, Zeikus R, McIntosh JM, Galyean R, Varga J, Gray WR, Rivier J. Biochemistry. 1987;26:2086. doi: 10.1021/bi00382a004. [DOI] [PubMed] [Google Scholar]
- 15.Fusetani N, Matsunaga S. Chem Rev. 1993;93:1793. [Google Scholar]
- 16.Bewley CA, Faulkner DJ. Angew Chem. 1998;110:2280. doi: 10.1002/(SICI)1521-3773(19980904)37:16<2162::AID-ANIE2162>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 1998;37:2162. [Google Scholar]
- 17.Dunlap WC, Battershill CN, Liptrot CH, Cobb RE, Bourne DG, Jaspars M, Long PF, Newman DJ. Methods. 2007;42:358. doi: 10.1016/j.ymeth.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 18.Takada K, Hamada T, Hirota H, Nakao Y, Matsunaga S, van Soest RW, Fusetani N. Chem Biol. 2006;13:569. doi: 10.1016/j.chembiol.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 19.Matsunaga S, Sakai R, Jimbo M, Kamiya H. ChemBioChem. 2007;8:1729. doi: 10.1002/cbic.200700305. [DOI] [PubMed] [Google Scholar]
- 20.Roder H, Elöve GA, Ramachandra Shastry MC. In: Frontiers in Molecular Biology: Mechanisms of Protein Folding. 2. Pain RH, editor. Oxford University Press; New York: 2000. p. 70. [Google Scholar]
- 21.Biemann K. Methods Enzymol. 1990;193:886. doi: 10.1016/0076-6879(90)93460-3. [DOI] [PubMed] [Google Scholar]
- 22.Marfey P. Carlsberg Res Commun. 1984;49:591. [Google Scholar]
- 23.Kröger N, Deutzmann R, Sumper M. Science. 1999;286:1129. doi: 10.1126/science.286.5442.1129. [DOI] [PubMed] [Google Scholar]
- 24.Kröger N, Deutzmann R, Bergsdorf C, Sumper M. Proc Natl Acad Sci USA. 2000;97:14133. doi: 10.1073/pnas.260496497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sumper M. Science. 2002;295:2430. doi: 10.1126/science.1070026. [DOI] [PubMed] [Google Scholar]
- 26.Gracy J, Le-Nguyen D, Gelly J-C, Kaas Q, Heitz A, Chiche L. Nucleic Acids Res. 2008;36:D314. doi: 10.1093/nar/gkm939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Norton RS, Pallaghy PK. Toxicon. 1998;36:1573. doi: 10.1016/s0041-0101(98)00149-4. [DOI] [PubMed] [Google Scholar]
- 28.Olivera BM, Rivier J, Scott JK, Hillyard DR, Cruz LJ. J Biol Chem. 1991;266:22067. [PubMed] [Google Scholar]
- 29.Liang S. Toxicon. 2004;43:575. doi: 10.1016/j.toxicon.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 30.Murphy DE, Snowhill EW, Williams M. Neurochem Res. 1987;12:775. doi: 10.1007/BF00971514. [DOI] [PubMed] [Google Scholar]
- 31.London ED, Coyle JT. Mol Pharmacol. 1979;15:492. [PubMed] [Google Scholar]
- 32.Sakai R, Swanson GT, Shimamoto K, Green T, Contractor A, Ghetti A, Tamura-Horikawa Y, Oiwa C, Kamiya H. J Pharmacol Exp Ther. 2001;296:650. [PubMed] [Google Scholar]
- 33.Nelson GJ. Biochim Biophys Acta Lipids Lipid Metab. 1967;144:221. doi: 10.1016/0005-2760(67)90152-x. [DOI] [PubMed] [Google Scholar]
- 34.Zasloff M. Nature. 2002;415:389. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- 35.Hancock REW, Diamond G. Trends Microbiol. 2000;8:402. doi: 10.1016/s0966-842x(00)01823-0. [DOI] [PubMed] [Google Scholar]
- 36.Takano T, Konoki K, Matsumori N, Murata M. Bioorg Med Chem. 2009;17:6301. doi: 10.1016/j.bmc.2009.07.036. [DOI] [PubMed] [Google Scholar]
- 37.Schägger H, von Jagow G. Anal Biochem. 1987;166:368. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- 38.Gill MB, Frausto S, Ikoma M, Sasaki M, Oikawa M, Sakai R, Swanson GT. Br J Pharmacol. 2010;160:1417. doi: 10.1111/j.1476-5381.2010.00784.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Banker GA, Cowan WM. Brain Res. 1977;126:397. doi: 10.1016/0006-8993(77)90594-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.