

Abstract
Antifibrinolytic drugs are widely used to reduce blood loss during surgery. One serious adverse effect of these drugs is convulsive seizures; however, the mechanisms underlying such seizures remain poorly understood. The antifibrinolytic drugs tranexamic acid (TXA) and ε-aminocaproic acid (EACA) are structurally similar to the inhibitory neurotransmitter glycine. Since reduced function of glycine receptors causes seizures, we hypothesized that TXA and EACA inhibit the activity of glycine receptors. Here we demonstrate that TXA and EACA are competitive antagonists of glycine receptors in mice. We also showed that the general anesthetic isoflurane, and to a lesser extent propofol, reverses TXA inhibition of glycine receptor–mediated current, suggesting that these drugs could potentially be used to treat TXA-induced seizures. Finally, we measured the concentration of TXA in the cerebrospinal fluid (CSF) of patients undergoing major cardiovascular surgery. Surprisingly, peak TXA concentration in the CSF occurred after termination of drug infusion and in one patient coincided with the onset of seizures. Collectively, these results show that concentrations of TXA equivalent to those measured in the CSF of patients inhibited glycine receptors. Furthermore, isoflurane or propofol may prevent or reverse TXA-induced seizures.
Introduction
Antifibrinolytic drugs are widely used to reduce blood loss in a variety of hemorrhagic conditions, including severe trauma (1), cardiac and non-cardiac surgery (2–5), and maternal hemorrhage (6). Safe and effective pharmacological blood conservation strategies are needed, as the risks and costs associated with allogenic blood transfusions continue to increase (7, 8). Such pharmacological treatments for hemorrhage are particularly important in developing nations, where blood banking facilities are limited, the risk of blood-borne infection is high, and the number of trauma-related deaths is rapidly rising (9).
Tranexamic acid (TXA) and ε-aminocaproic acid (EACA) are widely used antifibrinolytics (10). These two lysine analogs exert their antifibrinolytic effects by inhibiting the activation of plasminogen, thereby preventing degradation of fibrin and dissolution of clots (11). Another commonly used antifibrinolytic drug, aprotinin (a serine protease inhibitor derived from bovine lung), is structurally different from TXA and EACA. Aprotinin prevents blood loss by directly antagonizing plasmin (12). Aprotinin was suspended from the market because of reports of a higher incidence of death and renal dysfunction (13–15). Consequently, reliance on TXA and EACA will remain high for the foreseeable future.
TXA and EACA evoke seizures in both laboratory animals and patients, but the mechanisms underlying these seizures have not been clearly elucidated. Direct application of TXA to the cortex of cats (16) and intrathecal and intravenous administration of this drug to rats evoke convulsive and proconvulsive behaviors (17, 18). In patients, generalized tonic-clonic seizures have occurred after inadvertent intrathecal injection of TXA (19–21) and after intravenous administrations of EACA (22).
More recently, TXA has been associated with an increased incidence of postoperative seizures in cardiac patients (23, 24). Historically, postoperative seizures have occurred in about 0.5%–1% of cardiac patients (25, 26), but the use of higher doses of TXA has been associated with a higher incidence of seizures (2.7%–7.6%) (27, 28), primarily of the grand mal type (23, 29). The frequency of seizures is higher among patients with preoperative renal failure, patients undergoing open heart surgery, and older patients (30). These seizures typically occur within hours of the patient being transferred from the operating room to the intensive care unit, when the concentrations of anesthetic are declining rapidly and TXA levels remain high (29). Such postoperative seizures constitute a serious adverse effect, because they may be associated with an increased incidence of neurological complications (including delirium and stroke) (31), prolongation of recovery times, and higher mortality rates (25, 31). Currently, there are no mechanism-based treatments or prevention strategies for seizures associated with TXA or EACA.
Both TXA and EACA are structural analogs of the amino acid glycine, a major inhibitory neurotransmitter in the brain and spinal cord (Figure 1). Analogs of glycine may act as competitive antagonists, occupying the glycine-binding site and preventing glycine from binding to and activating its receptor. Glycine receptors are predominantly expressed in the spinal cord and brain stem but are also widely expressed in the prefrontal cortex, the hippocampus, and the amygdala (32). These receptors are pentameric chloride ion channels that are composed of α1–4 and β subunits (33). The subunit composition of each receptor determines its pharmacological properties, as well as its expression patterns in the CNS and the subcellular regions of neurons (34).
Figure 1. Molecular structures of glycine and the antifibrinolytic drugs TXA, EACA, and aprotinin.

Glycine receptors in the CNS mediate two distinct forms of inhibition: postsynaptic and tonic (32). Postsynaptic inhibitory currents are generated by glycine receptors clustered in the postsynaptic terminal, which are activated by the synchronous release of high concentrations of glycine (35). Tonic glycine currents mediate a “paracrine” form of inhibition that is generated by extrasynaptic glycine receptors, which are activated by low, ambient concentrations of glycine spilling over from the synaptic cleft or by glycine that is released by non-vesicular mechanisms (35). A tonic glycinergic inhibitory conductance has been identified in the hypothalamus, the hippocampus, and the dorsal horn of the spinal cord (36). The pharmacological and physiological properties of glycine receptors that generate postsynaptic and tonic inhibition may differ considerably (37).
Since drugs that inhibit glycine receptors are known to induce seizures, we tested the hypothesis that TXA and EACA, but not aprotinin, competitively inhibit glycine receptor function. Of the 3 antifibrinolytic drugs, we most thoroughly investigated the properties of TXA, as it is widely used. As predicted for a competitive antagonist, we anticipated that tonic inhibitory current, evoked by low concentrations of extracellular glycine, would be more sensitive than postsynaptic currents to TXA blockade. In addition, while the primary focus of this study was on TXA modulation of glycine receptors, we also studied the potency of TXA inhibition of γ-aminobutyric acid type A (GABAA) receptors. GABAA receptors are major mediators of inhibition in the CNS and were previously shown to be competitively inhibited by TXA when expressed recombinantly in human embryonic kidney cells (38). Here, we directly compare TXA blockade of native glycine and GABAA receptors in cortical and spinal cord neurons.
We also sought to identify a strategy to reverse the inhibitory effect of TXA on glycine receptors. Given that anesthetics, including isoflurane and propofol, allosterically potentiate glycine receptor activity (39–41), and seizures frequently occur during emergence from anesthesia (29), we hypothesized that these drugs would reverse or attenuate inhibition of glycine receptors by TXA. We also studied the proconvulsant properties of TXA in neocortical slices and anticipated that anesthetics would attenuate TXA-induced enhancement of network excitability.
Finally, to determine whether glycine receptors represent clinically relevant targets for TXA, we measured the concentration of TXA in the cerebrospinal fluid (CSF) of patients. CSF samples were obtained at different surgical time points from patients undergoing surgical repair of thoraco-abdominal aortic pathology. This surgical procedure was selected because it involves insertion of a catheter into the lumbar intrathecal space, to drain the CSF and thereby improve blood flow in the spinal cord and reduce neurological damage (42, 43). The results show that glycine receptors, particularly those activated by a low concentration of glycine, are inhibited by concentrations of TXA that occur in the CSF of patients.
Results
TXA is a competitive antagonist of glycine receptors.
TXA is a structural analog of glycine, which suggests that it may compete for the glycine-binding site and thereby inhibit glycine receptor function. The effects of TXA on glycine receptors were studied using mouse neurons grown in dissociated cell cultures. Primary cultures of mouse neurons are a suitable experimental model, because the glycine receptors of mice are remarkably similar to those of humans (32). All glycine-evoked currents were recorded in the presence of the GABAA receptor antagonist bicuculline (10 μM). First, the concentration of glycine that evoked the half-maximal current (EC50) in cortical neurons was determined to be 95.7 ± 16.2 μM (n = 4–6; Supplemental Figure 1A; supplemental material available online with this article; doi: 10.1172/JCI63375DS1). Next, the effect of TXA on current evoked by a glycine concentration near the EC50 (100 μM) was studied. Coapplication of TXA (0.01–30 mM) and glycine caused a rapid and reversible decrease in current amplitude (Figure 2A). The half-maximal inhibitory concentration of TXA (IC50) was 1.1 ± 0.1 mM (estimated Hill coefficient [nH] value = 1.4 ± 0.04, n = 6–8; Figure 2A).
Figure 2. TXA is a competitive antagonist of glycine receptors.

(A) TXA (1 mM) inhibits glycine (100 μM)–activated currents in cortical neurons. The corresponding concentration-response plot shows IC50 = 1.1 ± 0.1 mM (n = 6–8). Here, and in subsequent figures, pA and nA stand for picoamperes and nanoamperes, respectively. (B) Concentration-response plots for glycine current recorded in the absence and presence of TXA. All values were normalized to currents evoked by glycine (100 μM). The EC50 values and Hill coefficients for TXA (0, 0.1, 1, 10 mM) were 0.094 ± 0.007 mM and 1.4 ± 0.2; 0.079 ± 0.009 mM and 1.4 ± 0.2; 0.21 ± 0.038 mM and 1.2 ± 0.2; and 0.79 ± 0.013 mM and 1.7 ± 0.4, respectively (n = 6–7). The Schild plot (inset) was generated by calculating the dose-response ratios (DR-1) of the EC50 of TXA divided by the EC50 of glycine for each concentration of TXA. The plot is consistent with a slope near 1 (r2 = 0.81) and a dissociation constant of 0.73 mM. Data are mean ± SEM.
TXA inhibition of glycine current was similar whether TXA was pre- or coapplied with glycine (Supplemental Figure 2). Also, TXA alone did not elicit any current (data not shown), which indicates that TXA does not act as an agonist at glycine receptors. TXA caused a slight reduction in the rate and extent of current desensitization (Supplemental Figure 3). However, desensitization in the presence of TXA was similar to that observed when glycine receptors were activated by a lower concentration of glycine alone. This latter observation suggests that TXA does not directly modulate glycine receptor desensitization.
To further determine whether TXA acts as a competitive antagonist, glycine concentration response plots were generated for currents recorded in the absence or presence of TXA (Figure 2B). TXA (1 and 10 mM) shifted the glycine concentration response plot to the right without reducing the maximal response. More specifically, TXA (1 mM) increased the glycine EC50 from 93.9 ± 6.8 μM to 207.1 ± 38.9 μM (n = 6–7). TXA (10 mM) further increased the glycine EC50 to 788.6 ± 13.1 μM (n = 6–7). Schild regression analysis yielded a slope similar to unity (slope = 0.81, n = 6–7; Figure 2B, inset), which indicates that TXA is a competitive antagonist.
We next tested whether TXA inhibition of glycine receptors exhibited use or voltage dependence, as observed for noncompetitive antagonists that preferentially bind to the open state of the channel (44). The onset of TXA inhibition was not use dependent, and recovery from blockade was reversed immediately after TXA washout (Figure 3A). The degree of TXA inhibition was only slightly greater at negative holding potentials than positive holding potentials, as evidenced by the outward rectification of the current-voltage plot in the presence of TXA (Figure 3B). This apparent voltage sensitivity of TXA inhibition was attributed to the voltage sensitivity of glycine receptors being activated by lower concentrations of the agonist (45), because in the absence of TXA, a low concentration of glycine (30 μM) also produced a slight outward rectification of the current-voltage plot (Figure 3B). These results are consistent with the interpretation that TXA is a competitive antagonist of glycine receptors.
Figure 3. TXA inhibition of glycine receptors is not use or voltage dependent.

(A) Repeated application of TXA (1 mM) does not cause an increasing block of glycine (100 μM)–evoked currents. The bar graph shows mean peak amplitude of the current recorded with and without coapplication of TXA. Current recorded during the sequential application of TXA is identified in the bars as 1, 2, and 3 (n = 6). (B) Currents evoked by glycine (100 and 30 μM) in the absence and presence of TXA (1 mM) were recorded at two holding potentials. The current-voltage plot shows that TXA causes an outward rectification of glycine (100 μM) current-voltage plot and that glycine (30 μM) produced a similar modest outward rectification (n = 5–6). All values were normalized to glycine current activated by 100 μM at –60 mV. Data are mean ± SEM.
EACA, but not aprotinin, inhibits glycine receptors.
Like TXA, EACA may also antagonize glycine receptors. Coapplication of EACA (10 mM) and glycine (100 μM) rapidly and reversibly inhibited the glycine-evoked current (Figure 4A). The concentration-response plot for EACA blockade of glycine currents revealed an IC50 of 12.3 ± 0.9 mM and nH of 1.5 ± 0.1 (n = 5–6; Figure 4A). Notably, the potency of EACA was 10-fold lower than that of TXA (TXA IC50 = 1.1 ± 0.1 mM; Figure 2A). EACA caused a rightward shift in the glycine concentration-response plot without reducing the maximal response (Figure 4B).
Figure 4. EACA competitively inhibits glycine receptors, whereas aprotinin does not.

(A) Inhibition of glycine (100 μM)–activated current by EACA (10 mM) in cortical neurons. The corresponding concentration-response plot shows IC50 = 12.3 ± 0.9 mM (n = 5–6). (B) Plots for current recorded in the absence and presence of EACA (10 mM) show a rightward shift in the presence of EACA. The EC50 values and Hill coefficients for EACA were 134.1 ± 4.7 μM and 1.7 ± 0.2, respectively (n = 5–6). All values were normalized to currents evoked by glycine (100 μM). (C) Aprotinin (100 μM) has no effect on glycine (100 μM)–evoked current in cortical neurons. The graph shows that aprotinin (1–100 μM) failed to inhibit glycine-evoked current (n = 5). Data are mean ± SEM.
The effect of aprotinin on glycine receptor function was also investigated. Aprotinin was studied at lower concentrations, as its potency in terms of antifibrinolytic effect is greater than that of TXA or EACA (46). Aprotinin applied at concentrations as high as 100 μM did not inhibit glycine receptor function (Figure 4C).
TXA inhibits glycine receptors in spinal cord neurons.
Glycine receptors expressed in the spinal cord may exhibit pharmacological properties different from those of glycine receptors expressed in the cortex (35). Therefore, we next investigated the effect of TXA on glycine receptors in spinal cord neurons. In addition, cultures of spinal cord neurons offer the experimental advantage of a relatively high-throughput model to study the effects of TXA on postsynaptic glycinergic currents as well as currents activated by low and high concentrations of exogenous glycine (37). The EC50 of glycine in spinal cord neurons was determined to be 34.9 ± 14.6 μM (n = 5, Supplemental Figure 1A). TXA rapidly and reversibly inhibited the current evoked by glycine (30 μM) (IC50 = 1.4 ± 0.1 mM, nH = 1.5 ± 0.03, n = 6; Supplemental Figure 4).
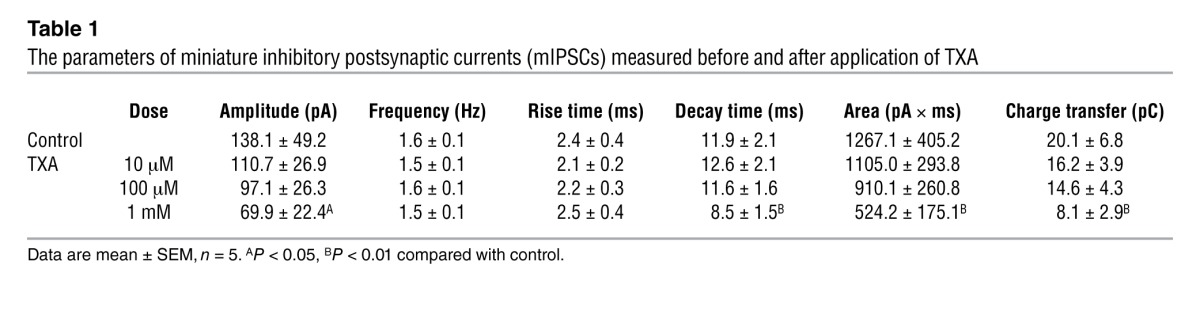
To evaluate inhibition by TXA of glycine receptors under more physiologically relevant conditions, we studied glycinergic miniature inhibitory postsynaptic currents (mIPSCs). The competitive glycine receptor antagonist strychnine (1 μM) completely abolished all mIPSCs (n = 6; Figure 5A), confirming that the currents were generated by glycine receptors, as shown previously (47). Relatively low concentrations of TXA (10 and 100 μM) did not inhibit the mIPSCs, whereas a higher concentration of TXA (1 mM) decreased the amplitude, decay time, area, and charge transfer of these currents (Figure 5 and Table 1).
Figure 5. TXA inhibits glycinergic mIPSCs at higher concentrations.

(A) Glycinergic mIPSCs recorded from a spinal cord neuron, in the absence and presence of TXA. Application of strychnine abolished all glycinergic mIPSCs. pC stands for picocoloumbs. (B) Cumulative frequency plots for mIPSCs show the effect of TXA on the amplitude and inter-event interval. TXA (1 mM) significantly decreased the amplitude of the mIPSCs (left panel) without affecting the inter-event interval (right panel). (C) Average traces from 100 individual mIPSCs before and after application of TXA (1 mM). Data for A–C were obtained from the same spinal cord neuron. (D) TXA (1 mM) significantly decreased charge transfer of glycinergic mIPSCs, whereas lower concentrations had no significant effect (n = 5). *P < 0.05, **P < 0.01 versus 0 TXA. Data are mean ± SEM.
Table 1.
The parameters of miniature inhibitory postsynaptic currents (mIPSCs) measured before and after application of TXA
Next, we investigated the effects of TXA on currents evoked by a low concentration of glycine (EC6 = 10 μM) under conditions intended to mimic the tonic glycine current recorded in spinal cord slices (33, 48, 49). Current amplitude was studied by applying strychnine (1 μM) and measuring the reduction in holding current, as described previously (36, 50). In the absence of TXA, the amplitude of the glycine-evoked current was 187.2 ± 54.3 pA (n = 12; Figure 6A). TXA caused a rapid, reversible, concentration-dependent decrease in the current (IC50 = 93.1 ± 2.7 μM, n = 6–8; Figure 6, A and B). Also, the variance of the baseline current noise regulates network excitability (51). Thus, we examined the effects of TXA on the baseline noise. TXA at concentrations as low as 10 μM reduced the variance of the noise (baseline: 20.2 ± 2.1 pA versus TXA: 16.1 ± 1.9 pA, n = 5–7; Figure 6C). In some recordings, a transient, small-amplitude, inward “rebound” current was observed after termination of TXA application. This rebound current was attributed to glycine receptors that were recovering from desensitization (52), as a similar rebound phenomenon has been reported for competitive antagonists of GABAA receptors (53). Thus, the current amplitude and the variance of the noise of the tonic glycine current were highly sensitive to TXA inhibition with the highest potency for glycine current evoked by low concentrations of agonist.
Figure 6. TXA, at clinical concentrations, inhibits current evoked by low concentrations of glycine.

(A) Inhibition of glycine (10 μM) currents by TXA (0.01–1 mM) in a spinal cord neuron. Application of strychnine (1 μM) completely inhibited the glycine current. (B) Concentration-response plot for TXA-mediated inhibition of glycine current in spinal cord neurons shows IC50 = 93.1 ± 2.7 μM (n = 6). (C) Analysis of the effect of TXA on noise shows that TXA (0.01–10 mM) significantly reduces receptor activity (n = 5–7). ***P < 0.001 versus 0 TXA. Data are mean ± SEM.
TXA inhibits GABAA receptors in cortical and spinal cord neurons.
Since TXA was previously shown to inhibit recombinant GABAA receptors expressed in human embryonic kidney cells (IC50 = 7.1 mM) (38), we next examined the effect of TXA on native GABAA receptors in both cortical and spinal cord neurons. First, the EC50 of GABA was determined to be 19.7 ± 2.3 μM (n = 4–6) in cortical neurons and 23.9 ± 2.4 μM (n = 5–7) in spinal cord neurons (Supplemental Figure 1B). Next, the IC50 for TXA inhibition of GABA currents evoked by concentrations near EC50 values were found to be 1.5 ± 0.1 mM (n = 4–6) in both cortical and spinal cord neurons (Figure 7A).
Figure 7. TXA inhibits currents evoked by high and low concentrations of GABA in cortical and spinal cord neurons.

(A) TXA (1 mM) inhibits currents evoked by high (EC50) concentrations of GABA in cortical neurons (20 μM) and spinal cord neurons (25 μM). The corresponding concentration-response plots show IC50 = 1.5 ± 0.1 mM (n = 4–6) in both the cortex and the spinal cord. (B) TXA (1 mM) also inhibits currents evoked by low concentrations of GABA (1 μM) in cortical and spinal cord neurons. The corresponding concentration-response plots show IC50 = 1.0 ± 0.1 mM (n = 5–7) in the cortex and IC50 = 0.9 ± 0.1 mM (n = 5–7) in the spinal cord. Application of bicuculline (BIC, 100 μM) completely inhibited GABA current. Data are mean ± SEM.
We next investigated the effect of TXA on GABAA receptors activated by a low concentration of GABA (EC4 = 1 μM), since a tonic GABAA receptor–mediated current has been described in the spinal cord (54), cortex (55), and several other regions in the CNS (56). This low concentration of GABA was selected because it is near the extracellular or ambient concentration of GABA measured previously (57). The amplitude of the “tonic current” was studied by applying bicuculline (100 μM) and measuring the reduction in holding current (56, 58). TXA inhibited currents evoked by a low concentration of GABA in both cortical neurons (IC50 = 1.0 ± 0.1 mM, n = 5–7; Figure 7B) and spinal cord neurons (IC50 = 0.9 ± 0.1 mM, n = 5–7; Figure 7B).
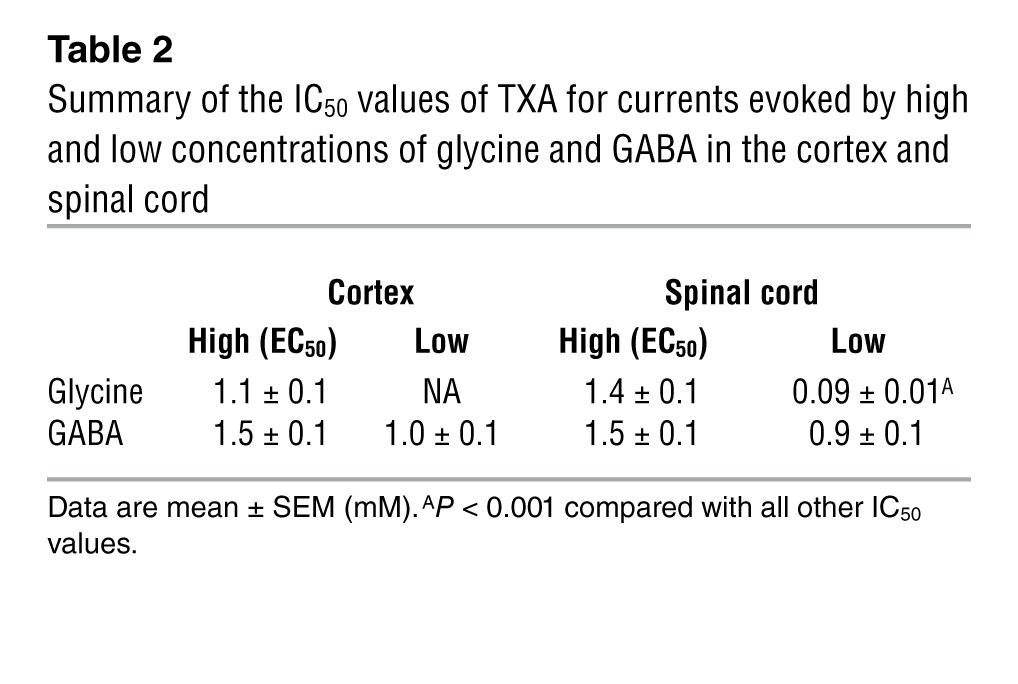
The potency of TXA for inhibition of GABAA receptors and glycine receptors in cortical and spinal cord neurons is summarized in Table 2. Notably, the potency of TXA for glycine-evoked current activated by a low concentration of glycine (EC6) was 10-fold higher than that for glycine- and GABA-evoked currents studied under all the other experimental conditions (F6,38 = 6.68, P < 0.0001, Table 2).
Table 2.
Summary of the IC50 values of TXA for currents evoked by high and low concentrations of glycine and GABA in the cortex and spinal cord
Isoflurane and propofol attenuate TXA inhibition of glycine receptors.
TXA-associated seizures occur most frequently as patients emerge from general anesthesia in the early postoperative period (23, 24, 27, 59). Thus, we postulated that the prototypic general anesthetics isoflurane (60) and propofol (39) would attenuate TXA inhibition of glycine receptors. Also, the benzodiazepine midazolam is frequently used as an anticonvulsant in the intensive care unit (61). Hence, the effect of midazolam, which selectively upregulates GABAA receptor activity but has no effect on glycine receptors (62), was studied as a negative control.
TXA (0.1 mM) reduced the current evoked by a low concentration of glycine to 59.7% ± 2.8% of control (n = 7; Figure 8). Coapplication of isoflurane, at clinically relevant concentrations (150 and 250 μM), reduced TXA inhibition as the current increased to 130.6% ± 4.5% and 176.2% ± 17.3% (n = 6–7) of control, respectively, whereas isoflurane at a lower concentration (50 μM; Figure 8C) had no effect. In the absence of TXA, isoflurane (150 μM and 250 μM) potentiated glycine current, whereas isoflurane (50 μM) had no effect on current amplitude (Figure 8, A and C).
Figure 8. Isoflurane and propofol attenuate TXA inhibition of current evoked by a low concentration of glycine.

(A and B) The effects of isoflurane (ISO, 150 and 250 μM) and propofol (Prop, 1 and 3 μM) on current evoked by glycine (10 μM) in the presence and absence of TXA (0.1 mM). (C and D) Isoflurane (150 and 250 μM) and propofol (3 and 10 μM) attenuated TXA-mediated inhibition of glycine current (n = 6–7). In the absence of TXA, isoflurane (150 and 250 μM) increased current amplitude to 182.2% ± 13.8% and 258.7 ± 21.8% of control, respectively. Similarly, in the absence of TXA, propofol (3 and 10 μM) increased current amplitude to 138.0% ± 2.6% and 260.1% ± 11.2% of control, respectively (n = 6–7). Lower concentrations of isoflurane (50 μM) or propofol (1 μM) had no effect on current amplitude. **P < 0.01, ***P < 0.001 versus control. Data are mean ± SEM.
Coapplication of propofol at a concentration approximating that occurring during anesthesia (1 μM) (63) had no significant effect on TXA inhibition (Figure 8, B and D), while a higher concentration (3 μM) reversed TXA inhibition to control levels (101.5% ± 2.3%, n = 6–7; Figure 8, B and D). The coapplication of propofol (10 μM) and TXA enhanced the current to 183.3% ± 4.6% of control (n = 6–7; Figure 8D). In the absence of TXA, propofol (1 μM) failed to increase the baseline current, whereas higher concentrations of propofol (3 μM and 10 μM) increased the current amplitude (Figure 8, B and D). Finally, midazolam at a high concentration (1 μM) had no effect on TXA-mediated inhibition (Supplemental Figure 5).
TXA increases the frequency of seizure-like events and enhances evoked field potentials in cortical slices.
Previous studies have shown that disinhibition, secondary to reduced glycine receptor function, can generate spontaneous epileptiform field potentials or seizure-like events (SLEs) in cortical slices (64, 65). We therefore studied whether TXA would increase the frequency of SLEs, as has been shown for other pro-epileptic drugs (66–68). We employed a widely used seizure model, wherein neocortical slices are perfused with a solution containing no extracellular magnesium (0 Mg2+) (69, 70). Under these conditions, extracellular recordings in layers II or III revealed SLEs that consisted primarily of “inter-ictal” discharges (Figure 9A). The slices were then perfused with a solution containing TXA at a concentration similar to that measured in the CSF of patients (200 μM, see below). Notably, protein binding of TXA is minimal (3%) (71); thus, the effective concentration of TXA measured in the CSF of patients was equivalent to that used to perfuse the slices. We found that TXA (200 μM) doubled the frequency of SLEs (control: 1.5 Hz ± 0.4 Hz versus TXA: 3.7 Hz ± 0.1 Hz, n = 4 slices; Figure 9A), which is consistent with the interpretation that TXA increases network excitability.
Figure 9. TXA enhances SLEs and evoked field potentials in cortical slices.

(A) SLEs recorded with 0 Mg in the extracellular fluid, in the absence and presence of TXA (200 μM). The bar graph shows that TXA increases the frequency but not the amplitude (control: 0.12 ± 0.001 mV, TXA: 0.13 ± 0.007 mV) of the SLEs (n = 4). (B) Evoked field potentials recorded in regular ACSF in the absence and presence of TXA (200 μM and 1 mM). The graphs show that both concentrations of TXA significantly increase the amplitude and the area of the evoked field responses (n = 6). *P < 0.05, **P < 0.01, ***P < 0.001 versus control. Data are mean ± SEM.
The proconvulsive properties of drugs can also be studied in cortical slices by recording field potentials in response to an excitatory electrical stimulus (72). Evoked field responses represent the integrated electrical activity of a large number of neurons, reflecting a combination of synchronous synaptic potentials and action potentials (73). This model provides useful information about the link between single neuron recordings and larger-scale neurophysiological signals such as electroencephalography (74). Increases in the amplitude or area of field responses in cortical networks are correlated with increased excitatory synaptic drive (73).
We investigated whether TXA enhanced the evoked field response in neocortical slices by recording field potential in layers II or III in response to electrical stimuli in layer VI. Under baseline conditions, the evoked field response was characterized by an initial stimulation artefact, followed by a brief negatively directed peak (Figure 9B), as described previously (73). Perfusion of the slice with TXA (200 μM, see below) increased the amplitude and area of the peak responses by 59.1% ± 8.0% and 85.9% ± 14.9%, respectively (n = 6; Figure 9B). TXA at 1 mM further increased the amplitude and the area of the evoked response (Figure 9B). The evoked response gradually returned to baseline after TXA washout. These results suggest that TXA causes an increase in excitatory synaptic drive.
Since glycine and GABAA receptors interact in a synchronized manner to regulate the excitability of neuronal networks, it is not possible to clearly delineate the specific contribution of each inhibitory receptor to the TXA-induced increase in network excitability. Nevertheless, we attempted to probe the relative role of glycine receptors versus GABAA receptors by perfusing the slices with either strychnine or bicuculline in the absence and presence of TXA.
First, to study the effect of TXA inhibition of glycine receptors on network excitability, we recorded evoked field responses in slices that were perfused with bicuculline (10 μM) to block GABAA receptors. Bicuculline alone increased the amplitude to 234.2% ± 5.3% and the area to 6074.0% ± 214.4% of control (n = 5; Supplemental Figure 6A). Subsequent coapplication of TXA (200 μM and 1 mM) further increased the peak amplitude of the field potentials by 10.7% ± 5.4% and 29.1% ± 3.7%, respectively (n = 5; Supplemental Figure 6A), whereas the area was unchanged.
To study the contribution of GABAA receptors, we recorded evoked field responses in slices that were perfused with strychnine (10 μM) to block glycine receptors. Strychnine increased the amplitude to 313.8% ± 7.9% and the area to 1082.2% ± 89.2% of control (n = 5; Supplemental Figure 6B). Coapplication of TXA (200 μM and 1 mM) further increased the area of the evoked field potentials by 190.6% ± 58.4% and 213.8% ± 35.7% of control, respectively (n = 5; Supplemental Figure 6B). The amplitude of the evoked response was not affected further by TXA. These results suggest that TXA-mediated inhibition of both glycine receptors and GABAA receptors increases excitatory synaptic drive in the cortex.
Isoflurane is more effective than propofol at reversing TXA enhancement of evoked field potentials in cortical slices.
Since we showed that isoflurane and propofol attenuate TXA-mediated inhibition of glycine receptors, we sought to determine whether these anesthetics attenuate the increase in excitatory synaptic drive induced by TXA. Coapplication of isoflurane (250 μM) with TXA (200 μM) fully restored the amplitude of the evoked field response to control levels and reduced the area by 61.8% ± 6.7% (n = 5; Figure 10A). Coapplication of propofol (1 μM) with TXA (200 μM) did not restore the amplitude of the evoked response but reversed the TXA-induced increase in area by 19.2% ± 5.5% (n = 5; Figure 10B). Perfusion of slices with isoflurane or propofol alone reduced both the amplitude and the area of the evoked field response (Supplemental Figure 7), as demonstrated previously (75, 76). Together these results suggest that isoflurane may be more effective than propofol at reversing TXA-induced increases in excitatory synaptic drive. However, the reversal of TXA-mediated excitability by anesthetics could be mediated by an attenuating effect on glycine receptors or GABAA receptors, by a combination of these inhibitory receptors, or by other anesthetic targets (77).
Figure 10. Isoflurane and propofol attenuate the enhancing effects of TXA on evoked field potentials in cortical slices.

(A) Coapplication of isoflurane (250 μM) with TXA (200 μM) reverses the effect of TXA on evoked field responses. The bar graphs show that isoflurane reverses the effect on amplitude and attenuates the effect on area of evoked field responses (n = 5). (B) Coapplication of propofol (1 μM) and TXA (200 μM) also attenuates the effect of TXA on the evoked field responses. The bar graphs show that propofol attenuates the enhancing effect on area but fails to reverse the effect on amplitude (n = 5). *P < 0.05, **P < 0.01 versus TXA. Data are mean ± SEM.
TXA concentrations in the serum and CSF of patients.
Only receptors that are sensitive to clinically relevant concentrations of TXA are plausible mediators of the adverse effects of this drug. Consequently, we sought to determine whether concentrations of TXA occurring in the CNS of patients who have received this drug would be sufficient to inhibit glycine receptors and GABAA receptors. Serum and CSF samples were obtained from patients who were undergoing repair of thoraco-abdominal pathology at certain times corresponding to procedural milestones. Serum and CSF concentrations of TXA for one patient who experienced a seizure are shown in Figure 11A. The average concentrations from the sample population are summarized in Figure 11B. The mean time points at which the serum and CSF samples were obtained and the total dose of TXA are presented in Table 3.
Figure 11. TXA concentrations measured in the serum and CSF of patients who underwent cardiopulmonary bypass and major vascular surgery.

(A) Time-dependent plot of TXA concentration in the serum and CSF of one patient. The timeline at the bottom of the figure represents key surgical events. (B) Average TXA concentration in the serum and CSF during key surgical events (n = 4). Data are mean ± SEM.
Table 3.
TXA doses and duration of the surgical events

Surprisingly, the temporal profiles for TXA concentrations in the serum and CSF were substantially different, as indicated by the samples from a single patient (Figure 11A). The peak concentration in the serum (1.9 ± 0.4 mM, n = 4; Figure 11B) occurred after cardiopulmonary bypass (CPB), whereas the peak concentration in the CSF (220.8 ± 116.7 μM, n = 4, Figure 11B) occurred after discontinuation of TXA infusion. Given that less than 3% of TXA is bound to protein (71), peak concentrations in the CSF were within the range required to inhibit glycine current evoked by a low concentration of agonist (IC50 = 93.1 ± 2.7 μM).
Discussion
To our knowledge, the study presented here is the first to identify glycine receptors as novel targets for inhibition by TXA and EACA, but not aprotinin. Because TXA is a competitive antagonist, inhibition mediated by this drug was expected to be highly dependent on the concentration of glycine applied, and this expectation was corroborated by the results. The potency of TXA was 10-fold higher for current evoked by a low concentration of glycine (IC50 = 93.1 μM) in spinal cord neurons than for postsynaptic glycinergic currents (IC50 = 1 mM) or currents activated by the EC50 of glycine in cortical neurons (IC50 = 1.1 mM) and spinal cord neurons (IC50 = 1.4 mM). Interestingly, the potency of TXA inhibition was similar for glycine receptors and GABAA receptors activated by the EC50 of agonist in cortical neurons and spinal cord neurons (Table 2).
We also show that TXA inhibition of glycine receptors activated by a low concentration of glycine (EC6) was reversed by isoflurane at clinically relevant concentrations. Propofol also reversed TXA-mediated glycine blockade, but only at concentrations 3-fold higher than those that occur during anesthesia (63). Consistent with these results, TXA-induced enhancement of excitatory synaptic drive in cortical slices was more effectively reversed by isoflurane than by propofol. Finally, we showed that the peak concentration of TXA in the CSF of patients undergoing major cardiovascular surgery (220.8 μM) occurred after CPB and termination of drug infusion. A similar concentration of TXA increased the frequency of SLEs and enhanced evoked field responses in cortical slices, which suggests that TXA has proconvulsive properties.
Inhibitors of glycine receptors are known to induce convulsions and proconvulsive behavior (78). For example, strychnine causes myoclonic spasms (79), twitching of muscles, convulsions, and hyperreactivity (78). Genetic disorders characterized by reduced expression of glycine receptors cause hyperekplexia, convulsions, and startle disorders in infants (80). In animals with naturally occurring mutations of the glycine receptor, the magnitude of the reduction in glycine receptor function directly correlates with the severity of the convulsive symptoms (81, 82). Thus, it is plausible that the reduction in function of these receptors caused by TXA leads to disinhibition and proconvulsive effects.
The potency of TXA was much greater for currents evoked by a low concentration of glycine than for synaptic glycinergic mIPSCs. The demonstration that TXA attenuates tonic current is of great interest, since a reduction in tonic current enhances neuronal excitability (36, 58). Tonic current reduces neuronal excitability via two mechanisms. First, this type of current allows for the transfer of large quantities of negative charge, a process that hyperpolarizes the cell membrane (36). Second, tonic conductance can attenuate the excitatory drive to generate action potentials by decreasing membrane resistance, thereby reducing the amplitude of excitatory postsynaptic potentials (83).
As anticipated for a competitive antagonist, the potency of TXA inhibition of glycine receptors was highly dependent on the concentration of agonist used in each experiment. However, this difference in potency might also have been due, at least in part, to the subunit composition of glycine receptors. Glycine receptors are composed of multiple α1–4 and β subunits, which form homomeric (5α) or heteromeric (2α3β) ion channels (32). Synaptic glycinergic currents are generated by heteromeric receptors that are clustered at the postsynaptic sites (35). Tonic glycinergic currents are mediated by homomeric receptors, which are located predominantly extrasynaptically (35). Glycine receptor antagonists, such as picrotoxin, show higher potency for homomeric than heteromeric glycine receptors (37). Post-transitional factors such as glycine receptor phosphorylation and the clustering of receptors in the postsynaptic domain can also alter the pharmacological properties of glycine receptors (52, 84). TXA potency in relation to various combinations of glycine receptor subunits as well as the effects of cytosolic regulatory factors on TXA potency are topics worthy of future study.
Our results and those of others (38) showed that TXA is a competitive antagonist of GABAA receptors in both cortical and spinal cord neurons (IC50 = 1.5 ± 0.1 mM for both). Interestingly, the potency of TXA for current evoked by a low concentration of GABA (EC4) in cortical (IC50 = 1.0 mM) and spinal cord (IC50 = 0.9 mM) neurons was similar to its potency for current evoked by higher concentrations of GABA (EC50). This result was unexpected and may reflect differences in the subunit composition of receptors activated by the various concentrations of GABA (56, 57, 85, 86). In addition, our results showed that TXA inhibition of GABAA receptors increase evoked field responses in neocortical slices in the presence of strychnine. Thus, TXA inhibition of GABAA receptors may increase network excitability.
A previous study showed that TXA has an IC50 value of 7.1 ± 3.1 mM for recombinant GABAA receptors (α1β2γ2), transfected into human embryonic kidney cells when receptors are activated by GABA 30 μM (EC70) (38). Also, binding assays for TXA with [3H]muscimol showed that the IC50 value for TXA binding to GABAA receptors was 2.1 ± 0.2 mM (38). In our study, the potency of TXA for GABA currents evoked by EC50 concentrations of agonist was lower. This difference may be attributable to our use of cell cultures with native receptors rather than recombinantly expressed receptors (87).
The peak TXA concentration in the CSF as measured in the current study was higher than that measured in the CSF of 4 patients undergoing CPB in a previous study (220.8 μM versus 31 μM) (88). In the earlier study, the TXA dose was significantly lower, and samples were taken at only two time points: 15 and 90 minutes after the start of TXA infusion (88). The discrepancy in results is probably related to two findings from the current study, specifically, that serum concentrations of TXA do not correlate well with CSF concentrations and that TXA concentrations peak in the CSF long after the start of drug infusion. Thus, the previous study may not have detected the true peak concentrations of TXA in the CSF.
The timing and magnitude of peak TXA concentrations after CPB may be attributable to increased permeability of the blood-brain barrier during and after surgery (89). In a study that used a protein marker (S100) to measure damage to the blood-brain barrier during CPB, the highest levels of the marker occurred after termination of the bypass (90). The blood-brain barrier appears to be particularly “leaky” at the end of CPB, which could account for the delayed rise of TXA concentrations in the CSF. Thus, the CSF may act as a slow compartment, which could explain the observed time course of TXA concentrations. A detailed pharmacokinetic analysis of TXA in the plasma and CSF would be of interest in future studies.
The results of this translational preclinical study have important clinical implications. First, we have shown that TXA is a competitive antagonist of both glycine and GABAA receptors. Thus, higher brain concentrations of TXA likely increase the risk of seizure. Indeed a higher incidence of seizures occurs in patients with preoperative renal failure (91) and those receiving higher doses of TXA (30). Currently, there is no consensus regarding optimum TXA dosing, and studies are urgently needed to determine the minimal effective dose of this drug (92).
Second, our results also suggest that isoflurane or propofol may prevent or treat TXA-induced seizures in the early postoperative period. Given the variable clinical presentation and incidence of TXA-associated seizures, clinical trials comparing the efficacy of various anticonvulsant treatments are unlikely to be feasible. Our results suggest that it may be possible to use isoflurane or propofol to prevent or treat TXA-induced seizures in the early postoperative period. Inhaled anesthetics, including isoflurane, are already being used for sedation of intubated patients in the intensive care unit (93). It must be emphasized, however, that anticonvulsants with no direct effects on glycine receptors, such as midazolam, may nonetheless be effective for treating TXA-associated seizures.
Third, because glycine receptors regulate numerous functions in the CNS, their inhibition by TXA could have adverse behavioral consequences other than seizures. One potential implication is that TXA and EACA may have pro-nociceptive properties, since antagonists of glycine receptors increase nociceptive responses in both laboratory animals and patients (94). Consistent with this prediction, patients with subarachnoid hemorrhage who were treated with TXA and EACA required higher doses of analgesics than placebo-treated patients (95). Also, patients with menorrhagia treated with TXA experienced a higher incidence of headaches, abdominal pain, and back pain than placebo-treated patients (96). Furthermore, inhibition of glycine receptors causes excessive or involuntary motor activity (hyperkinesia) in laboratory animals (97) and patients (98). Myoclonic activity observed in some patients treated with TXA may result from inhibition of glycine receptors (99).
In summary, these results show that TXA and EACA inhibit glycine receptors and suggest a novel mechanism for seizures associated with cardiovascular surgery. Isoflurane may be effective for preventing or treating TXA-induced excitability in the early postoperative period. As the indications for TXA and EACA increase, we hope that these results will aid in the prevention and management of serious neurological side effects associated with these drugs.
Methods
Whole-cell patch clamp recording in cell culture.
Primary cultures of embryonic cortical and spinal cord neurons were prepared from Swiss white mice. Briefly, fetal pups (E18) were removed from mice sacrificed by cervical dislocation. The cortex or spinal cord of each fetus was collected and placed in an ice-cooled culture dish. The neurons were then dissociated by mechanical titration using two Pasteur pipettes (tip diameter, 150–200 μm) and plated on 35-mm culture dishes at a density of about 1 × 106 cells/ml. The culture dishes were coated with collagen or poly-d-lysine (Sigma-Aldrich). For the first 5 days in vitro, cells were maintained in MEM supplemented with 10% fetal bovine serum and 10% horse serum (both from Life Technologies). The neurons were cultured at 37°C in a 5% CO2/95% air environment. After the background cells had grown to confluence, 0.1 ml of a mixture of 4 mg 5-fluorodeoxyuridine and 10 mg uridine in 20 ml MEM was added to the extracellular solution to reduce the number of dividing cells. The medium was further supplemented with 10% horse serum and was changed every 3 or 4 days. Cells were maintained in culture for 12–16 days before use. For all reported results, data were acquired from cells from at least 3 different dissections.
These experiments were performed at room temperature. Whole-cell currents were recorded under voltage clamp (–60 mV) conditions using an Axopatch 1D amplifier (Molecular Devices) controlled with pClamp 8.0 software (Molecular Devices) via a Digidata 1322 interface (Molecular Devices). Patch pipettes were pulled from thin-walled borosilicate glass capillary tubes; these had open tip resistances of 3–5 M⁄. Patch electrodes were filled with an intracellular solution containing (in mM) 140 CsCl, 10 HEPES, 11 EGTA, 2 MgCl2, 1 CaCl2, 4 MgATP, and 2 TEA (pH 7.3 with CsOH, 285–295 mOsm). The extracellular solution contained (in mM) 140 NaCl, 1.3 CaCl2, 2 KCl, 1 MgCl2, 25 HEPES, and 28 glucose. pH was adjusted to 7.4 with NaOH, and osmolarity was adjusted to 320–330 mOsm. The extracellular solution was applied to neurons by a computer-controlled, multi-barrel perfusion system (SF-77B, Warner Instruments). Membrane capacitance was measured according to the membrane test protocol in the pClamp 8.0 software. Access resistance was monitored throughout the experiments by applying a brief 10-mV hyperpolarization during the experiments. Cells were eliminated from further analysis if the access resistance changed by about 20% or more. Tetrodotoxin (TTX, 300 nM) was added to the extracellular solution to block voltage-sensitive sodium channels, and 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, 10 μM) and 2-amino-4-phosphonovalericacid (D-AP5, 40 μM) were added to inhibit the ionotropic glutamate receptors. Bicuculline (10 μM) was added to block GABAA receptors when glycine current was recorded. Strychnine (1 μM) was added to block glycine receptors when GABA current was measured.
To study synaptic glycine receptor–mediated currents, we added sucrose to the extracellular solution to stimulate glycine release and increase the frequency of glycinergic mIPSCs recorded from cultured spinal cord neurons. To increase the amplitude of tonic current and to facilitate its pharmacological characterization, we added glycine (EC6 = 10 μM) or GABA (EC4 = 1 μM) to the extracellular solution. These concentrations are similar to extracellular concentrations of glycine (48) and GABA (57) in the CNS.
Extracellular field potentials recorded in neocortical slices.
Mice (C57BL/6J and Sv129Ev cross) at P21–P28 were anesthetized with isoflurane and decapitated. Coronal neocortical slices (400 μm) were prepared using a vibratome (Leica VT 1200S) and placed in ice-cold, oxygenated (95% O2, 5% CO2) artificial cerebrospinal fluid (ACSF) containing (in mM) 124 NaCl, 3 KCl, 1.3 MgCl2, 2.6 CaCl2, 1.25 NaH2PO4, 26 NaHCO3, and 10 d-glucose, with osmolarity adjusted to 300–310 mOsm. After a recovery period of 1 hour in the oxygenated ACSF, the slices were transferred to a submersion recording chamber and perfused with oxygenated ACSF, maintained at 33–34°C, at a perfusion rate of 3–4 ml/min.
To demonstrate that TXA exhibits proconvulsant properties, we used the widely accepted 0 Mg+2 seizure model (69, 70). Slices were perfused with ACSF from which MgCl2 was omitted (designated 0 Mg2+). Recording electrodes were filled with ACSF and placed in layer II or III of the cortex. SLEs, or spontaneous high-frequency rhythmic potentials, which are typically observed in this model (100), were recorded under gap-free conditions using a Multiclamp 700A amplifier (Molecular Devices) in the current-clamp configuration. SLEs are traditionally divided into “ictal” states, defined as the onset of high-frequency rhythmic field discharges, and “interictal” states, defined as the low-frequency discharges that occur in the period between ictal states (100, 101). It is not uncommon for SLEs recorded in the neocortex to lack full ictal events (101). Indeed, we were only able to record interictal events in this preparation.
Evoked field responses were recorded in layers II or III of the neocortex with recording electrodes filled with standard ACSF and were monitored in each slice for 20 minutes or until the response stabilized. A concentric bipolar stimulating electrode (Rhodes Medical Instruments) was used to stimulate layer VI of the neocortex. Responses were evoked at 0.05 Hz at an intensity that yielded a half-maximal field potential amplitude. Evoked field responses were recorded in standard ACSF.
Data analysis.
Currents were analyzed with pClamp 10 software (Molecular Devices). The glycine concentration-response plot was fitted to the modified Michaelis-Menten equation: I = Imax / [1 + (EC50/c)nH],where I is the current amplitude, c is the concentration of agonist, EC50 is the concentration of agonist that produces currents with 50% of maximal amplitude, and nH is the estimated Hill coefficient. The reversal potential for Cl– was calculated with the Nernst equation: ECl– = 58 log[Cl–]inside/[Cl–]outside. Schild regression analysis was performed by calculating the dose-response ratios of antagonist EC50 to agonist EC50 for each concentration of TXA; the log of the concentration-response ratio was then plotted against the log of TXA concentration. The slope of the Schild plot was used to determine the nature of antagonism, given that competitive antagonists would generate a slope of 1. The x-intercept of the fitted regression line was used to calculate the equilibrium dissociation constant, which estimates the affinity of TXA for the glycine receptor (102).
To assess the effect of the drugs on synaptic glycinergic conductance, mIPSCs were analyzed with Mini Analysis software (Synaptosoft). For detection of mIPSCs, the threshold was set at approximately 3 times the level of baseline noise (~3.6 pA). However, compound events (i.e., those with multiple peaks) were excluded from the analysis. Manual inspection of each file was also performed, to allow rejection of false events caused by noise and inclusion of events that were not detected automatically. At least 100 individual mIPSC events were detected under each experimental condition. The peak amplitude, frequency, rise time, decay time (τ), area, and charge transfer were analyzed. Peak amplitude refers to the maximum height of the mIPSC, and the frequency was calculated by determining the inter-event interval. The rise time refers to the time elapsed between 10% and 90% of the peak amplitude response. The decay τ was determined using the biexponential equation I(t) = A1exp(–t/ τ1) + A2exp(–t/ τ2), where I is the current amplitude at any given time (t); A1 and A2 are the amplitudes of the fast and slow decay components, respectively; and τ1 and τ2 are their respective decay time constants. The weighted time constant of current decay was determined by the equation τdecay = ∑Aiτi/∑Ai. Area was determined by integrating the area under the mIPSC curve. Charge transfer was determined by multiplying the frequency by the area of the mIPSCs (51). Cumulative distribution plots of mIPSC amplitudes and frequency were generated in GraphPad Prism 5 (GraphPad Software Inc.).
The amplitude of tonic glycine current was measured as the difference in the holding current before and during application of strychnine (1 μM), while the amplitude of tonic GABA current was measured by application of bicuculline (100 μM). Drug efficacy was reported as percentage of control, where control was equal to the amplitude of the tonic current revealed by application of either strychnine or bicuculline. The variance of baseline noise, measured for tonic glycine current, was determined by the root mean square deviation from the recording segments that lacked mIPSCs, as previously described (51, 58).
SLEs and evoked field potentials were analyzed using GraphPad Prism 5. The frequency of SLEs was determined using pClamp 10 data acquisition and analysis software (Molecular Devices). The threshold for event detection was set at ~0.5 mV.
Drugs and chemicals.
TXA and EACA were obtained from Sigma-Aldrich, while aprotinin (Trasylol) was purchased from Bayer. Propofol (Diprivan) and midazolam were purchased from AstraZeneca, while isoflurane was obtained from Abbott Laboratories. Tetrodotoxin was purchased from Alomone Laboratories; CNQX, D-AP5, bicuculline, strychnine, and glycine were obtained from Sigma-Aldrich. Propofol and midazolam were each diluted in extracellular solution as previously described (103). Isoflurane solutions were prepared and applied as previously described (104). The highest concentration of isoflurane in the undiluted aqueous solution was 2,500 μM. The aqueous phase concentration of isoflurane equivalent to minimal alveolar concentrations in vitro studies at room temperature is 253–280 μM (105, 106). Stock solutions of all other reagents were prepared with distilled water.
Patients and surgical procedures.
Patients included in the study were ages 19–85, inclusive, and scheduled for open thoraco-abdominal aortic surgery, with planned placement of a lumbar CSF drain and intraoperative use of TXA. This is a complex procedure that is performed in a selected number of specialized surgical centers. Due to the known variability in duration of the procedure and operative techniques, and since the amount of CSF available for study is limited, samples were obtained at predetermined procedural milestones. TXA dosing was in accordance with recommended protocols (13), with a loading dose of 30 mg/kg plus additional 2 mg/kg added to the CPB prime followed by 16 mg/kg/h infusion until chest closure. TXA concentrations in serum and CSF were quantified using a modified liquid chromatography and a tandem mass spectrometry assay (88, 107). At a functional coefficient of variance of 20%, the minimum detectable level of TXA is 0.05 μg/ml.
Statistics.
GraphPad Prism 5 was used to generate the bar graphs. Results are presented as mean ± SEM. Differences between groups were determined using a 1-tailed Student t test, 1-way ANOVA with a Dunnett multiple comparison post hoc test, or 2-way ANOVA with Bonferroni post hoc test. Cumulative distribution plots of mIPSCs were analyzed with the Kolmogorov-Smirnov test. A P value of less than 0.05 was considered significant.
Study approval.
All animal experiments were approved by the Animal Care Committee of the University of Toronto. The human study protocol was approved by the Research Ethics Board of St. Michael’s Hospital, Toronto, Ontario, Canada. Written and informed consent was obtained from all patients.
Supplementary Material
Acknowledgments
This work was supported by operating grants from the Canadian Institute of Health Research (MOP-38028, MOP-79428) and a grant from the Canadian Anaesthesiologists’ Society to B.A. Orser. I. Lecker was supported by a Savoy Foundation studentship. B.A. Orser holds a Canada Research Chair. We are grateful to Laura A. Burns and Sanjay Yagnik for patient enrollment, data collection, and data analysis. We thank Ella Czerwinski for her assistance with the cell cultures. The authors would also like to thank Liang Zhang for his advice and unwavering support.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2012;122(12):4654–4666. doi:10.1172/JCI63375.
See the related Commentary beginning on page 4339.
References
- 1.Shakur H, et al. Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH-2): a randomised, placebo-controlled trial. Lancet. 2010;376(9734):23–32. doi: 10.1016/S0140-6736(10)60835-5. [DOI] [PubMed] [Google Scholar]
- 2.Zufferey PJ, et al. Tranexamic acid in hip fracture surgery: a randomized controlled trial. Br J Anaesth. 2010;104(1):23–30. doi: 10.1093/bja/aep314. [DOI] [PubMed] [Google Scholar]
- 3.Rannikko A, Petas A, Taari K. Tranexamic acid in control of primary hemorrhage during transurethral prostatectomy. Urology. 2004;64(5):955–958. doi: 10.1016/j.urology.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 4.Neilipovitz DT. Tranexamic acid for major spinal surgery. Eur Spine J. 2004;13(suppl 1):S62–S65. doi: 10.1007/s00586-004-0716-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rahman Z, Hoque R, Ali A, Rahman M, Rahman MS. Blood conservation strategies for reducing peri-operative blood loss in open heart surgery. Mymensingh Med J. 2011;20(1):45–53. [PubMed] [Google Scholar]
- 6.Ducloy-Bouthors AS, et al. High-dose tranexamic acid reduces blood loss in postpartum haemorrhage. Crit Care. 2011;15(2):R117. doi: 10.1186/cc10143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koch CG, et al. Morbidity and mortality risk associated with red blood cell and blood-component transfusion in isolated coronary artery bypass grafting. Crit Care Med. 2006;34(6):1608–1616. doi: 10.1097/01.CCM.0000217920.48559.D8. [DOI] [PubMed] [Google Scholar]
- 8.Blanchette CM, et al. Cost and utilization of blood transfusion associated with spinal surgeries in the United States. Eur Spine J. 2007;16(3):353–363. doi: 10.1007/s00586-006-0066-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martinez MC. Management of scarce resources in blood services in developing countries. Vox Sang. 2002;83(suppl 1):137–140. doi: 10.1111/j.1423-0410.2002.tb05287.x. [DOI] [PubMed] [Google Scholar]
- 10.Henry DA, et al. Anti-fibrinolytic use for minimising perioperative allogeneic blood transfusion. Cochrane Database Syst Rev. 2011;3(3):CD001886. doi: 10.1002/14651858.CD001886.pub4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iwamoto M. Plasminogen-plasmin system IX. Specific binding of tranexamic acid to plasmin. Thromb Diath Haemorrh. 1975;33(3):573–585. [PubMed] [Google Scholar]
- 12.McEvoy MD, Reeves ST, Reves JG, Spinale FG. Aprotinin in cardiac surgery: a review of conventional and novel mechanisms of action. Anesth Analg. 2007;105(4):949–962. doi: 10.1213/01.ane.0000281936.04102.9f. [DOI] [PubMed] [Google Scholar]
- 13.Fergusson DA, et al. A comparison of aprotinin and lysine analogues in high-risk cardiac surgery. N Engl J Med. 2008;358(22):2319–2331. doi: 10.1056/NEJMoa0802395. [DOI] [PubMed] [Google Scholar]
- 14.Mangano DT, Tudor IC, Dietzel C. The risk associated with aprotinin in cardiac surgery. N Engl J Med. 2006;354(4):353–365. doi: 10.1056/NEJMoa051379. [DOI] [PubMed] [Google Scholar]
- 15.Shaw AD, et al. The effect of aprotinin on outcome after coronary-artery bypass grafting. N Engl J Med. 2008;358(8):784–793. doi: 10.1056/NEJMoa0707768. [DOI] [PubMed] [Google Scholar]
- 16.Pellegrini A, Giaretta D, Chemello R, Zanotto L, Testa G. Feline generalized epilepsy induced by tranexamic acid (AMCA). Epilepsia. 1982;23(1):35–45. doi: 10.1111/j.1528-1157.1982.tb05051.x. [DOI] [PubMed] [Google Scholar]
- 17.Schlag MG, Hopf R, Redl H. Convulsive seizures following subdural application of fibrin sealant containing tranexamic acid in a rat model. Neurosurgery. 2000;47(6):1463–1467. doi: 10.1097/00006123-200012000-00048. [DOI] [PubMed] [Google Scholar]
- 18.Schlag MG, Hopf R, Zifko U, Redl H. Epileptic seizures following cortical application of fibrin sealants containing tranexamic acid in rats. Acta Neurochir (Wien). 2002;144(1):63–69. doi: 10.1007/s701-002-8275-z. [DOI] [PubMed] [Google Scholar]
- 19.de Leede-van der Maarl MG, Hilkens P, Bosch F. The epileptogenic effect of tranexamic acid. J Neurol. 1999;246(9):843. doi: 10.1007/s004150050466. [DOI] [PubMed] [Google Scholar]
- 20.Mohseni K, Jafari A, Nobahar MR, Arami A. Polymyoclonus seizure resulting from accidental injection of tranexamic acid in spinal anesthesia. Anesth Analg. 2009;108(6):1984–1986. doi: 10.1213/ane.0b013e3181a04d69. [DOI] [PubMed] [Google Scholar]
- 21.Yeh HM, Lau HP, Lin PL, Sun WZ, Mok MS. Convulsions and refractory ventricular fibrillation after intrathecal injection of a massive dose of tranexamic acid. Anesthesiology. 2003;98(1):270–272. doi: 10.1097/00000542-200301000-00042. [DOI] [PubMed] [Google Scholar]
- 22.Feffer SE, Parray HR, Westring DW. Seizure after infusion of aminocaproic acid. JAMA. 1978;240(22):2468. doi: 10.1001/jama.1978.03290220080026. [DOI] [PubMed] [Google Scholar]
- 23.Murkin JM, et al. High-dose tranexamic acid is associated with nonischemic clinical seizures in cardiac surgical patients. Anesth Analg. 2010;110(2):350–353. doi: 10.1213/ANE.0b013e3181c92b23. [DOI] [PubMed] [Google Scholar]
- 24.Keyl C, et al. High-dose tranexamic acid is related to increased risk of generalized seizures after aortic valve replacement. Eur J Cardiothorac Surg. 2011;39(5):e114–e121. doi: 10.1016/j.ejcts.2010.12.030. [DOI] [PubMed] [Google Scholar]
- 25.Goldstone AB, et al. Predictors and outcomes of seizures after cardiac surgery: a multivariable analysis of 2,578 patients. Ann Thorac Surg. 2011;91(2):514–518. doi: 10.1016/j.athoracsur.2010.10.090. [DOI] [PubMed] [Google Scholar]
- 26.Roach GW, et al. Adverse cerebral outcomes after coronary bypass surgery. Multicenter Study of Perioperative Ischemia Research Group and the Ischemia Research and Education Foundation Investigators. N Engl J Med. 1996;335(25):1857–1863. doi: 10.1056/NEJM199612193352501. [DOI] [PubMed] [Google Scholar]
- 27.Sander M, et al. Mortality associated with administration of high-dose tranexamic acid and aprotinin in primary open-heart procedures: a retrospective analysis. Crit Care. 2010;14(4):R148. doi: 10.1186/cc9216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martin K, et al. Seizures after open heart surgery: comparison of ε-aminocaproic acid and tranexamic acid. J Cardiothorac Vasc Anesth. 2011;25(1):20–25. doi: 10.1053/j.jvca.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 29.Manji RA, et al. Seizures following cardiac surgery: the impact of tranexamic acid and other risk factors. Can J Anaesth. 2012;59(1):6–13. doi: 10.1007/s12630-011-9618-z. [DOI] [PubMed] [Google Scholar]
- 30.Kalavrouziotis D, Voisine P, Mohammadi S, Dionne S, Dagenais F. High-dose tranexamic acid is an independent predictor of early seizure after cardiopulmonary bypass. Ann Thorac Surg. 2012;93(1):148–154. doi: 10.1016/j.athoracsur.2011.07.085. [DOI] [PubMed] [Google Scholar]
- 31.Hunter GR, Young GB. Seizures after cardiac surgery. J Cardiothorac Vasc Anesth. 2011;25(2):299–305. doi: 10.1053/j.jvca.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 32.Betz H, Laube B. Glycine receptors: recent insights into their structural organization and functional diversity. J Neurochem. 2006;97(6):1600–1610. doi: 10.1111/j.1471-4159.2006.03908.x. [DOI] [PubMed] [Google Scholar]
- 33.Colquhoun D, Sivilotti LG. Function and structure in glycine receptors and some of their relatives. Trends Neurosci. 2004;27(6):337–344. doi: 10.1016/j.tins.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 34.Lynch JW. Molecular structure and function of the glycine receptor chloride channel. Physiol Rev. 2004;84(4):1051–1095. doi: 10.1152/physrev.00042.2003. [DOI] [PubMed] [Google Scholar]
- 35.Muller E, Le-Corronc H, Legendre P, Muller E, Le-Corronc H, Legendre P. Extrasynaptic and postsynaptic receptors in glycinergic and GABAergic neurotransmission: a division of labor? Front Mol Neurosci. 2008;1:3. doi: 10.3389/neuro.02.003.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takazawa T, MacDermott AB. Glycinergic and GABAergic tonic inhibition fine tune inhibitory control in regionally distinct subpopulations of dorsal horn neurons. J Physiol. 2010;588(pt 14):2571–2587. doi: 10.1113/jphysiol.2010.188292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lynch JW. Native glycine receptor subtypes and their physiological roles. Neuropharmacology. 2009;56(1):303–309. doi: 10.1016/j.neuropharm.2008.07.034. [DOI] [PubMed] [Google Scholar]
- 38.Furtmuller R, et al. Tranexamic acid, a widely used antifibrinolytic agent, causes convulsions by a γ-aminobutyric acidA receptor antagonistic effect. . J Pharmacol Exp Ther. 2002;301(1):168–173. doi: 10.1124/jpet.301.1.168. [DOI] [PubMed] [Google Scholar]
- 39.Hales TG, Lambert JJ. The actions of propofol on inhibitory amino acid receptors of bovine adrenomedullary chromaffin cells and rodent central neurones. Br J Pharmacol. 1991;104(3):619–628. doi: 10.1111/j.1476-5381.1991.tb12479.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ahrens J, et al. A transmembrane residue influences the interaction of propofol with the strychnine-sensitive glycine α1 and α1β receptor. Anesth Analg. 2008;107(6):1875–1883. doi: 10.1213/ane.0b013e3181875a31. [DOI] [PubMed] [Google Scholar]
- 41.Downie DL, Hall AC, Lieb WR, Franks NP. Effects of inhalational general anaesthetics on native glycine receptors in rat medullary neurones and recombinant glycine receptors in Xenopus oocytes. Br J Pharmacol. 1996;118(3):493–502. doi: 10.1111/j.1476-5381.1996.tb15430.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fedorow CA, Moon MC, Mutch WA, Grocott HP. Lumbar cerebrospinal fluid drainage for thoracoabdominal aortic surgery: rationale and practical considerations for management. Anesth Analg. 2010;111(1):46–58. doi: 10.1213/ANE.0b013e3181ddddd6. [DOI] [PubMed] [Google Scholar]
- 43.Cina CS, et al. Cerebrospinal fluid drainage to prevent paraplegia during thoracic and thoracoabdominal aortic aneurysm surgery: a systematic review and meta-analysis. J Vasc Surg. 2004;40(1):36–44. doi: 10.1016/j.jvs.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 44.MacDonald JF, Miljkovic Z, Pennefather P. Use-dependent block of excitatory amino acid currents in cultured neurons by ketamine. J Neurophysiol. 1987;58(2):251–266. doi: 10.1152/jn.1987.58.2.251. [DOI] [PubMed] [Google Scholar]
- 45.Jin X, Covey DF, Steinbach JH. Kinetic analysis of voltage-dependent potentiation and block of the glycine α3 receptor by a neuroactive steroid analogue. J Physiol. 2009;587(pt 5):981–997. doi: 10.1113/jphysiol.2008.159343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Later AF, et al. Tranexamic acid and aprotinin in low- and intermediate-risk cardiac surgery: a non-sponsored, double-blind, randomised, placebo-controlled trial. Eur J Cardiothorac Surg. 2009;36(2):322–329. doi: 10.1016/j.ejcts.2008.11.038. [DOI] [PubMed] [Google Scholar]
- 47.Caraiscos VB, et al. Insulin increases the potency of glycine at ionotropic glycine receptors. Mol Pharmacol. 2007;71(5):1277–1287. doi: 10.1124/mol.106.033563. [DOI] [PubMed] [Google Scholar]
- 48.Whitehead KJ, Manning JP, Smith CG, Bowery NG. Determination of the extracellular concentration of glycine in the rat spinal cord dorsal horn by quantitative microdialysis. Brain Res. 2001;910(1–2):192–194. doi: 10.1016/S0006-8993(01)02684-1. [DOI] [PubMed] [Google Scholar]
- 49.Meier JC, et al. RNA editing produces glycine receptor α3(P185L), resulting in high agonist potency. Nat Neurosci. 2005;8(6):736–744. doi: 10.1038/nn1467. [DOI] [PubMed] [Google Scholar]
- 50.Ataka T, Gu JG. Relationship between tonic inhibitory currents and phasic inhibitory activity in the spinal cord lamina II region of adult mice. Mol Pain. 2006;2:36. doi: 10.1186/1744-8069-2-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bai D, et al. Distinct functional and pharmacological properties of tonic and quantal inhibitory postsynaptic currents mediated by γ-aminobutyric acidA receptors in hippocampal neurons. . Mol Pharmacol. 2001;59(4):814–824. doi: 10.1124/mol.59.4.814. [DOI] [PubMed] [Google Scholar]
- 52.Legendre P, et al. Desensitization of homomeric α1 glycine receptor increases with receptor density. Mol Pharmacol. 2002;62(4):817–827. doi: 10.1124/mol.62.4.817. [DOI] [PubMed] [Google Scholar]
- 53.Chang Y, Ghansah E, Chen Y, Ye J, Weiss DS. Desensitization mechanism of GABA receptors revealed by single oocyte binding and receptor function. J Neurosci. 2002;22(18):7982–7990. doi: 10.1523/JNEUROSCI.22-18-07982.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Takahashi A, Mashimo T, Uchida I. GABAergic tonic inhibition of substantia gelatinosa neurons in mouse spinal cord. Neuroreport. 2006;17(12):1331–1335. doi: 10.1097/01.wnr.0000230515.86090.bc. [DOI] [PubMed] [Google Scholar]
- 55.Vardya I, Drasbek KR, Dosa Z, Jensen K. Cell type-specific GABAA receptor-mediated tonic inhibition in mouse neocortex. . J Neurophysiol. 2008;100(1):526–532. doi: 10.1152/jn.01224.2007. [DOI] [PubMed] [Google Scholar]
- 56.Caraiscos VB, et al. Tonic inhibition in mouse hippocampal CA1 pyramidal neurons is mediated by α5 subunit-containing γ-aminobutyric acid type A receptors. Proc Natl Acad Sci U S A. 2004;101(10):3662–3667. doi: 10.1073/pnas.0307231101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yeung JY, et al. Tonically activated GABAA receptors in hippocampal neurons are high-affinity, low-conductance sensors for extracellular GABA. Mol Pharmacol. 2003;63(1):2–8. doi: 10.1124/mol.63.1.2. [DOI] [PubMed] [Google Scholar]
- 58.Bonin RP, Martin LJ, MacDonald JF, Orser BA. α5GABAA receptors regulate the intrinsic excitability of mouse hippocampal pyramidal neurons. . J Neurophysiol. 2007;98(4):2244–2254. doi: 10.1152/jn.00482.2007. [DOI] [PubMed] [Google Scholar]
- 59.Casati V, Romano A, Novelli E, D’Angelo A. Tranexamic acid for trauma. Lancet. 2010;376(9746):1049–1050; author reply 1050–1041. doi: 10.1016/S0140-6736(10)61477-8. [DOI] [PubMed] [Google Scholar]
- 60.Lobo IA, Harris RA. Sites of alcohol and volatile anesthetic action on glycine receptors. Int Rev Neurobiol. 2005;65:53–87. doi: 10.1016/S0074-7742(04)65003-3. [DOI] [PubMed] [Google Scholar]
- 61.Bleck TP. Intensive care unit management of patients with status epilepticus. Epilepsia. 2007;48(suppl 8):59–60. doi: 10.1111/j.1528-1167.2007.01352.x. [DOI] [PubMed] [Google Scholar]
- 62.Kohno T, et al. Actions of midazolam on GABAergic transmission in substantia gelatinosa neurons of adult rat spinal cord slices. Anesthesiology. 2000;92(2):507–515. doi: 10.1097/00000542-200002000-00034. [DOI] [PubMed] [Google Scholar]
- 63.Rehberg B, Duch DS. Suppression of central nervous system sodium channels by propofol. Anesthesiology. 1999;91(2):512–520. doi: 10.1097/00000542-199908000-00026. [DOI] [PubMed] [Google Scholar]
- 64.Kirchner A, Breustedt J, Rosche B, Heinemann UF, Schmieden V. Effects of taurine and glycine on epileptiform activity induced by removal of Mg2+ in combined rat entorhinal cortex-hippocampal slices. . Epilepsia. 2003;44(9):1145–1152. doi: 10.1046/j.1528-1157.2003.01603.x. [DOI] [PubMed] [Google Scholar]
- 65.Straub H, Kohling R, Speckmann EJ. Strychnine-induced epileptiform activity in hippocampal and neocortical slice preparations: suppression by the organic calcium antagonists verapamil and flunarizine. Brain Res. 1997;773(1–2):173–180. doi: 10.1016/S0006-8993(97)00933-5. [DOI] [PubMed] [Google Scholar]
- 66.Alger BE, Nicoll RA. Epileptiform burst afterhyperolarization: calcium-dependent potassium potential in hippocampal CA1 pyramidal cells. Science. 1980;210(4474):1122–1124. doi: 10.1126/science.7444438. [DOI] [PubMed] [Google Scholar]
- 67.Schwartzkroin PA, Prince DA. Changes in excitatory and inhibitory synaptic potentials leading to epileptogenic activity. Brain Res. 1980;183(1):61–76. doi: 10.1016/0006-8993(80)90119-5. [DOI] [PubMed] [Google Scholar]
- 68.Dingledine R, Gjerstad L. Reduced inhibition during epileptiform activity in the in vitro hippocampal slice. J Physiol. 1980;305:297–313. doi: 10.1113/jphysiol.1980.sp013364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mody I, Lambert JD, Heinemann U. Low extracellular magnesium induces epileptiform activity and spreading depression in rat hippocampal slices. J Neurophysiol. 1987;57(3):869–888. doi: 10.1152/jn.1987.57.3.869. [DOI] [PubMed] [Google Scholar]
- 70.Walther H, Lambert JD, Jones RS, Heinemann U, Hamon B. Epileptiform activity in combined slices of the hippocampus, subiculum and entorhinal cortex during perfusion with low magnesium medium. Neurosci Lett. 1986;69(2):156–161. doi: 10.1016/0304-3940(86)90595-1. [DOI] [PubMed] [Google Scholar]
- 71.McCormack PL. Tranexamic acid: a review of its use in the treatment of hyperfibrinolysis. Drugs. 2012;72(5):585–617. doi: 10.2165/11209070-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 72.Schwartzkroin PA, Prince DA. Penicillin-induced epileptiform activity in the hippocampal in vitro prepatation. Ann Neurol. 1977;1(5):463–469. doi: 10.1002/ana.410010510. [DOI] [PubMed] [Google Scholar]
- 73.David SV, Malaval N, Shamma SA. Decoupling action potential bias from cortical local field potentials. Comput Intell Neurosci. 2010;2010:393019. doi: 10.1155/2010/393019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Buzsaki G, Anastassiou CA, Koch C. The origin of extracellular fields and currents-EEG, ECoG, LFP and spikes. Nat Rev Neurosci. 2012;13(6):407–420. doi: 10.1038/nrn3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Becker K, et al. Low dose isoflurane exerts opposing effects on neuronal network excitability in neocortex and hippocampus. PLoS One. 2012;7(6):e39346. doi: 10.1371/journal.pone.0039346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ohmori H, Sato Y, Namiki A. The anticonvulsant action of propofol on epileptiform activity in rat hippocampal slices. Anesth Analg. 2004;99(4):1095–1101. doi: 10.1213/01.ANE.0000130356.22414.2B. [DOI] [PubMed] [Google Scholar]
- 77.Hemmings HC., Jr Sodium channels and the synaptic mechanisms of inhaled anaesthetics. Br J Anaesth. 2009;103(1):61–69. doi: 10.1093/bja/aep144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Parker AJ, Lee JB, Redman J, Jolliffe L. Strychnine poisoning: gone but not forgotten. Emerg Med J. 2011;28(1):84. doi: 10.1136/emj.2009.080879. [DOI] [PubMed] [Google Scholar]
- 79.Young AB, Snyder SH. Strychnine binding associated with glycine receptors of the central nervous system. Proc Natl Acad Sci U S A. 1973;70(10):2832–2836. doi: 10.1073/pnas.70.10.2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chung SK, et al. Pathophysiological mechanisms of dominant and recessive GLRA1 mutations in hyperekplexia. . J Neurosci. 2010;30(28):9612–9620. doi: 10.1523/JNEUROSCI.1763-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gundlach AL, et al. Deficit of spinal cord glycine/strychnine receptors in inherited myoclonus of Poll Hereford calves. Science. 1988;241(4874):1807–1810. doi: 10.1126/science.2845573. [DOI] [PubMed] [Google Scholar]
- 82.Graham BA, Schofield PR, Sah P, Margrie TW, Callister RJ. Distinct physiological mechanisms underlie altered glycinergic synaptic transmission in the murine mutants spastic, spasmodic, and oscillator. J Neurosci. 2006;26(18):4880–4890. doi: 10.1523/JNEUROSCI.3991-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mitchell SJ, Silver RA. Shunting inhibition modulates neuronal gain during synaptic excitation. Neuron. 2003;38(3):433–445. doi: 10.1016/S0896-6273(03)00200-9. [DOI] [PubMed] [Google Scholar]
- 84.Caraiscos VB, Mihic SJ, MacDonald JF, Orser BA. Tyrosine kinases enhance the function of glycine receptors in rat hippocampal neurons and human α1β glycine receptors. J Physiol. 2002;539(pt 2):495–502. doi: 10.1113/jphysiol.2001.013508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liang J, Cagetti E, Olsen RW, Spigelman I. Altered pharmacology of synaptic and extrasynaptic GABAA receptors on CA1 hippocampal neurons is consistent with subunit changes in a model of alcohol withdrawal and dependence. . J Pharmacol Exp Ther. 2004;310(3):1234–1245. doi: 10.1124/jpet.104.067983. [DOI] [PubMed] [Google Scholar]
- 86.Walker MC. GABAA receptor subunit specificity: a tonic for the excited brain. . J Physiol. 2008;586(4):921–922. doi: 10.1113/jphysiol.2007.150581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mathers DA. Activation and inactivation of the GABAA receptor: insights from comparison of native and recombinant subunit assemblies. . Can J Physiol Pharmacol. 1991;69(7):1057–1063. doi: 10.1139/y91-157. [DOI] [PubMed] [Google Scholar]
- 88.Abou-Diwan C, et al. Plasma and cerebral spinal fluid tranexamic acid quantitation in cardiopulmonary bypass patients. J Chromatogr B Analyt Technol Biomed Life Sci. 2011;879(7–8):553–556. doi: 10.1016/j.jchromb.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 89.Gillinov AM, et al. Cardiopulmonary bypass and the blood-brain barrier. An experimental study. J Thorac Cardiovasc Surg. 1992;104(4):1110–1115. [PubMed] [Google Scholar]
- 90.Grocott HP, Arrowsmith JE. Serum S100 protein as a marker of cerebral damage during cardiac surgery. Br J Anaesth. 2001;86(2):289–290. [PubMed] [Google Scholar]
- 91.Montes FR, et al. Risk factors associated with postoperative seizures in patients undergoing cardiac surgery who received tranexamic acid: a case-control study. Ann Card Anaesth. 2012;15(1):6–12. doi: 10.4103/0971-9784.91467. [DOI] [PubMed] [Google Scholar]
- 92.Lecker I, Orser BA, Mazer CD. “Seizing” the opportunity to understand antifibrinolytic drugs. Can J Anaesth. 2012;59(1):1–5. doi: 10.1007/s12630-011-9621-4. [DOI] [PubMed] [Google Scholar]
- 93.Sackey PV, Martling CR, Granath F, Radell PJ. Prolonged isoflurane sedation of intensive care unit patients with the Anesthetic Conserving Device. Crit Care Med. 2004;32(11):2241–2246. doi: 10.1097/01.ccm.0000145951.76082.77. [DOI] [PubMed] [Google Scholar]
- 94.Sivilotti L, Woolf CJ. The contribution of GABAA and glycine receptors to central sensitization: disinhibition and touch-evoked allodynia in the spinal cord. . J Neurophysiol. 1994;72(1):169–179. doi: 10.1152/jn.1994.72.1.169. [DOI] [PubMed] [Google Scholar]
- 95.Kassell NF, Torner JC, Adams HP., Jr Antifibrinolytic therapy in the acute period following aneurysmal subarachnoid hemorrhage. Preliminary observations from the Cooperative Aneurysm Study. J Neurosurg. 1984;61(2):225–230. doi: 10.3171/jns.1984.61.2.0225. [DOI] [PubMed] [Google Scholar]
- 96.Lukes AS, Freeman EW, Van Drie D, Baker J, Adomako TL. Safety of tranexamic acid in women with heavy menstrual bleeding: an open-label extension study. Women’s Health (Lond Engl). 2011;7(5):591–598. doi: 10.2217/whe.11.55. [DOI] [PubMed] [Google Scholar]
- 97.Gundlach AL. Disorder of the inhibitory glycine receptor: inherited myoclonus in Poll Hereford calves. FASEB J. 1990;4(10):2761–2766. doi: 10.1096/fasebj.4.10.2165010. [DOI] [PubMed] [Google Scholar]
- 98.Simon ES. Phenotypic heterogeneity and disease course in three murine strains with mutations in genes encoding for α1 and β glycine receptor subunits. Mov Disord. 1997;12(2):221–228. doi: 10.1002/mds.870120213. [DOI] [PubMed] [Google Scholar]
- 99.Hui AC, Wong TY, Chow KM, Szeto CC. Multifocal myoclonus secondary to tranexamic acid. J Neurol Neurosurg Psychiatry. 2003;74(4):547. doi: 10.1136/jnnp.74.4.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhang ZJ, et al. Transition to seizure: ictal discharge is preceded by exhausted presynaptic GABA release in the hippocampal CA3 region. J Neurosci. 2012;32(7):2499–2512. doi: 10.1523/JNEUROSCI.4247-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Trevelyan AJ, Sussillo D, Watson BO, Yuste R. Modular propagation of epileptiform activity: evidence for an inhibitory veto in neocortex. J Neurosci. 2006;26(48):12447–12455. doi: 10.1523/JNEUROSCI.2787-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wyllie DJ, Chen PE. Taking the time to study competitive antagonism. Br J Pharmacol. 2007;150(5):541–551. doi: 10.1038/sj.bjp.0706997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.McAdam LC, MacDonald JF, Orser BA. Isobolographic analysis of the interactions between midazolam and propofol at GABAA receptors in embryonic mouse neurons. . Anesthesiology. 1998;89(6):1444–1454. doi: 10.1097/00000542-199812000-00022. [DOI] [PubMed] [Google Scholar]
- 104.Joo DT, et al. Blockade of AMPA receptors and volatile anesthetics: reduced anesthetic requirements in GluR2 null mutant mice for loss of the righting reflex and antinociception but not minimum alveolar concentration. Anesthesiology. 2001;94(3):478–488. doi: 10.1097/00000542-200103000-00020. [DOI] [PubMed] [Google Scholar]
- 105.Franks NP, Lieb WR. Which molecular targets are most relevant to general anaesthesia? Toxicol Lett. 1998;100:1–8. doi: 10.1016/S0378-4274(98)00158-1. [DOI] [PubMed] [Google Scholar]
- 106.Caraiscos VB, et al. Selective enhancement of tonic GABAergic inhibition in murine hippocampal neurons by low concentrations of the volatile anesthetic isoflurane. J Neurosci. 2004;24(39):8454–8458. doi: 10.1523/JNEUROSCI.2063-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chang Q, Yin OQ, Chow MS. Liquid chromatography-tandem mass spectrometry method for the determination of tranexamic acid in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2004;805(2):275–280. doi: 10.1016/j.jchromb.2004.03.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.