

Abstract
Cardiac natriuretic peptides (NP) are major activators of human fat cell lipolysis and have recently been shown to control brown fat thermogenesis. Here, we investigated the physiological role of NP on the oxidative metabolism of human skeletal muscle. NP receptor type A (NPRA) gene expression was positively correlated to mRNA levels of PPARγ coactivator-1α (PGC1A) and several oxidative phosphorylation (OXPHOS) genes in human skeletal muscle. Further, the expression of NPRA, PGC1A, and OXPHOS genes was coordinately upregulated in response to aerobic exercise training in human skeletal muscle. In human myotubes, NP induced PGC-1α and mitochondrial OXPHOS gene expression in a cyclic GMP–dependent manner. NP treatment increased OXPHOS protein expression, fat oxidation, and maximal respiration independent of substantial changes in mitochondrial proliferation and mass. Treatment of myotubes with NP recapitulated the effect of exercise training on muscle fat oxidative capacity in vivo. Collectively, these data show that activation of NP signaling in human skeletal muscle enhances mitochondrial oxidative metabolism and fat oxidation. We propose that NP could contribute to exercise training–induced improvement in skeletal muscle fat oxidative capacity in humans.
Introduction
The cardiac hormones, atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP), play a major role in the regulation of fluid homeostasis and cardiac physiology (1). Natriuretic peptide–mediated (NP-mediated) biological responses are largely mediated through cyclic GMP (cGMP) produced by the guanylyl cyclase domain of NP receptor type A (NPRA) (2). Although classically considered as cardiovascular hormones, we have shown that NP display a potent lipolytic effect in human adipocytes (3). They promote a rapid and sustained rise of intracellular cGMP that activates a cGMP-dependent protein kinase, PRKG1, which then phosphorylates perilipin 1 and hormone-sensitive lipase, necessary steps to initiate lipolysis (4). The potent lipolytic effect of NP is restricted to primates. In contrast, murine adipocytes exhibit a predominance of the clearance receptor NP receptor type C (NPR-C) and a very low expression of the biologically active NPRA (5). Interestingly, the lipolytic effect of NP is fully rescued in adipocytes of NPR-C (also known as Npr3) knockout mice. Moreover, NP induce a “browning” of human white adipocytes (6). This finding may be physiologically relevant considering the presence of functional brown fat in humans (7). Together, these studies suggest that NP plays a potent metabolic role in human adipose tissue. Recent data suggest that mice overexpressing Nppb and Prkg1 are protected from high-fat diet–induced obesity and insulin resistance and show increased energy expenditure (8). This phenotype could be explained by significant changes in skeletal muscle fat oxidative capacity. The physiological relevance and molecular mechanisms of this finding have yet to be addressed in humans. In this study, we first show in vivo the relevance of the relationship between NPRA expression and oxidative metabolism in human skeletal muscle before and after 8 weeks of exercise training. We next establish that NP, in physiologically relevant concentrations, enhanced oxidative capacity in human skeletal muscle cells and unravel the molecular mechanisms.
Results and Discussion
Exercise training upregulates NPRA and oxidative phosphorylation genes in human skeletal muscle.
In human skeletal muscle, we observed a positive relationship between the expression of NPRA and several oxidative phosphorylation (OXPHOS) genes (Supplemental Table 1; supplemental material available online with this article; doi: 10.1172/JCI64526DS1). PPARγ coactivator-1α (PGC1A), a master regulator of OXPHOS genes (9), was also correlated with NPRA expression (r = 0.58, P = 0.01) (Supplemental Figure 1). We next investigated the potential link between NP signaling and skeletal muscle mitochondrial oxidative metabolism during an 8-week aerobic exercise training program in obese men. Exercise training improved maximal oxygen uptake (VO2max) (+10%) and resting metabolic rate (RMR) (+8.9%). A 6.6% higher RMR was observed after adjustment for fat-free mass (Table 1). The observed increase in RMR, independent of fat-free mass, reflects a state of metabolic inefficiency, in agreement with other longitudinal exercise training studies (10, 11). This physiological adaptation could partly involve mitochondrial uncoupling in skeletal muscle (12). Pathway analysis of skeletal muscle microarray data revealed pathways related to mitochondrial dysfunction and OXPHOS as top-ranking biological functions (Padjusted < 0.002) in response to exercise training. Exercise training increased skeletal muscle OXPHOS protein expression (+32%, overall effect P = 0.0018) (Figure 1, A and B) independently of substantial changes in mitochondrial DNA (mtDNA) content (Figure 1C). These data are consistent with previous findings and suggest that exercise training can improve mitochondrial function per se (13). A concomitant upregulation of NPRA and total PGC1A transcripts was shown in human skeletal muscle in response to 8 weeks of aerobic exercise training (Figure 1D). No significant change in resting plasma ANP and BNP concentrations was observed after exercise training (data not shown). These data suggest a physiological link between NP/NPRA signaling and mitochondrial oxidative capacity in human skeletal muscle.
Table 1.
Clinical and metabolic variables at baseline and after 8 weeks of exercise training
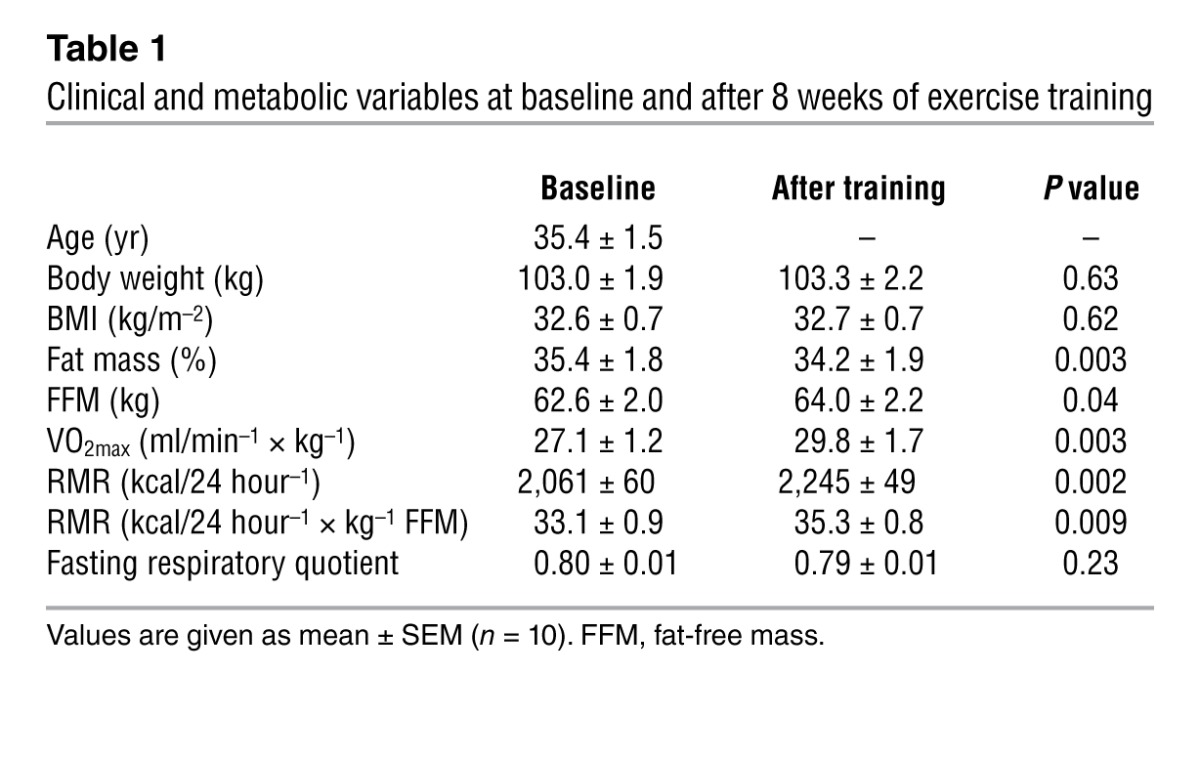
Figure 1. Functional link between NPRA expression and oxidative markers in human skeletal muscle.

(A) Representative blot of OXPHOS proteins in skeletal muscle of one subject at baseline (Bas) and after training (Post). (B) OXPHOS complex protein expression, (C) mtDNA content, and (D) NPRA and PGC1A gene expression in skeletal muscle of sedentary subjects at baseline and after 8 weeks of aerobic exercise training. (E and F) Gene expression of PRKG1 and PDE5A in primary human myoblasts during the time course of differentiation (n = 3–5). (G) Effect of acute ANP and BNP (1 μM) treatment, and (H) dose-response effect of ANP, on intracellular cGMP levels in differentiated human primary myotubes (n = 4–15). *P < 0.05, **P < 0.01 versus baseline.
NP/cGMP signaling activates PGC1A and OXPHOS expression in human myotubes.
The functional and mechanistic link between NP and mitochondrial oxidative metabolism was explored in human primary skeletal muscle cells. The NP receptor NPRA was expressed in differentiated myotubes, and the expression of key NP signaling components, PRKG1 and the cGMP-specific phosphodiesterase PDE5A, increased during differentiation into myotubes (Figure 1, E and F). ANP and BNP dose dependently increased intracellular cGMP concentrations in human myotubes (Figure 1, G and H). A physiologically relevant concentration of ANP (10 nM) induced PGC1A mRNA levels (2.5 fold, P < 0.01), while PPARD gene expression remained unaffected after 48 hours of treatment (Figure 2, A and B). Similar results were obtained with BNP (2.4 fold, P < 0.01), the stable cGMP analog 8-bromo-cGMP (2.1 fold, P < 0.01), and the selective PDE5 inhibitor sildenafil (2.7 fold, P < 0.01) (Figure 2, C and D). This finding is consistent with recent data showing a transcriptional regulation of PGC-1α by NP-mediated activation of p38 MAPK and activating transcription factor-2 (ATF2) in human adipocytes (6). The lack of transcriptional regulation of PPARD by the NP/cGMP signaling pathway is in agreement with previous findings using cAMP analogs in C2C12 myoblasts (14). Upregulation of PGC1A by NP was paralleled by a concomitant upregulation of several genes involved in OXPHOS complex I and IV, energy uncoupling, and transcriptional control of these pathways, such as nuclear respiratory factor 1 (NRF1) (Figure 2E). NRF1 controls the expression of nuclear genes encoding respiratory chain subunits and other proteins required for mitochondrial function (15). Similar data were obtained with BNP (Supplemental Figure 2) and sildenafil (Supplemental Figure 3). Chronic ANP treatment induced PGC-1α protein (+34%, P = 0.06) (Figure 3, A and B) and OXPHOS proteins from complex I (+24%, P < 0.01) and complex IV (+24%, P < 0.01) (Figure 3, A–C). These functional adaptations occurred without significant changes in the mitochondrial transcription factor A (TFAM) gene expression (Figure 2E), mtDNA content (Figure 3D), and mass (Figure 3E). No change in mtDNA occurred in response to 8-bromo-cGMP either (Supplemental Figure 4).
Figure 2. NP induces PGC-1α and mitochondrial oxidative genes in human primary myotubes.

Time course of (A) PGC1A and (B) PPARD gene expression in response to treatment with 10 nM ANP (n = 6). (C) PGC1A and (D) PPARD gene expression in response to 10 nM ANP/BNP, 1 μM sildenafil, and 1 mM 8-bromo-cGMP after 48-hour treatment (n = 6). (E) Changes in transcription factors and OXPHOS and energy uncoupling genes in response to 48-hour treatment with 10 nM ANP (n = 6). *P < 0.05, **P < 0.01, ***P < 0.001 versus control.
Figure 3. NP increases mitochondrial oxidative metabolism in human primary myotubes.

(A) Representative blots of PGC-1α and OXPHOS proteins and (B) quantitative bar graphs of PGC-1α protein and (C) OXPHOS proteins after 72-hour treatment with 100 and 10 nM ANP, respectively (n = 6). C, control. (D) mtDNA content, (E) mass, and (F) palmitate oxidation were measured after 72-hour treatment with 10 nM ANP (n = 4–6). (G) Oxygen consumption rate (OCR) after 72-hour treatment with 10 nM ANP or vehicle (control) at baseline and after injection of oligomycin (ATP synthase inhibitor), FCCP (uncoupling agent), and rotenone (complex I inhibitor) plus myxothiazol (complex III inhibitor) (n = 8 experiments; control columns, mean ± SEM of 75 wells; ANP columns, mean ± SEM of 72 wells). (H) Changes in fatty acid transport genes in response to 72-hour treatment with 10 nM ANP (n = 6). #P = 0.06, *P < 0.05, **P < 0.01, ***P < 0.001 versus control.
NP/cGMP signaling increases fat oxidation and maximal respiration in human myotubes.
Chronic ANP treatment enhanced palmitate oxidation (+27%, P < 0.05) (Figure 3F). Palmitate oxidation also increased in BNP-treated myotubes (+20%, P < 0.05) (Supplemental Figure 5). In addition, we show that 72 hours of ANP treatment of human myotubes increased basal (+15%), uncoupled (+10% and +18% for oligomycin and rotenone, respectively), and maximal (+38% for FCCP) respiration in human myotubes (2-way ANOVA overall treatment effect P = 0.007) (Figure 3G and Supplemental Figure 6). In contrast, acute ANP treatment had no effect on mitochondrial respiration (data not shown). ANP-mediated fat oxidation could be related to increased fatty acid transporter expression (Figure 3H), while mRNA levels of fat oxidation genes were not significantly affected (Supplemental Figure 7). ANP-induced energy uncoupling may be partly mediated through induction of uncoupling protein-3 (UCP3) and ATP/ADP translocase (ANT1) (Figure 2E). ANT1 controls mitochondrial proton conductance, and downregulation of ANT1 in human skeletal muscle has been linked to improved OXPHOS efficiency (higher P/O ratio) (16).
Together, our data point toward a major physiological role of NP in the regulation of skeletal muscle oxidative capacity in humans. Interestingly, NP enhances mitochondrial fat oxidative capacity and respiration in skeletal muscle in vitro similarly to exercise training in vivo. It is tempting to speculate that increased NP signaling in skeletal muscle may contribute, at least in part, to exercise training–induced improvement in fat oxidative capacity. Indeed, physical exercise is a strong physiological stimulus for cardiac ANP release. We have previously reported the physiological role of ANP in the control of lipolysis during exercise in humans (17). Interestingly, acute infusion of ANP in lean healthy men during ingestion of a high-fat meal substantially raised postprandial energy expenditure and lipid oxidation (18). Here, we demonstrate for the first time to our knowledge that chronic NP treatment increases oxidative capacity and energy uncoupling in human skeletal muscle. The molecular mechanism involves a transcriptional activation of PGC1A and subsequent induction of OXPHOS genes and mitochondrial respiration.
Our data indicate that enhanced NP signaling in skeletal muscle through exercise may trigger favorable metabolic adaptations to increase fat oxidation. This is in agreement with recent data showing a protective role of NP and PRKG1 against high-fat diet–induced obesity and glucose intolerance in mice (8). Transgenic mice overexpressing Nppb (also known as BNP) and Prkg1 display higher whole-body energy expenditure and fat oxidation, lower fat mass, and higher expression of mitochondrial oxidative genes in skeletal muscle. Interestingly, rectal temperature was significantly elevated in Prkg1 transgenic mice, suggesting an increased metabolic rate. Similarly, high-fat diet–fed mice chronically treated with sildenafil for 12 weeks have reduced fat mass, higher energy expenditure, and insulin sensitivity (19). Our findings may be relevant for conditions associated with reduced circulating NP concentrations, such as obesity (20). The reduced NP availability in obesity may result from upregulation of NPR-C clearance receptor in adipose tissue (21). Reduced circulating NP concentrations predispose to insulin resistance and type 2 diabetes, and a genetic polymorphism in the BNP gene promoter region affects circulating BNP levels as well as type 2 diabetes risk (22). It is tempting to speculate that obesity-associated NP deficiency could promote metabolic disease and that physical exercise could counteract this effect.
In conclusion, exercise interventions and pharmacological treatments enhancing circulating NP levels and/or NP signaling in skeletal muscle could improve skeletal muscle fat oxidative capacity and alleviate obesity-related metabolic disorders.
Methods
Clinical study.
Eighteen middle-aged healthy obese subjects were enrolled in the clinical trial (NCT01083329 and EudraCT-2009-012124-85) and ten participated in the training program (Table 1). The participants were asked to refrain from vigorous physical activity 48 hours before presenting to the clinical investigation center and ate a weight-maintaining diet consisting of 35% fat, 16% protein, and 49% carbohydrates 2 days before the experiment. Muscle biopsy of vastus lateralis, weighing 60–100 mg, was obtained using the Bergstrom technique, blotted, cleaned, and snap-frozen in liquid nitrogen (23). RMR and respiratory quotient were measured in lying position for 1 hour, while VO2max was measured on a bicycle ergometer, by indirect calorimetry after a 10-hour overnight fast. The subjects were investigated at baseline and after 8 weeks of aerobic exercise training, at least 48 to 72 hours after the last acute exercise bout. Further experimental details are provided in Supplemental Methods and Supplemental Table 2.
Skeletal muscle cell culture.
Satellite cells from rectus abdominis of healthy male subjects (age 34.3 ± 2.5 years, BMI 26.0 ± 1.4 kg/m2, fasting glucose 5.0 ± 0.2 mM) were cultured as previously described (24). Differentiation of myoblasts into myotubes was initiated at approximately 80%–90% confluence by switching to α-MEM with antibiotics, 2% FBS, and fetuin. The medium was changed every other day, up to 5 to 6 days.
Cellular respiration.
Human primary skeletal muscle cells were cultured on 24-well culture microplates (40,000 cells per well) (Seahorse Bioscience). Differentiated myotubes were treated for 72 hours with 1, 10, or 100 nM ANP or vehicle, and oxygen consumption was measured using the XF24-3 extracellular flux analyzer (Seahorse Bioscience). After 7 baseline measurements, cells were treated sequentially with 1 μM oligomycin to inhibit ATP synthase for 5 measurements, then with 0.25 μM FCCP to induce uncoupling of the cells for 5 measurements, and then with 1 μM rotenone plus 0.1 μM myxothiazol to inhibit complex I and III for 5 measurements.
Statistics.
Statistical analyses were performed using GraphPad Prism 5.0 for Windows (GraphPad Software Inc.). Normal distribution and homogeneity of variance of the data were tested using Shapiro-Wilk and F tests, respectively. One-way ANOVA followed by Tukey’s post-hoc tests and paired Student’s t tests were performed to determine differences between treatments and the effect of exercise training (Table 1). Two-way ANOVA were applied to determine the effect of training and ANP on gene expression, OXPHOS proteins, and cellular respiration. Linear regression was performed after log transformation of nonparametric data. For pathway analysis and correlations, adjusted P values were calculated using the Benjamini-Hochberg method. All values in figures and tables are presented as mean ± SEM. Statistical significance was set at P < 0.05.
Study approval.
The study was approved by the institutional review board of the Toulouse University Hospitals, and all volunteers gave written informed consent.
Supplementary Material
Acknowledgments
The authors are very grateful to François Crampes and Max Lafontan (I2MC, Toulouse) for helpful discussions, to the Toulouse Clinical Investigation Center staff, and to the study participants. The work was supported by grants from the National Research Agency ANR-09-JCJC-0019-01 (to C. Moro) and ANR-09-GENO-0018-01 (to D. Langin), European Federation for the Study of Diabetes/Novo Nordisk (to C. Moro), Inserm DHOS Recherche Translationnelle 2009 (to C. Thalamas and D. Langin), AOL-0816302 Hôpitaux de Toulouse (to C. Thalamas and D. Langin), Glaxo Smith Kline (to D. Langin), the Deutsche Forschungsgemeinschaft (Bl 1292/4-1), German Hypertension League (to A.L. Birkenfeld) and (JO 284/5-2) (to J. Jordan), and a collaborative research grant of the European Commission (SICA-HF, FP7 241558) (to S. Engeli and J. Jordan).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2012;122(12):4675–4679. doi:10.1172/JCI64526.
References
- 1.Gardner DG. Natriuretic peptides: markers or modulators of cardiac hypertrophy? Trends Endocrinol Metab. 2003;14(9):411–416. doi: 10.1016/s1043-2760(03)00113-9. [DOI] [PubMed] [Google Scholar]
- 2.Kuhn M. Structure, regulation, and function of mammalian membrane guanylyl cyclase receptors, with a focus on guanylyl cyclase-A. Circ Res. 2003;93(8):700–709. doi: 10.1161/01.RES.0000094745.28948.4D. [DOI] [PubMed] [Google Scholar]
- 3.Lafontan M, Moro C, Berlan M, Crampes F, Sengenes C, Galitzky J. Control of lipolysis by natriuretic peptides and cyclic GMP. Trends Endocrinol Metab. 2008;19(4):130–137. doi: 10.1016/j.tem.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 4.Sengenès C, et al. Involvement of a cGMP-dependent pathway in the natriuretic peptide-mediated hormone-sensitive lipase phosphorylation in human adipocytes. J Biol Chem. 2003;278(49):48617–48626. doi: 10.1074/jbc.M303713200. [DOI] [PubMed] [Google Scholar]
- 5.Sengenes C, et al. Natriuretic peptide-dependent lipolysis in fat cells is a primate specificity. Am J Physiol Regul Integr Comp Physiol. 2002;283(1):R257–R265. doi: 10.1152/ajpregu.00453.2001. [DOI] [PubMed] [Google Scholar]
- 6.Bordicchia M, et al. Cardiac natriuretic peptides act via p38 MAPK to induce the brown fat thermogenic program in mouse and human adipocytes. J Clin Invest. 2012;122(3):1022–1036. doi: 10.1172/JCI59701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Enerback S. Human brown adipose tissue. Cell Metab. 2010;11(4):248–252. doi: 10.1016/j.cmet.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 8.Miyashita K, et al. Natriuretic peptides/cGMP/cGMP-dependent protein kinase cascades promote muscle mitochondrial biogenesis and prevent obesity. Diabetes. 2009;58(12):2880–2892. doi: 10.2337/db09-0393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Handschin C, Spiegelman BM. PGC-1 coactivators and the regulation of skeletal muscle fiber-type determination. Cell Metab. 2011;13(4):351. doi: 10.1016/j.cmet.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 10.Potteiger JA, Kirk EP, Jacobsen DJ, Donnelly JE. Changes in resting metabolic rate and substrate oxidation after 16 months of exercise training in overweight adults. Int J Sport Nutr Exerc Metab. 2008;18(1):79–95. doi: 10.1123/ijsnem.18.1.79. [DOI] [PubMed] [Google Scholar]
- 11.Tremblay A, Fontaine E, Poehlman ET, Mitchell D, Perron L, Bouchard C. The effect of exercise-training on resting metabolic rate in lean and moderately obese individuals. Int J Obes. 1986;10(6):511–517. [PubMed] [Google Scholar]
- 12.Cline GW, Vidal-Puig AJ, Dufour S, Cadman KS, Lowell BB, Shulman GI. In vivo effects of uncoupling protein-3 gene disruption on mitochondrial energy metabolism. J Biol Chem. 2001;276(23):20240–20244. doi: 10.1074/jbc.M102540200. [DOI] [PubMed] [Google Scholar]
- 13.Menshikova EV, Ritov VB, Toledo FG, Ferrell RE, Goodpaster BH, Kelley DE. Effects of weight loss and physical activity on skeletal muscle mitochondrial function in obesity. Am J Physiol Endocrinol Metab. 2005;288(4):E818–E825. doi: 10.1152/ajpendo.00322.2004. [DOI] [PubMed] [Google Scholar]
- 14.Holst D, Luquet S, Nogueira V, Kristiansen K, Leverve X, Grimaldi PA. Nutritional regulation and role of peroxisome proliferator-activated receptor delta in fatty acid catabolism in skeletal muscle. Biochim Biophys Acta. 2003;1633(1):43–50. doi: 10.1016/s1388-1981(03)00071-4. [DOI] [PubMed] [Google Scholar]
- 15.Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest. 2006;116(3):615–622. doi: 10.1172/JCI27794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Larsen FJ, et al. Dietary inorganic nitrate improves mitochondrial efficiency in humans. Cell Metab. 2011;13(2):149–159. doi: 10.1016/j.cmet.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 17.Moro C, et al. Atrial natriuretic peptide contributes to physiological control of lipid mobilization in humans. FASEB J. 2004;18(7):908–910. doi: 10.1096/fj.03-1086fje. [DOI] [PubMed] [Google Scholar]
- 18.Birkenfeld AL, et al. Atrial natriuretic peptide induces postprandial lipid oxidation in humans. Diabetes. 2008;57(12):3199–3204. doi: 10.2337/db08-0649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ayala JE, Bracy DP, Julien BM, Rottman JN, Fueger PT, Wasserman DH. Chronic treatment with sildenafil improves energy balance and insulin action in high fat-fed conscious mice. Diabetes. 2007;56(4):1025–1033. doi: 10.2337/db06-0883. [DOI] [PubMed] [Google Scholar]
- 20.Wang TJ, et al. Impact of obesity on plasma natriuretic peptide levels. Circulation. 2004;109(5):594–600. doi: 10.1161/01.CIR.0000112582.16683.EA. [DOI] [PubMed] [Google Scholar]
- 21.Sarzani R, et al. Natriuretic peptide clearance receptor alleles and susceptibility to abdominal adiposity. Obes Res. 2004;12(2):351–356. doi: 10.1038/oby.2004.44. [DOI] [PubMed] [Google Scholar]
- 22.Magnusson M, et al. Low plasma level of atrial natriuretic peptide predicts development of diabetes: the prospective malmo diet and cancer study. J Clin Endocrinol Metab. 2011;97(2):638–645. doi: 10.1210/jc.2011-2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bergstrom J. Percutaneous needle biopsy of skeletal muscle in physiological and clinical research. Scand J Clin Lab Invest. 1975;35(7):609–616. doi: 10.3109/00365517509095787. [DOI] [PubMed] [Google Scholar]
- 24.Badin PM, et al. Altered skeletal muscle lipase expression and activity contribute to insulin resistance in humans. Diabetes. 2011;60(6):1734–1742. doi: 10.2337/db10-1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.