

Abstract
Congenital diarrheal disorders (CDDs) are a collection of rare, heterogeneous enteropathies with early onset and often severe outcomes. Here, we report a family of Ashkenazi Jewish descent, with 2 out of 3 children affected by CDD. Both affected children presented 3 days after birth with severe, intractable diarrhea. One child died from complications at age 17 months. The second child showed marked improvement, with resolution of most symptoms at 10 to 12 months of age. Using exome sequencing, we identified a rare splice site mutation in the DGAT1 gene and found that both affected children were homozygous carriers. Molecular analysis of the mutant allele indicated a total loss of function, with no detectable DGAT1 protein or activity produced. The precise cause of diarrhea is unknown, but we speculate that it relates to abnormal fat absorption and buildup of DGAT substrates in the intestinal mucosa. Our results identify DGAT1 loss-of-function mutations as a rare cause of CDDs. These findings prompt concern for DGAT1 inhibition in humans, which is being assessed for treating metabolic and other diseases.
Introduction
Congenital diarrheal disorders (CDDs) are rare and heterogeneous enteropathies, often with severe clinical manifestations (1, 2). Those whose cause has been identified typically result from autosomal recessive mutations. Affected genes include those related to disaccharidase deficiency, ion or nutrient transport defects, pancreatic insufficiency, or lipid trafficking (2). Some types of CDDs can be treated with dietary modification, but many present challenging clinical conditions, often requiring chronic nutritional support.
Here, we identified and characterized a rare DGAT1 mutation in a family with CDD. DGAT1 encodes 1 of 2 acyl CoA:diacylglycerol acyltransferases (DGATs), which catalyze the final step in triglyceride (TG) synthesis (3). DGAT1 is expressed ubiquitously, with highest expression in human intestine (4). Mice lacking DGAT1 have normal fat absorption, although absorption is delayed and more fat reaches distal intestinal regions (5). Because of the favorable metabolic phenotype of DGAT1-knockout mice (6), DGAT1 inhibitors have been developed (7–9) and proven efficacious in animal studies (8, 10). Several are being evaluated in clinical trials (11–13). However, mutations in human DGAT1 have not been reported, and information about human DGAT1 deficiency is limited.
Results and Discussion
Clinical summary.
The affected family is a nonconsanguineous couple of Ashkenazi Jewish descent with 3 children from full-term, uncomplicated pregnancies. The first child, a boy, was unaffected. The second child (case 1), a girl, weighed 3.18 kg at birth and was fed with breast milk and cow’s milk formula. Three days after birth, she developed vomiting, colicky pain, and nonbloody, watery diarrhea, 8–10 times daily. She was treated with oral rehydration solution, and her formula was changed to soy-based formula, but diarrhea continued. Cultures for bacterial pathogens, rotavirus, and adenovirus were negative. She exhibited protein-losing enteropathy, with stool α1 antitrypsin of 8 to 20 mg/g (normal, <3 mg/g stool) and hypoalbuminemia. She required total parenteral nutrition and intermittent infusions of albumin. Stomach, duodenum, and colon biopsies were negative for chronic granulomatous disease, autoimmune enteropathy, food protein-induced enterocolitis, microvillous inclusion disease, and tufting enteropathy (Supplemental Figure 1; supplemental material available online with this article; doi: 10.1172/JCI64873DS1). Neuroendocrine cells were present in intestinal biopsies. Congenital lymphangiectasia, a cause of protein-losing enteropathy, was excluded by CT scan and histology. There was evidence of dystrophic microvilli in the duodenum. Immunological tests were unremarkable except for slightly decreased IgG (275 mg/dl) with normal subclasses.
The child exhibited hyperlipidemia, with a fasting serum TG level of 325 mg/dl at 1 month of age (subsequently, 81–631 mg/dl; mean, 264 mg/dl [n = 55]) (Table 1). The father had elevated fasting TG (118–481 mg/dl) and total cholesterol levels (140–260 mg/dl), with a HDL cholesterol level of 33 to 39 mg/dl. The mother also had elevated fasting TG (144–229 mg/dl) and total cholesterol levels (174–220 mg/dl), with a HDL cholesterol level of 39 to 42 mg/dl.
Table 1.
Blood lipids
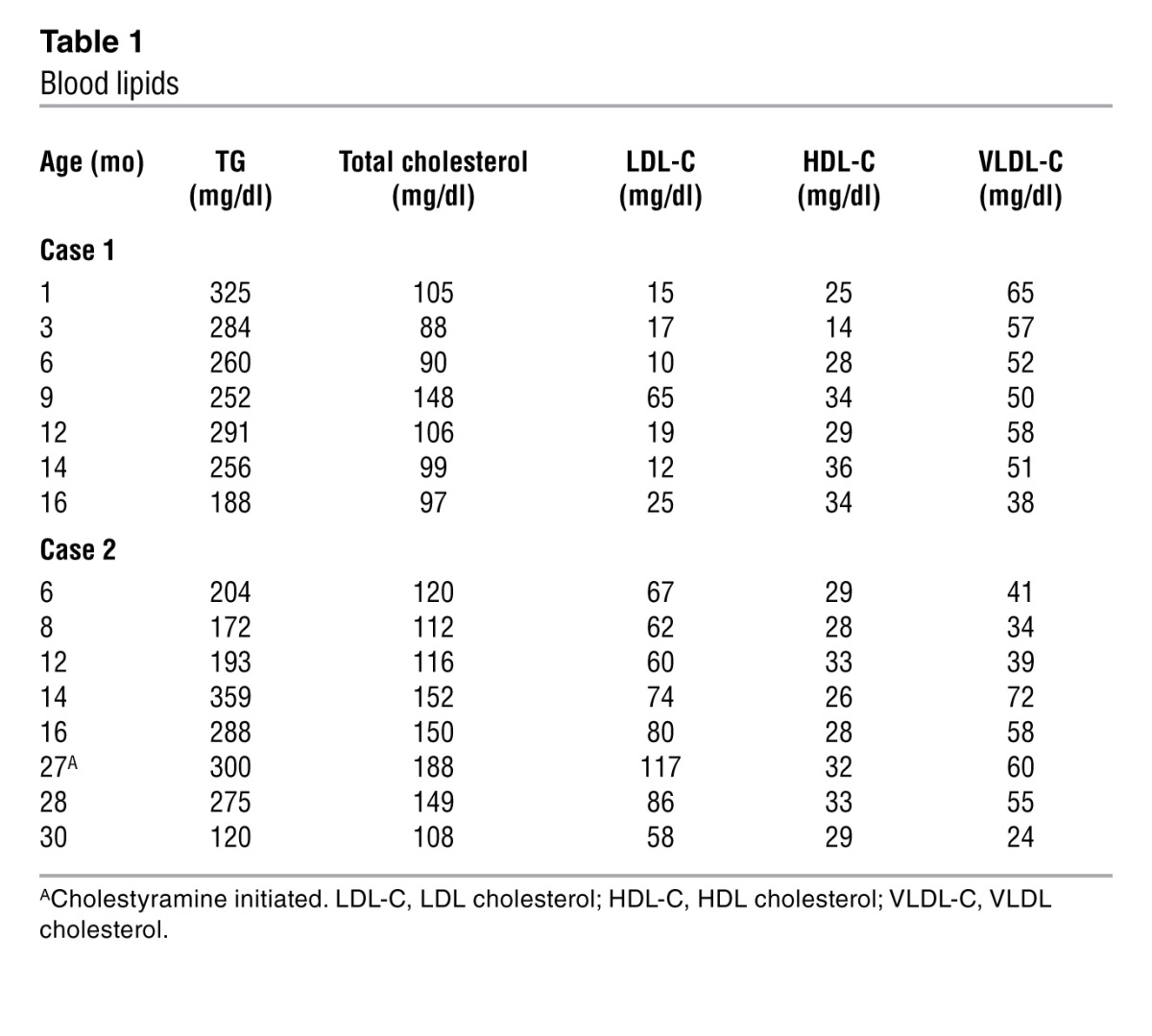
At 14 months of age, the child was below the first percentile for weight, despite parenteral nutrition and feeding per gastrostomy tube. She had recurrent episodes of sepsis, presumably related to a venous catheter. Eventually, she tolerated tube feedings (80 kcal/kg/d) with amino acid–based formula containing maltodextrin and medium chain TGs but did not gain weight and continued to lose protein in her stool. She died at 17 months of age from complications of malnutrition and sepsis.
The third child (case 2), a boy, weighed 3.7 kg at birth. He initially tolerated breast milk and soy formula but developed diarrhea 3 days after birth. His stools were nonbloody, 4–6 times daily, and watery. Six days after birth, he was admitted for dehydration, metabolic acidosis, and hyponatremia. Stool cultures were negative for bacterial pathogens, rotavirus, and adenovirus. Stool sodium and potassium levels were normal. Serum IgG was normal, but fecal α-1 antitrypsin was increased to 4.7 to 7.9 mg/g. He required intravenous albumin to correct the protein-losing enteropathy. Light and electron microscopy of duodenum at 2 months of age revealed findings similar to those for case 1 (Supplemental Figure 2, A and B). With nutritional support of amino acid–based formula and parenteral nutrition, he began to gain weight and by 10 months of age was no longer losing protein in his stool. A second duodenal biopsy, performed when his diarrhea improved (age 13 months), showed improved enterocyte morphology (Supplemental Figure 2, C and D) and increased eosinophils. Because of combined hyperlipidemia (elevated serum total cholesterol and TG levels; Table 1), he was treated with cholestyramine at 27 months of age, and fasting serum lipid levels decreased. He was thriving at 46 months of age on an unrestricted diet.
Mutation identification and characterization.
The identical phenotype and onset in 2 affected siblings suggested a recessive mutation (Figure 1A). To identify candidates, exome sequencing was performed on DNA from both parents and case 2 (Supplemental Methods). Using published sequence databases (14, 15), we searched for rare alleles (frequency, <1%) that were predicted to be loss of function (nonsense, splice, frameshift) or were nonsynonymous and predicted to be deleterious by PolyPhen2 (16). A single candidate gene (DGAT1) was identified. The child was homozygous (and both parents heterozygous) for a splice variant (chromosome 8, 145541756 A → G) in the splice donor site 3′ of exon 8, altering the invariant GT to GC. We identified 3 individuals in approximately 12,500 control exomes as carriers for this mutation and estimate probability of homozygosity of 1 in approximately 50 to 100 million births, revealing a novel and severe recessive disorder.
Figure 1. Mutation in DGAT1 segregates with CDD.

(A) Pedigree of the affected family, indicating diarrheal phenotype and DGAT1 genotype. T, wild type; C, mutant. (B) RFLP assay of genomic DNA confirming mutation presence. PCR product digested with Fnu4HI yields 165 and 32 bp for reference and 122, 43, and 32 bp for mutant allele. Numbers refer to the family members indicated in A. (C) Predicted splicing patterns for wild-type and mutant alleles. (D) RT-PCR analysis of mRNA from blood of the proband, unaffected sibling, and parents. Numbers refer to exons. The asterisk indicates the mutated nucleotide (“T” for wild type and “C” for the mutant allele). Mutant DGAT1 allele yields Δ8 mRNA. Numbers refer to the family members indicated in A. (E) DGAT1 protein diagram showing predicted transmembrane domains (gray), putative catalytic residues (*), and Δ8 region (Δ, diagonal lines).
Using PCR and RFLP, we confirmed both affected children as homozygous carriers of the mutation. The maternal grandmother, paternal grandfather, both parents, and the unaffected child were heterozygous (Figure 1B). The known founder effect likely explains why two unrelated families from the Ashkenazi Jewish subpopulation carry such a rare mutation (17).
The predicted result of the mutation is skipping of exon 8 (18), yielding an in-frame deletion of 75 bp (Figure 1C). Using RT-PCR with primers flanking exon 8 (Figure 1D), we confirmed cDNA products corresponding to the wild-type and exon 8–deleted (Δ8) allele in the parents and unaffected child, with only the mutant in the affected child (Figure 1D). No other gene-specific products were detected. Exon 8 deletion removes 25 amino acids from the highly conserved MBOAT domain of DGAT1 but does not affect putative active site residues (ref. 19 and Figure 1E).
To determine the mutation’s effects on DGAT1 activity, we expressed full-length and Δ8 cDNAs (FLAG-tagged and untagged) in mouse embryonic fibroblasts (MEFs) lacking Dgat1 (20) (DGAT1-KO MEFs). Although both mRNA species were present, neither DGAT1 protein nor in vitro DGAT1 activity was detected in cells expressing Δ8 cDNA (Figure 2A). We also expressed the constructs in MEFs lacking both DGAT1 and DGAT2 (DGAT1,2-KO MEFs) (20). DGAT1,2-KO MEFs expressing wild-type DGAT1 and incubated with [14C]-oleate accumulated TGs, but cells expressing Δ8 did not (Figure 2B). These results indicate that DGAT1 protein and activity are lost with the Δ8 mutation.
Figure 2. Δ8 mutant allele yields an unstable protein, resulting in loss of DGAT1 activity.

DGAT1 cDNAs were expressed in DGAT1-KO or DGAT1,2-KO MEFs. (A) DGAT1 activity (accumulation of [14C]-TG) was absent in lysates from Δ8-expressing cells, despite the presence of mRNA. All lysates synthesized [14C]-diacylglycerol ([14C]-DAG) (internal control). (B) Intact Δ8-expressing cells do not accumulate TG, as measured by thin-layer chromatography in DGAT1,2-KO cell lines after incubation with oleic acid (200 mM). Free cholesterol (FC) was used as a loading control. (C) Immunoblot showing that proteasome inhibition (MG132) rescues Δ8 protein expression in DGAT1,2-KO MEFs. (D) MG132-treated DGAT1,2-KO MEFs expressing Δ8 do not accumulate [14C]-TG after a 2-hour treatment with 200 mM [14C]-oleic acid. (E) qPCR showing that humans lack DGAT2 expression in the small intestine. Act., activity; Duo., duodenum; Jeju., jejunum; Ile., ileum.
To determine whether Δ8 protein is rapidly degraded, we used MG132 to inhibit proteasomal degradation and, indeed, found that Δ8 protein was detected after 4 hours (Figure 2C). Nevertheless, DGAT1,2-KO cells treated with MG132 accumulated TG only when expressing wild-type DGAT1 but not Δ8 (Figure 2D). Thus, the Δ8 allele yields an unstable protein that, even when present, is likely inactive.
Our findings show that homozygous DGAT1 loss of function is associated with CDD. The reported mutation results in deletion of exon 8 and a null allele. The frequency of randomly occurring homozygous or compound heterozygous mutations in the exome database indicate it is highly likely (P < 0.01) that the Δ8 mutation causes CDD. DGAT1 function in the intestine and potential relevance to the phenotypes supports this conclusion.
CDD can be caused by mutations in APOB, MTP, and SAR1B (21), which function in chylomicron assembly and export. These mutations yield defective intestinal fat absorption and consequent steatorrhea. These defects differ from those in this report in that they occur distal to TG synthesis, whereas, with DGAT1 deficiency, the defect is in TG synthesis itself.
The phenotype of DGAT1 deficiency in humans differs from that of DGAT1-KO mice, which exhibit reduced body fat, increased energy expenditure, improved glucose tolerance, resistance to diet-induced obesity, and extended longevity (6, 22). The mice also have delayed fat absorption and decreased postprandial excursions of plasma TG but not excess fecal fat or diarrhea (5). These findings suggest species-specific differences for DGAT1 deficiency. In mammals, DGAT1 and DGAT2 account for nearly all TG synthesis (20). Using quantitative PCR, we found that both Dgat1 and Dgat2 are expressed in murine intestine, but only DGAT1 is expressed highly in human intestine (Figure 2E), as we reported (6, 23). Thus, human intestine may be more sensitive to DGAT1 inhibition, owing to lack of DGAT2 expression.
How human DGAT1 deficiency causes diarrhea is unclear, but buildup of DGAT1 lipid substrates in the intestinal mucosa or lumen likely contributes. Excess diacylglycerols or fatty acids could become toxic by acting as bioactive signaling lipids or by detergent-like behavior of fatty acid moieties. Toxicity to enterocytes could result in protein-losing enteropathy. Alternatively, bile acid malabsorption can cause diarrhea, and DGAT1 deficiency could affect bile acid metabolism. Unfortunately, no information on fecal bile acid levels in the affected individuals was available.
The reasons for the different clinical outcomes of the affected children remains unclear. Case 2 suggests that there may be aging-associated increases in intestinal DGAT2 expression or other adaptation to DGAT1 deficiency. Without complications from sepsis, the case 1 individual might also have improved. The two children were different sexes, but relative levels of DGAT2 expression in male and female children are unknown. Treatment with a bile acid–binding resin might have helped in case 2. However, timing of this treatment did not correspond with clinical improvement.
Moderate hyperlipidemia, including hypertriglyceridemia, occurred in the affected children and parents. Whether this is causally linked to the DGAT1 mutation or an independent trait is unclear. Mice lacking DGAT1 have normal fasting levels of serum TGs (6) and reduced TGs after fat intake (5), thus hypertriglyceridemia linked to DGAT1 deficiency would be specific to humans. If hypertriglyceridemia is associated, overcompensation from hepatic DGAT2 may contribute via lipoprotein-mediated secretion of TGs. Alternatively, interruption of bile acid absorption in the distal small intestine is associated with VLDL overproduction and hypertriglyceridemia (24). The identification of more individuals with homozygous DGAT1 deficiency could shed light on this issue.
Notably, recent studies of DGAT1 inhibitors in humans reported dose-related adverse effects of mild-to-moderate diarrhea in some patients (25). Our findings suggest this effect results from DGAT1 inhibition, rather than being off target. A better understanding of DGAT1’s role in the intestine and how DGAT1 deficiency causes diarrhea will be useful for developing or improving therapies with DGAT1 inhibitors.
Methods
Experimental procedures are described in detail in Supplemental Methods.
Genetic screening and verification.
Exome capture was performed with the Agilent Whole Exome SureSelect v2 Kit. Single-nucleotide-variant search parameters are described in the text (see Mutation identification and characterization). For RFLP analysis, PCR products were digested with Fnu4-HI. Bands are 165, 32, and 3 bp (wild type) and 122, 43, 32, and 3 bp (mutant).
Molecular characterization.
MEFs were isolated and immortalized (26). DGAT activity was assayed (4) with conditions specific for DGAT1 (50 mM MgCl2). Total RNA was isolated with TRIzol (Invitrogen) and reverse-transcribed with the iScript cDNA Synthesis Kit (Bio-Rad). Antibodies were FLAG antibody M2 (1:2,000, Sigma-Aldrich), DGAT1 antibody NB110-41487 (1:1,000, Novus), and HSP90 (1:2,000, BD Biosciences). HRP secondary antibodies (1:5,000, Amersham) were used with ECL (Pierce).
Statistics.
The probability of Δ8 mutation causing CDD was empirically determined from the frequency of any homozygous or compound heterozygous, deleterious mutations occurring in the reference exome sets (14, 15).
Study approval.
Written informed consent was obtained from all adult participants and from the parents of the children. These genetic studies were approved by the Partners Institutional Review Board, Boston, Massachusetts, USA.
Supplementary Material
Acknowledgments
We thank Sekar Kathiresan for advice, Daryl Jones for manuscript preparation, Delphine Eberlé for helpful comments, Gary Howard and Anna Lisa Lucido for editorial assistance, and Chaim Jalas (Center for Rare Jewish Genetic Disorders) for assistance in sample acquisition. This work was supported by NIH grant 2R56DK56084 (to R.V. Farese, Jr.), animal facilities grant NIH/NCRR CO6 RR018928, the Gladstone Institutes, the National Science Foundation (to J.T. Haas), the Pediatric IBD Foundation (to H.S. Winter), and funding from Martin Schlaff (to H.S. Winter).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2012;122(12):4680–4684. doi:10.1172/JCI64873.
References
- 1.Terrin G, et al. Congenital diarrheal disorders: an updated diagnostic approach. Int J Mol Sci. 2012;13(4):4168–4185. doi: 10.3390/ijms13044168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berni Canani R, Terrin G, Cardillo G, Tomaiuolo R, Castaldo G. Congenital diarrheal disorders: improved understanding of gene defects is leading to advances in intestinal physiology and clinical management. J Pediatr Gastroenterol Nutr. 2010;50(4):360–366. doi: 10.1097/MPG.0b013e3181d135ef. [DOI] [PubMed] [Google Scholar]
- 3.Yen CL, Stone SJ, Koliwad S, Harris C, Farese RV. Thematic review series: glycerolipids. DGAT enzymes and triacylglycerol biosynthesis. J Lipid Res. 2008;49(11):2283–2301. doi: 10.1194/jlr.R800018-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cases S, et al. Identification of a gene encoding an acyl CoA:diacylglycerol acyltransferase, a key enzyme in triacylglycerol synthesis. Proc Natl Acad Sci U S A. 1998;95(22):13018–13023. doi: 10.1073/pnas.95.22.13018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buhman KK, et al. DGAT1 is not essential for intestinal triacylglycerol absorption or chylomicron synthesis. J Biol Chem. 2002;277(28):25474–25479. doi: 10.1074/jbc.M202013200. [DOI] [PubMed] [Google Scholar]
- 6.Smith SJ, et al. Obesity resistance and multiple mechanisms of triglyceride synthesis in mice lacking Dgat. Nat Genet. 2000;25(1):87–90. doi: 10.1038/75651. [DOI] [PubMed] [Google Scholar]
- 7.Zhao G, et al. Validation of diacyl glycerolacyltransferase I as a novel target for the treatment of obesity and dyslipidemia using a potent and selective small molecule inhibitor. J Med Chem. 2008;51(3):380–383. doi: 10.1021/jm7013887. [DOI] [PubMed] [Google Scholar]
- 8.Cao J, et al. Targeting Acyl-CoA:diacylglycerol acyltransferase 1 (DGAT1) with small molecule inhibitors for the treatment of metabolic diseases. J Biol Chem. 2011;286(48):41838–41851. doi: 10.1074/jbc.M111.245456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakada Y, et al. Novel acyl coenzyme A (CoA): diacylglycerol acyltransferase-1 inhibitors: synthesis and biological activities of diacylethylenediamine derivatives. Bioorg Med Chem. 2010;18(7):2785–2795. doi: 10.1016/j.bmc.2010.01.067. [DOI] [PubMed] [Google Scholar]
- 10.McLaren DG, et al. The use of stable-isotopically labeled oleic acid to interrogate lipid assembly in vivo: assessing pharmacological effects in preclinical species. J Lipid Res. 2011;52(6):1150–1161. doi: 10.1194/jlr.M011049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Birch AM, Buckett LK, Turnbull AV. DGAT1 inhibitors as anti-obesity and anti-diabetic agents. Curr Opin Drug Discov Devel. 2010;13(4):489–496. [PubMed] [Google Scholar]
- 12.Yin W, et al. Plasma lipid profiling across species for the identification of optimal animal models of human dyslipidemia. J Lipid Res. 2012;53(1):51–65. doi: 10.1194/jlr.M019927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cao G, Konrad RJ, Li SD, Hammond C. Glycerolipid acyltransferases in triglyceride metabolism and energy homeostasis-potential as drug targets. Endocr Metab Immune Disord Drug Targets. 2012;12(2):197–206. doi: 10.2174/187153012800493459. [DOI] [PubMed] [Google Scholar]
- 14. National Institute of Environmental Health Sciences. Environmental Genome Project Exome Variant Server. NIEHS Web site. http://evs.gs.washington.edu/niehsExome/ . Updated April 22, 2012. Accessed September 28, 2012.
- 15.Project G. A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adzhubei IA, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Slatkin M. A population-genetic test of founder effects and implications for Ashkenazi Jewish diseases. Am J Hum Genet. 2004;75(2):282–293. doi: 10.1086/423146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krawczak M, et al. Single base-pair substitutions in exon-intron junctions of human genes: nature, distribution, and consequences for mRNA splicing. Hum Mutat. 2007;28(2):150–158. doi: 10.1002/humu.20400. [DOI] [PubMed] [Google Scholar]
- 19.Hofmann K. A superfamily of membrane-bound O-acyltransferases with implications for wnt signaling. Trends Biochem Sci. 2000;25(3):111–112. doi: 10.1016/S0968-0004(99)01539-X. [DOI] [PubMed] [Google Scholar]
- 20.Harris CA, et al. DGAT enzymes are required for triacylglycerol synthesis and lipid droplets in adipocytes. J Lipid Res. 2011;52(4):657–667. doi: 10.1194/jlr.M013003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zamel R, Khan R, Pollex RL, Hegele RA. Abetalipoproteinemia: two case reports and literature review. Orphanet J Rare Dis. 2008;3:19. doi: 10.1186/1750-1172-3-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Streeper RS, et al. Deficiency of the lipid synthesis enzyme, DGAT1, extends longevity in mice. Aging (Albany NY). 2012;4(1):13–27. doi: 10.18632/aging.100424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cases S, et al. Cloning of DGAT2, a second mammalian diacylglycerol acyltransferase, and related family members. J Biol Chem. 2001;276(42):38870–38876. doi: 10.1074/jbc.M106219200. [DOI] [PubMed] [Google Scholar]
- 24.Beil U, Crouse JR, Einarsson K, Grundy SM. Effects of interruption of the enterohepatic circulation of bile acids on the transport of very low density-lipoprotein triglycerides. Metabolism. 1982;31(5):438–444. doi: 10.1016/0026-0495(82)90231-1. [DOI] [PubMed] [Google Scholar]
- 25. Novartis. DGAT1 inhibitor clinical trial results. Clinical Trial Results Database. Novartis Web site. http://www.novctrd.com/ctrdWebApp/clinicaltrialrepository/displayFile.do?trialResult=4313 . Updated June 21, 2010. Accessed September 28, 2012.
- 26.Willnow TE, Herz J. Genetic deficiency in low density lipoprotein receptor-related protein confers cellular resistance to Pseudomonas exotoxin A. Evidence that this protein is required for uptake and degradation of multiple ligands. J Cell Sci. 1994;107(pt 3):719–726. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.