

Abstract
Ulcerative colitis (UC) is a chronic disease associated with long periods of quiescent disease followed by fulminant exacerbation. Imminent relapse in UC is associated with high mucosal expression of suppressor of cytokine signaling 3 (SOCS3); hence, knowledge of the mechanisms driving mucosal SOCS3 expression may provide important clues as to rational therapy. Thus, here we aim to characterize the molecular forces driving SOCS3 expression in the mucosal compartment, focusing on druggable pathways. The colon epithelial cell line Caco-2 was stimulated with interferon (IFN)-γ, interleukin (IL)-4 or prostaglandin E2 (PGE2) to allow correlations between SOCS3 expression with signal transducer and activator of transcription 1 (STAT1), STAT6 and adenosine 3′,5′-cyclic monophosphate (cAMP) signaling, respectively. The physiological relevance of the findings obtained was assessed by immunohistochemical staining for the activated forms of STAT1, STAT6, protein kinase A (PKA)-Cγ and cAMP response element-binding protein (CREB) in biopsies from inactive UC patients and controls. Stimulation with IFN-γ, IL-4 or PGE2 induced activation of STAT1, STAT6 and cAMP, respectively, in colonic cells, without any signs of concomitant STAT3 activation. Forced activation of all these signaling pathways was sufficient for SOCS3 expression. Biopsies from patients with inactive UC showed significant increase of phosphorylated STAT1 (p-STAT1) (p < 0.0001), p-STAT6 (p = 0.0001), p-PKA-Cγ (p = 0.0003) and p-CREB (p = 0.0025) expression compared with controls. STAT3-independent SOCS3 induction in inactive UC involves multiple proinflammatory signaling pathways and contradicts the usefulness of pathway-specific antiinflammatory drugs for preventing relapse. Our findings suggest that broad-spectrum antiinflammatory drugs are essential to counteract increases in SOCS3 expression and exacerbation of disease. Our results highlight the multifactorial nature of the factors that cause exacerbation in UC.
INTRODUCTION
Ulcerative colitis (UC) is a chronic inflammatory bowel disease, characterized by alternating periods of remission and relapse. Although the majority of studies focus on the pathways that drive active inflammation, it can be argued that the development of strategies focusing on remission maintenance is the larger clinical challenge in the management of this disease (1). Nevertheless, the insights into the molecular mechanisms that cause the colonic mucosa to relapse after prolonged periods of quiescent disease remain largely unknown, prompting further investigation.
We previously reported that an increase of mucosal suppressor of cytokine signaling 3 (SOCS3) protein levels in colonic epithelium in biopsies otherwise devoid of signs of a mounting inflammatory reaction is a highly reliable predictor of upcoming relapse (2). This result suggests a causative role of increased SOCS3 expression for disease exacerbation. The main biochemical target for SOCS3 appears to be signal transducer and activator of transcription 3 (STAT3), a pivotal immunoregulatory molecule in intestinal epithelial cells (IECs) involved in controlling proliferation during homeostasis as well as tissue repair (3–6). Characterization of the molecular mechanisms that drive SOCS3 expression in the mucosal epithelium of UC patients is of utmost importance for both our fundamental understanding of processes driving UC relapse and for devising rational strategies for its prevention.
Analysis of the molecular structure of the genetic elements in the SOCS3 promoter that drive transcription of the gene can already provide important clues as to mechanisms potentially involved here. The SOCS3 promoter contains two putative STAT-response elements (SREs): a GC-rich region and an APETALA1/adenosine 3′,5′-cyclic monophosphate (cAMP)-response element (AP1/CRE) region (Figure 1) (7,8). It is known that in response to interferon (IFN)-γ, a Th1 cytokine expressed during UC remission (9), Janus kinases associated with the IFN-γ receptor complex phosphorylate the latent transcription factor STAT1 (phosphorylated STAT1 [p-STAT1]) at plasma membrane and then translocate to the nucleus on homodimerization. In the nucleus, p-STAT1 transactivates a variety of gene products, including SOCS3 expression, through binding to the SREs such as those present in the SOCS3 promoter (10,11). The importance of STAT1 activation for epithelial SOCS3 expression, both in colonic epithelium in general and during UC in particular has, however, not been addressed. The SREs in general are also capable of binding p-STAT6 homodimers, which are generated as a cellular response to Th2 cytokines such as IL-4 or IL-13 (12). Because UC is regarded as a Th2-mediated disease, STAT6 activation would be especially responsible for SOCS3 induction, but again this has not been investigated. In addition, p-STAT5 homodimers should be able bind and transactivate SREs, but the methylation of STAT5 observed in UC patients suggests that it is an unlikely activator of SOCS3 expression (13). In total, however, the existing body of biomedical literature suggests different STATs are potential mediators for driving SOCS3 expression in UC, provoking relapse, and their function needs to be addressed.
Figure 1.
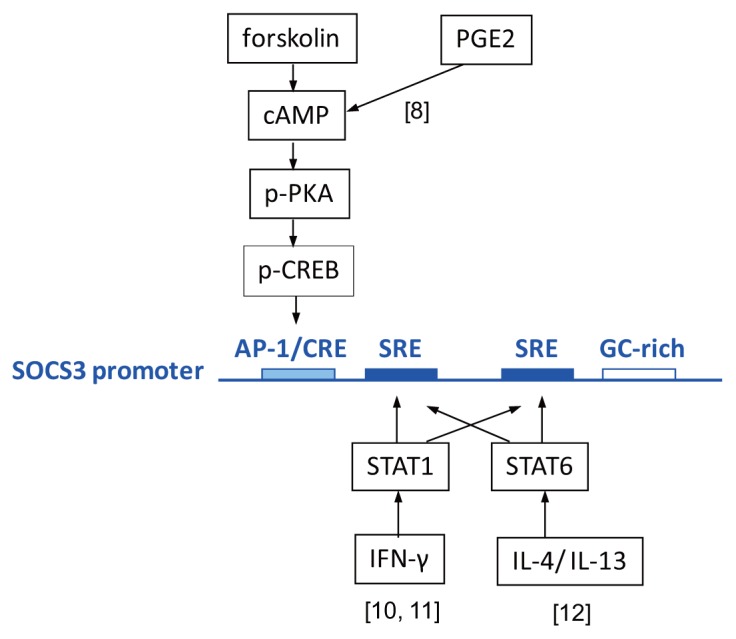
The signaling pathways reported to induce SOCS3 expression (3,4,6,8) in STAT3-independent manners. PGE2- or forskolin-induced cAMP signaling can activate PKA and CREB binding to an AP1/CRE element on an SOCS3 promoter. IFN-γ–induced STAT1 signaling and IL-4/IL-13–induced STAT6 signaling stimulate the SOCS3 promoter by binding to SRE elements. [8], [10, 11], and [12]: references (8), (10,11), and (12), respectively.
Beside SREs, the transcription factors that bind to the AP-1/CRE binding element of the CG-rich element could also be involved in increased IEC SOCS3 expression. Recently, prostaglandin E2 (PGE2) was demonstrated to induce SOCS3 expression in breast cancer cells via the activation of cAMP and then the AP-1/CRE region in the SOCS3 promoter (8,14). In the latter pathway, the downstream cAMP-dependent protein kinase A (PKA) is activated by the binding of cAMP to the regulatory subunit and releasing of catalytic subunits (PKA-C) (15,16). In UC patients and experimental colitis, PGE2 expression was increased and related to the severity of colitis (17,18), which may contribute to the SOCS3 induction.
Because SOCS3 expression is an essential step in the sequence of events leading to relapse of UC, preventing SOCS3 expression seems to be a rational strategy in the clinical management of inflammatory bowel disease. Devising such a strategy requires knowledge as to which signaling pathways are dominant in about-to-relapse UC patients with respect to SOCS3 induction. The promoter analysis of SOCS3 in the context of the current body of biomedical literature suggests that the STAT1 pathway, the STAT6 pathway or cyclooxygenase-dependent cAMP signaling may be important. Importantly, all of these pathways are druggable and thus offer the possibilities for therapeutic intervention. Hence, our aim was to investigate the pathways involved in the induction of SOCS3 in IECs and their functionality in quiescent UC.
MATERIALS AND METHODS
Patients and Biopsies
This study was approved by the ethical committee of the Erasmus MC Rotterdam. After gaining informed consent, biopsies from UC patients were obtained during colonoscopies from March to June 2009. Only patients with a diagnosis of UC over more than 1 year and in remission for at least 3 months were included. The diagnosis of UC was based on conventional clinical, endoscopic and histological criteria, as described by Lennard-Jones (19). Inactive UC was defined as patients in 3-month clinical remission without endoscopic abnormalities and with a Geboes histology score of 0 or 1 (20). Demographics and current medical therapy were noted. Colonic biopsies from non-UC patients without abnormalities at colonoscopy and no history of gastrointestinal disease served as controls. Patient characteristics are summarized in Table 1.
Table 1.
Patient characteristics.
| Controls (n = 9) | Inactive UC (n = 17) | P | |
|---|---|---|---|
| Sex | 0.429 | ||
| Male | 6 | 8 | |
| Female | 3 | 9 | |
| Mean age (years) (± SD) | 39.6 ± 17.8 | 45.0 ± 12.0 | 0.366 |
| Mean duration of disease (years) (± SD) | — | 17.7 ± 9.8 | — |
| Extent of disease | — | ||
| Extensive colitis | — | 0 | |
| Left side colitis | — | 11 | |
| Proctitis | — | 6 | |
| Therapy | — | ||
| Aminosalicylates | — | 11 | |
| Corticosteroids | — | 7 | |
| Immunosuppressive drugs | — | 3 | |
| Biological drugs | — | 2 | |
| None | — | 3 |
Cells and Reagents
Caco-2 cells were grown into a fully differentiated and confluent monolayer in transwells for 18–21 d. They were cultured in Dulbecco’s modified Eagle medium, supplemented with fetal calf serum and 10% (v/v) penicillin and streptomycin, at 37°C in a humidified atmosphere containing 5% CO2. Cells were stimulated with IL-6 (100 ng/mL; Cell Signaling, Danvers, MA, USA; #8904), IL-4 (10 ng/mL; R&D, Minneapolis, MN, USA; 204-IL), PGE2 (250 ng/mL; Sigma-Aldrich, St. Louis, MO, USA; P5640) or IFN-γ (1,000 U/mL; R&D). Each condition was repeated in three independent experiments.
RNA Isolation and RT-PCR
Total RNA was isolated from cell lines using TRIzol (Sigma-Aldrich). RNA pellets were resuspended in RNAse free water, and total RNA concentrations were determined by nanodrop. RNA (1 μg) was reversely transcribed using an iScript™ cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA). Quantitative polymerase chain reactions (qPCRs) were performed by using a real-time PCR kit, SensiMix™ SYBR & Fluorescein Kit (Quantace, London, UK), in a total volume of 25 μL containing 10-pmol primers. qPCR was performed in an IQ5 (Bio-Rad) with 40 cycles of amplification (15 s at 95°C, 15 s at 60°C and 15 s at 72°C). GAPDH served as a household gene. The primers 5′-TCTGT CGGAA GACCG TCAAC-3′ (sense) and 5′-GGTCC AGGAA CTCCC GAATG-3′ (antisense) were used to amplify SOCS3. The primers 5′-GCATT GCCCT CAACG ACCAC-3′ (sense) and 5′-CCACC ACCCT GTTGC TGTAG-3′ (antisense) were used to amplify GAPDH (both from Isogen Biosolutions, Hyderabad, India). Presented data are the ratio between SOCS3 and GAPDH mRNA deltaCt. For the cytokine measurements in Figure 2, biopsies were directly lysed in Tripure (Roche, Basel, Switzerland) for RNA extraction, according the manufacturer’s protocol. After the Tripure extraction, the RNA samples were purified using the RNA II extract kit from Macherey Nagel (Bioke, Leiden, the Netherlands) according to the manufacturer’s protocol. cDNA was created from 500 ng RNA using the iScript cDNAse kit (Bio-Rad). Gene expression of Ywaz (forward, ACTTT TGGTA CATTG TGGCT TCAA; reverse, CCGCC AGGAC AAACC AGTAT), IL-8 (forward, CACTG CGCCA ACACA GAAAT TA; reverse, ACTTC TCCAC AACCC TCTGC AC) and IL-1β (forward, CCCTA AACAG ATGAA GTGCT CCTT; reverse, GTAGC TGGAT GCCGC CAT) were measured by using qPCR (IQ5; Bio-Rad). Gene expression is plotted as fold change by using the deltaCt method. The data from patients with nine pieces of colonic biopsies were averaged. All qPCRs were at least performed in duplicate.
Figure 2.

Different cytokines are able to activate their canonical signaling pathways in IECs. (A) In Caco-2 cells, IFN-γ induced the phosphorylation of STAT1 without affecting STAT6 or STAT3. (B) IL-4 induced the phosphorylation of STAT6 without affecting STAT1 or STAT3. (C) IL-6 was used as a positive control and increased STAT3 as well as STAT1 phosphorylation. (D–E) Stimulation with PGE2 induced p-PKA and p-CREB expression in Caco-2 cells without any signs of STAT3 phosphorylation, demonstrating specificity of the results. (F) Cytokine mRNA expression in colonic biopsies. The graph represents the average IL-8 and IL-1β mRNA relative expression (deltaCt) of whole colonic biopsies from healthy individuals and inactive and active UC patients. The error bar equals standard error of the mean (SEM). *p < 0.001, control compared with active UC.
Western Blot
Total cell lysates were prepared, and Western blot was performed as described earlier (21). Transfer membranes (Immobilon-FL, pore size 0.45 μm) were probed with primary antibodies specific for SOCS3 (rabbit polyclonal; Abcam, Cambridge, UK; ab16030), p-STAT3 (Cell Signaling; #9145), p-STAT1 (Cell Signaling; #9167), p-STAT6 (Cell Signaling; #9361), p-PKA-Cγ (Abcam; ab75991) and p–cAMP response element-binding protein (p-CREB) (Cell Signaling; #9198). β-Actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA; E0610) was used as a reference protein. Horseradish peroxidase (HRP)- labeled goat anti-rabbit (channel 680 and 800) and goat anti-mouse antibodies were used as secondary antibodies (LI-COR Biosciences, Lincoln, NE, USA). Transfer membranes were incubated with antibodies and washed in 50-mL sterile centrifuge tubes and light protection centrifuge tubes (Greiner Bio-One, San Francisco, CA, USA). Protein expression was visualized using a fluorescence Odyssey system (LI-COR Biosciences). Quantitative expression was determined by Odyssey 3.0 software and normalized by using β-actin and α-tubulin as a reference.
Immunohistochemistry Staining and Scoring
All biopsies were fixed in 10% formalin and embedded in paraffin. Hematoxylin and eosin staining on paraffin-embedded biopsies was performed to confirm lack of inflammation. Sections (4 μm) were mounted onto Superfrost Plus slides (Thermo Scientific, Waltham, MA, USA), deparaffinized in xylene and rehydrated. The endogenous peroxidase activity was blocked by 3% H2O2 in methanol for 15 min at room temperature. Antigen retrieval was performed by boiling in a preheated buffer (EDTA, pH 8.0, for p-STAT1 and p-STAT6; Tris/EDTA, pH 9.0, for PKA-Cγ; citric acid, pH 6.0, for p-CREB) for 15 min at 200 W in a microwave. Next, slides were transferred to Shandon chambers and blocked by 5% goat serum in Tris-buffered saline (TBS) and 0.1% Tween 20 for p-STAT1, p-STAT6 and p-CREB; 10% normal human serum and 10% goat serum in phosphate-buffered saline (PBS), pH 7.4, was used for PKA-Cγ for 1 h at room temperature. The primary antibodies rabbit polyclonal anti-p-STAT1 (1:400; Cell Signaling; #9167), anti-p-STAT6 (1:100; Abcam; ab28829), rabbit monoclonal anti-PKA-Cγ (1:250; Abcam; ab75991) and rabbit monoclonal anti-p-CREB (1:400; Cell Signaling; #9198) were added and incubated at 4°C overnight. Envision goat anti-rabbit/mouse-HRP (DAKO, Glostrup, Denmark) was used as the secondary antibody. Immunoreactions were detected by using 3-3-diaminobenzidine (10 mg/mL) in 0.05 mol/L Tris-HCl, pH 7.6, containing imidazole stock (0.068 g/10 mL) and 0.03% H2O2, followed by counterstaining with hematoxylin. Negative controls were performed by using only the secondary antibody. Two experienced people scored the staining of epithelial cells in a blinded manner; discrepancies were reassessed to reach agreement. The percentage of cells that stained positive (immunoreactivity above background) in the area consistent with that used for diagnoses was quantified. P-STAT1, p-STAT6, PKA-Cγ and p-CREB were counted for nuclear staining in epithelial cells.
Statistical Analysis
Statistical analysis was performed by using SPSS software, version 15.0. Data were presented as means ± standard deviation (SD). The difference between each two groups was determined by a Mann-Whitney U test. The relationship between p-STAT1–, p-STAT6– and PKA-Cγ–positive IECs were determined by Spearman correlation testing. p Values <0.05 were considered statistically significant.
All supplementary materials are available online at www.molmed.org.
RESULTS
Patient Characteristics
In total, biopsies from 17 patients with inactive UC were included. There were eight men (47.1%) and nine women (52.9%) with a mean age of 45.0 (± 12.0) years and a disease duration of 17.7 (± 9.8) years. The control group consisted of six men and three women with a mean age of 39.6 (± 17.8) years. There were no significant differences in patient sex and mean age between groups of control and inactive UC patients (Table 1). We concluded that our patient cohort is suitable for making meaningful comparisons between UC and controls with respect to the factors driving SOCS3 expression.
STAT1, STAT6 and cAMP Signaling Induce STAT3-Independent SOCS3 Expression In Vitro
We aimed to identify druggable pathways driving mucosal SOCS3 expression in UC that is about to relapse (Supplementary Figure S1), taking cues from the genomic makeup of the human SOCS3 promoter (7,8,10,12) (see Figure 1). Theoretically, SRE in the SOCS3 promoter can bind and become transactivated by p-STAT1 homodimers as well as p-STAT6 homodimers, but whether they can do so in the context of the human intestinal epithelium has not been investigated. Hence, Caco-2 cells were treated with IFN-γ (the canonical activator of STAT1 signaling) and IL-4 (the classical stimulus upstream of STAT6), respectively (see Figure 1). First, we confirmed that the different cytokines activated their specific signaling pathways in IECs. IFN-γ induced the phosphorylation of STAT1 (p < 0.05) without affecting STAT6 or STAT3 (Figures 2A–C). IL-4 induced the phosphorylation of STAT6 (p < 0.05) without affecting STAT1 or STAT3 (see Figures 2A–C). IL-6 was used as a positive control and increased STAT3 as well as STAT1 phosphorylation (p < 0.05) (see Figures 2A–C). In addition to the SREs, the promoter region of SOCS3 also contains an AP-1/CRE binding site, which is a well-known downstream target of cAMP signaling. To see whether cAMP-mediated signal transduction could induce SOCS3 expression, Caco-2 cells were stimulated with PGE2 (see Figure 1). Stimulation with PGE2 induced p-PKA and p-CREB expression (p < 0.05) (Figure 2D). The specificity of this effect was highlighted by the apparent absence of a concomitant effect on STAT3 phosphorylation (Figure 2E). Importantly, IFN-γ and IL-4 as well as PGE2 all provoked increased SOCS3 expression at both the mRNA level and the protein level (p < 0.05) (Figures 3A, B), and this result corresponded with lower levels of IL-8 and virtually unchanged levels of IL-1β in the inactive UC biopsies (p < 0.05) compared with active ones. These cytokines are not involved in SOCS3 induction in the inactive stage. Thus, multiple signaling pathways are capable of driving SOCS3 expression in IECs in vitro, and experiments were initiated to address the importance of these pathways for SOCS3 expression in UC in vivo.
Figure 3.

SOCS3 expression induced by different UC-associated factors and signaling pathways. (A, B) Stimulation of IFN-γ and IL-4 as well as the PGE2 increases SOCS3 expression at both mRNA and protein levels.
Active STAT1 and STAT6 Signaling in IEC of Patients with Inactive UC
The assess whether STAT1 and/or the STAT6 signaling pathways are active in UC biopsies, biopsies displaying a high level of SOCS3 expression were selected and compared with biopsies with low SOCS3 expression with respect to p-STAT1 and p-STAT6 levels (Figures 4A, B). Quantification of the percentage of positive IECs in each biopsy showed a significantly increased expression of both p-STAT1 (p < 0.0001) and p-STAT6 (p = 0.0001) in biopsies with high SOCS3 expression (Figures 4C, D). Thus, these data provide evidence that active STAT1 and STAT6 signaling might be involved in the high SOCS3 expression in IEC of inactive UC patients.
Figure 4.

IHC staining of p-STAT1 (A) and p-STAT6 (B) IEC-positive cells between patients with inactive UC and controls (nuclear staining). Quantification of the percentage positive IEC in each biopsy showed a significantly increased percentages of p-STAT1–positive (*p < 0.0001) (C) and p-STAT6–positive (**p = 0.0001) (D) IEC in biopsies from patients with inactive UC compared with control biopsies.
Active cAMP Signaling in IECs of Patient with Inactive UC
The same biopsies were also stained for p-PKA-Cγ and p-CREB as markers of active cAMP signaling (Figures 5A, B). Analysis of this staining showed significantly increased percentages of p-PKA-Cγ–positive (p = 0.0003) and p-CREB–positive (p = 0.0025) IECs in biopsies from patients with high SOCS3 expression compared with control biopsies (Figures 5C, D). These data show that the cAMP signaling pathway may also be involved in high SOCS3 expression in IECs of inactive UC patients with about-to-relapse disease.
Figure 5.

IHC staining of p-PKA-Cγ (A) and p-CREB (B) IEC-positive cells between patients with inactive UC and controls (nuclear staining). Quantification of the percentage positive IECs in each biopsy showed significantly increased percentages of p-PKA-Cγ–positive (***p = 0.0003) (C) and p-CREB–positive (****p = 0.0025) (D) IECs in biopsies from patients with inactive UC compared with control biopsies.
No Significant Correlation between Three Signaling Pathways and Medicine Use
To check whether the signaling pathways were mutually exclusive or had corresponding expression, we correlated the expression between the different stainings within the biopsy from the same patient. In the biopsies of inactive UC, no significant correlation was found between the different parameters (all p > 0.05) (data not shown). We further compared the medication use with the signaling pathways that were dominant in each patient. However, when we sub-identified the signaling activities according to different medicine use in these patients, no significant difference was found between these signaling activities and different medicine users (data now shown).
DISCUSSION
In the current study, we show that SOCS3 expression can be induced in IECs via the STAT1, STAT6, or cAMP pathway independent of STAT3. We found that the same pathways were active in IECs of colonic biopsies from UC patients with inactive disease and with high SOCS3 expression. These data provide new insight into the mechanism involved in induction of SOCS3 expression.
Mice lacking IL-6/gp130/p-STAT3 signaling were more sensitive to chemically induced colitis because of reduced IEC survival and proliferation (4,5). In addition, transgenic SOCS3 expression in epithelial cells negatively affected STAT3-dependent wound healing (22). These data suggest that because of the high SOCS3 expression, normal STAT3 signaling is impaired and could lead to enhanced vulnerability of IECs (23), which could promote the development of UC or at least enhance the risk for UC relapse, as we have recently seen (2).
By determining the STAT response element involved in the activation of the SOCS3 promoter, it was reported that the induction of the SOCS3 reporter gene by IFN-γ and IL-4 depended on a STAT-binding sequence because STAT-specific mutation or knockdown by siRNA totally abolished the responsiveness of SOCS3 to IFN-γ and IL-4 (10,24). Our observation of enhanced p-STAT1 and SOCS3 expression in biopsies of patients with inactive UC and the IFN-γ–induced SOCS3 expression via p-STAT1 in vitro are consistent with the previous detection of increased levels of IFN-γ and STAT1 in UC pouchitis patients without clinical and endoscopic evidence (9). It was shown in vascular endothelial cells that IFN-γinduced SOCS3 expression selectively inhibits IL-6–induced STAT3 activation and IL-6–dependent expression of antiinflammatory, but not proinflammatory, target genes. In turn, other proinflammatory genes are even more increased (25). This IFN-γ–induced proinflammatory phenotype via the activation of SOCS3 represents a novel mechanism of IEC inflammation. However, IFN-γ was not shown to be upregulated in any other studies looking at gene or protein expression in biopsies of patients with inactive UC. In addition, not much is known about their prediction during remission, which would be interesting to investigate.
CONCLUSION
In our study, we showed for the first time that IL-4 induced STAT6 phosphorylation and subsequent SOCS3 expression in IEC, and p-STAT6 combined with high SOCS3 is expressed in the biopsies from UC patients with inactive disease. These data fit with the STAT6-dependent induction of SOCS3 by stimulation keratinocytes by combining IL-4 and IL-13 (24). Various reports have mentioned a balancing effect between STAT1 and STAT6 signaling, where IL-4 and IL-13 inhibit IFN-γ via the induction of SOCS1 and SOCS3 and vice versa (24,26). Unfortunately, because of the small patient groups, we did not see this effect in our biopsies.
In addition to STAT signaling, we also saw that PGE2-induced cAMP signaling can induce SOCS3 upregulation in IECs. Phosphorylation of PKA-Cγ and CREB was used as a readout for cAMP signaling, as demonstrated previously (8). Our data also showed for the first time that cAMP signaling is active in IECs of UC patients with inactive inflammation, as detected by enhanced expression of p-PKA-Cγ and p-CREB expression. Although PGE2 worked well as a positive control in our in vitro assays, it may not be the most likely candidate to induce the observed cAMP activity in biopsies. A recent study showed that PGE2 was highly expressed in actively inflamed tissue (27). In the same study, the pro-resolution mediator prostaglandin D2 (PGD2) and its receptor DP1 were expressed in biopsies of UC patients in remission (27). This observation combine with the even stronger induction of cAMP by PGD2 compared with PGE2 make the PGD2 a good candidate for the increased SOCS3 expression in some of the UC patients (28).
No significant correlations were found between the different pathways, suggesting that multiple pathways could be active in the same in IECs in specific biopsies, obviously hampering targeted therapy. Nevertheless, our data provide evidence that STAT1, STAT6 and cAMP signaling activation may all be involved in SOCS3 expression in IECs. Because SOCS3 expression precedes exacerbation of disease in otherwise quiescent UC, these pathways provide interesting therapeutic targets for maintaining UC remission, but our findings that multiple pathways target the SOCS3 promoter raises important questions as to the pathway that actually mediates induction of SOCS3 in inactive colitis and is the best target for pharmacological intervention. IL-6 is a potent inducer of SOCS3 via STAT3, but previously we showed that neither IL-6 nor STAT3 was markedly upregulated in inactive colitis (23), focusing attention on the alternative pathways investigated in the present study. In a prospective study by Lauritsen et al.(29), in 23 patients with completely inactive disease, concentrations of PGE2 were determined. In the group of patients with purified fecal and rectal dialysates at entry, no case with normal rectal PGE2 concentration was associated with relapse in the short term, whereas in a group of five patients with increased PGE2, four displayed relapse of disease. Although the number of patients is small, this result would imply that PGE2-cAMP is a major contributor to the relapse marker SOCS3 induction. Our immunohistochemistry using a phosphorylation state–specific antibody that recognizes only the activated form of CREB would support that notion. However, whereas the strong STAT6 signaling observed in inactive UC indicates an important role for IL-4 signaling (a notion that is supported by the observed Th2 profile of inactive UC), levels of IL-4 are not increased in quiescent UC (30,31), suggesting that this pathway has a more contributing rather than critical action in relapse induction. Finally, although we observe strong expression of STAT1 in inactive UC, IFN-γ levels are associated with remission rather than active disease. Thus, in total, a picture emerges that not a single pathway is responsible for the driving relapse through SOCS3 expression but that this expression brought about the action of multiple pathways. In agreement, a study using a mushroom extract showed a potential benefit in suppressing STAT1 and STAT6 associated with IFN-γ and IL-4 expression in a model for UC, suggesting that targeting multiple pathways is a strategy forward (32). Our findings suggest that broad-spectrum antiinflammatory drugs are essential to counteract increases in SOCS3 expression and exacerbation of disease.
Supplemental Data
ACKNOWLEDGMENTS
Part of this study was supported by the State Scholarship Fund from the Chinese Scholarship Council (2007101714).
This study was conducted with the approval of the relevant institutional ethics board.
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
REFERENCES
- 1.Peyrin-Biroulet L, Lemann M. Review article: remission rates achievable by current therapies for inflammatory bowel disease. Aliment Pharmacol Ther. 2011;33:870–9. doi: 10.1111/j.1365-2036.2011.04599.x. [DOI] [PubMed] [Google Scholar]
- 2.Li Y, et al. Increased suppressor of cytokine signaling-3 expression predicts mucosal relapse in ulcerative colitis. Inflamm Bowel Dis. 2012 doi: 10.1002/ibd.22992. [DOI] [PubMed] [Google Scholar]
- 3.Li Y, de Haar C, Peppelenbosch MP, van der Woude CJ. New insights into the role of STAT3 in IBD. Inflamm Bowel Dis. 2012;18:1177–83. doi: 10.1002/ibd.21884. [DOI] [PubMed] [Google Scholar]
- 4.Grivennikov S, et al. IL-6 and stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103–13. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bollrath J, et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell. 2009;15:91–102. doi: 10.1016/j.ccr.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 6.Li Y, de Haar C, Peppelenbosch MP, van der Woude CJ. SOCS3 in immune regulation of inflammatory bowel disease and inflammatory bowel disease-related cancer. Cytokine Growth Factor Rev. 2012;23:127–38. doi: 10.1016/j.cytogfr.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 7.He B, et al. Cloning and characterization of a functional promoter of the human SOCS-3 gene. Biochem Biophys Res Commun. 2003;301:386–91. doi: 10.1016/s0006-291x(02)03071-1. [DOI] [PubMed] [Google Scholar]
- 8.Bousquet C, Chesnokova V, Kariagina A, Ferrand A, Melmed S. cAMP neuropeptide agonists induce pituitary suppressor of cytokine signaling-3: novel negative feedback mechanism for corticotroph cytokine action. Mol Endocrinol. 2001;15:1880–90. doi: 10.1210/mend.15.11.0733. [DOI] [PubMed] [Google Scholar]
- 9.Ferguson LR, et al. Genetic factors in chronic inflammation: single nucleotide polymorphisms in the STAT-JAK pathway, susceptibility to DNA damage and Crohn’s disease in a New Zealand population. Mutat Res. 2010;690:108–15. doi: 10.1016/j.mrfmmm.2010.01.017. [DOI] [PubMed] [Google Scholar]
- 10.Gatto L, et al. Analysis of SOCS-3 promoter responses to interferon gamma. J Biol Chem. 2004;279:13746–54. doi: 10.1074/jbc.M308999200. [DOI] [PubMed] [Google Scholar]
- 11.Kovarik A, et al. Interferon-gamma, but not interferon-alpha, induces SOCS 3 expression in human melanoma cell lines. Melanoma Res. 2005;15:481–8. doi: 10.1097/00008390-200512000-00001. [DOI] [PubMed] [Google Scholar]
- 12.Ehret GB, et al. DNA binding specificity of different STAT proteins: comparison of in vitro specificity with natural target sites. J Biol Chem. 2001;276:6675–88. doi: 10.1074/jbc.M001748200. [DOI] [PubMed] [Google Scholar]
- 13.Betts G, et al. Suppression of tumour- specific CD4+ T cells by regulatory T cells is associated with progression of human colorectal cancer. Gut. 2012;61:1163–71. doi: 10.1136/gutjnl-2011-300970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barclay JL, Anderson ST, Waters MJ, Curlewis JD. Characterization of the SOCS3 promoter response to prostaglandin E2 in T47D cells. Mol Endocrinol. 2007;21:2516–28. doi: 10.1210/me.2007-0030. [DOI] [PubMed] [Google Scholar]
- 15.Lohmann SM, Walter U. Regulation of the cellular and subcellular concentrations and distribution of cyclic nucleotide-dependent protein kinases. Adv Cyclic Nucleotide Protein Phosphorylation Res. 1984;18:63–117. [PubMed] [Google Scholar]
- 16.Kammer GM. The adenylate cyclase-cAMP-protein kinase A pathway and regulation of the immune response. Immunol Today. 1988;9:222–9. doi: 10.1016/0167-5699(88)91220-0. [DOI] [PubMed] [Google Scholar]
- 17.Wiercinska-Drapalo A, Flisiak R, Prokopowicz D. Plasma and mucosal prostaglandin E2 as a surrogate marker of ulcerative colitis activity. Rocz Akad Med Bialymst. 2001;46:60–8. [PubMed] [Google Scholar]
- 18.Razavi A, Khodadadi A, Eslami MB, Eshraghi S, Mirshafiey A. Therapeutic effect of sodium alginate in experimental chronic ulcerative colitis. Iran J Allergy Asthma Immunol. 2008;7:13–8. [PubMed] [Google Scholar]
- 19.Lennard-Jones JE. Classification of inflammatory bowel disease. Scand J Gastroenterol Suppl. 1989;170:2–6. doi: 10.3109/00365528909091339. discussion 16–19. [DOI] [PubMed] [Google Scholar]
- 20.Geboes K, et al. A reproducible grading scale for histological assessment of inflammation in ulcerative colitis. Gut. 2000;47:404–9. doi: 10.1136/gut.47.3.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang M, et al. Deficiency of TNFR1 protects myocardium through SOCS3 and IL-6 but not p38 MAPK or IL-1beta. Am J Physiol Heart Circ Physiol. 2007;292:H1694–9. doi: 10.1152/ajpheart.01063.2006. [DOI] [PubMed] [Google Scholar]
- 22.Wang YC, et al. Notch signaling determines the M1 versus M2 polarization of macrophages in antitumor immune responses. Cancer Res. 2010;70:4840–9. doi: 10.1158/0008-5472.CAN-10-0269. [DOI] [PubMed] [Google Scholar]
- 23.Braconi C, Huang N, Patel T. MicroRNA-dependent regulation of DNA methyltransferase-1 and tumor suppressor gene expression by interleukin-6 in human malignant cholangiocytes. Hepatology. 2009;51:881–90. doi: 10.1002/hep.23381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Albanesi C, et al. IL-4 and IL-13 negatively regulate TNF-alpha- and IFN-gamma-induced beta-defensin expression through STAT-6, suppressor of cytokine signaling (SOCS)-1, and SOCS-3. J Immunol. 2007;179:984–92. doi: 10.4049/jimmunol.179.2.984. [DOI] [PubMed] [Google Scholar]
- 25.Bluyssen HA, et al. IFN gamma-dependent SOCS3 expression inhibits IL-6-induced STAT3 phosphorylation and differentially affects IL-6 mediated transcriptional responses in endothelial cells. Am J Physiol Cell Physiol. 2010;299:C354–62. doi: 10.1152/ajpcell.00513.2009. [DOI] [PubMed] [Google Scholar]
- 26.Venkataraman C, Leung S, Salvekar A, Mano H, Schindler U. Repression of IL-4-induced gene expression by IFN-gamma requires Stat1 activation. J Immunol. 1999;162:4053–61. [PubMed] [Google Scholar]
- 27.Vong L, Ferraz JG, Panaccione R, Beck PL, Wallace JL. A pro-resolution mediator, prostaglandin D(2), is specifically up-regulated in individuals in long-term remission from ulcerative colitis. Proc Natl Acad Sci U S A. 2010;107:12023–7. doi: 10.1073/pnas.1004982107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Craven PA, Saito R, DeRubertis FR. Role of local prostaglandin synthesis in the modulation of proliferative activity of rat colonic epithelium. J Clin Invest. 1983;72:1365–75. doi: 10.1172/JCI111093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lauritsen K, Laursen LS, Bukhave K, Rask-Madsen J. Use of colonic eicosanoid concentrations as predictors of relapse in ulcerative colitis: double blind placebo controlled study on sulphasalazine maintenance treatment. Gut. 1988;29:1316–21. doi: 10.1136/gut.29.10.1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roberts-Thomson IC, Fon J, Uylaki W, Cummins AG, Barry S. Cells, cytokines and inflammatory bowel disease: a clinical perspective. Expert Rev Gastroenterol Hepatol. 2011;5:703–16. doi: 10.1586/egh.11.74. [DOI] [PubMed] [Google Scholar]
- 31.Inoue S, et al. Characterization of cytokine expression in the rectal mucosa of ulcerative colitis: correlation with disease activity. Am J Gastroenterol. 1999;94:2441–6. doi: 10.1111/j.1572-0241.1999.01372.x. [DOI] [PubMed] [Google Scholar]
- 32.Lim BO. Coriolus versicolor suppresses inflammatory bowel disease by inhibiting the expression of STAT1 and STAT6 associated with IFN-gamma and IL-4 expression. Phytother Res. 2011;25:1257–61. doi: 10.1002/ptr.3378. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.