

Abstract
Myelodysplastic syndromes represent particularly challenging hematologic malignancies that arise from a large spectrum of genetic events resulting in a disease characterized by a range of different presentations and outcomes. Despite efforts to classify and identify the key genetic events, little improvement has been made in therapies that will increase patient survival. Animal models represent powerful tools to model and study human diseases and are useful pre-clinical platforms. In addition to enforced expression of candidate oncogenes, gene inactivation has allowed the consequences of the genetic effects of human myelodysplastic syndrome to be studied in mice. This review aims to examine the animal models expressing myelodysplastic syndrome-associated genes that are currently available and to highlight the most appropriate model to phenocopy myelodysplastic syndrome disease and its risk of transformation to acute myelogenous leukemia.
Introduction
Myelodysplastic syndromes (MDS) are thought to arise from a malignant hematopoietic stem cell (HSC) that has sustained irreversible genetic or epigenetic events resulting in dominance of the abnormal clone. Approximately 30% of MDS cases progress to acute myelogenous leukemia (AML). Dysmyelopoiesis and increased apoptosis are the main features of MDS. Bone marrow (BM) isolated from MDS patients exhibits dysplasia of at least one lineage (according to the World Health Organization (WHO) classification),1,2 hypercellularity, and increased apoptosis correlating with peripheral blood cytopenia.3,4 Because MDS patients present with a range of different characteristics, classification has become important, and prognostic scoring techniques have been developed to address patient management and treatment.2,5,6 Recently, these classification and scoring techniques have also taken into account the presence of cytogenetic abnormalities. Cytogenetic abnormalities are relatively frequent in MDS; deletions and rare chromosomal reciprocal translocations have been identified, while DNA sequencing and micro-arrays (RNA, CGH) have confirmed additional mutations and deletions.7,8 Thus, genetic and epigenetic abnormalities are linked to MDS pathogenesis.9 Efforts have been made to develop accurate animal models of MDS that would help us understand the pathogenesis of the disease and the mechanisms of its transformation to AML. Various strategies have been used to express MDS-associated candidate genes in animal models, either by transduction of candidate genes under the control of retroviral promoters and subsequent transplantation of transduced mouse BM cells into syngenic mice, or by the establishment of transgenic mice expressing the candidate genes under the control of myeloid-specific promoters (Figure 1). Indications of function have also been obtained by analysis of knock-in (KI) and knock-out (KO) models of various genes. Recently, analyses of xenografts of human MDS cells have yielded promising results. Hematopoietic analysis of these animal models has revealed a great variety of hematologic abnormalities, some of which are not necessarily reminiscent of either the human MDS or MDS/myeloproliferative neoplasms (MDS/MPN). This review focuses on the animal models expressing the most frequent genetic abnormalities associated with MDS and MDS/MPN, including chronic myelomonocytic leukemia (CMML) and juvenile myelomonocytic leukemia (JMML), as well as those models that resemble the human MDS disease regardless of the expressed or deleted genes (Table 1). The WHO classification of human MDS and MDS/MPN is shown in the Online Supplementary Tables S1 and S2.1,2 Our working model of disease progression is illustrated in Figure 2. Starting with an initiation event targeting the stem cell, an abnormal clone arises which has a selective growth advantage and expands. This is the phase that we recognize as pre-leukemia or MDS. Several events then follow until a leukemic clone arises and accelerates the disease to give rise to acute leukemia. The genes that drive this progression are those that code for differentiation, proliferation and apoptosis. A block in differentiation can occur in any compartment; although in MDS it is thought to occur at a very immature stage, giving rise to leukemic stem cells (LSC), so-called because they are cells that have acquired self-renewal capacity. Early MDS is often characterized as pro-apoptotic, and as disease progresses, the cells become more anti-apoptotic. There is abnormal proliferation throughout the disease process.
Figure 1.

Mouse models to study malignant hematologic disease. Several strategies can be employed to create mouse models of disease. Candidate genes can be introduced into the germline under the control of appropriate promoters to drive expression in certain cell compartments. Knock-out strategies create gene deficient mice, whilst knock-in strategies use the endogenous promoters; but again these tend to be conditional systems requiring specific promoters to determine in which cell the genes are introduced. Transduction of bone marrow and reinfusion is a powerful tool. Xenograft models are theoretically the best if the techniques involved can be improved.
Table 1.
Gene alterations found in MDS patients and their corresponding mouse models with corresponding hematologic disease entities according to WHO classification 5.
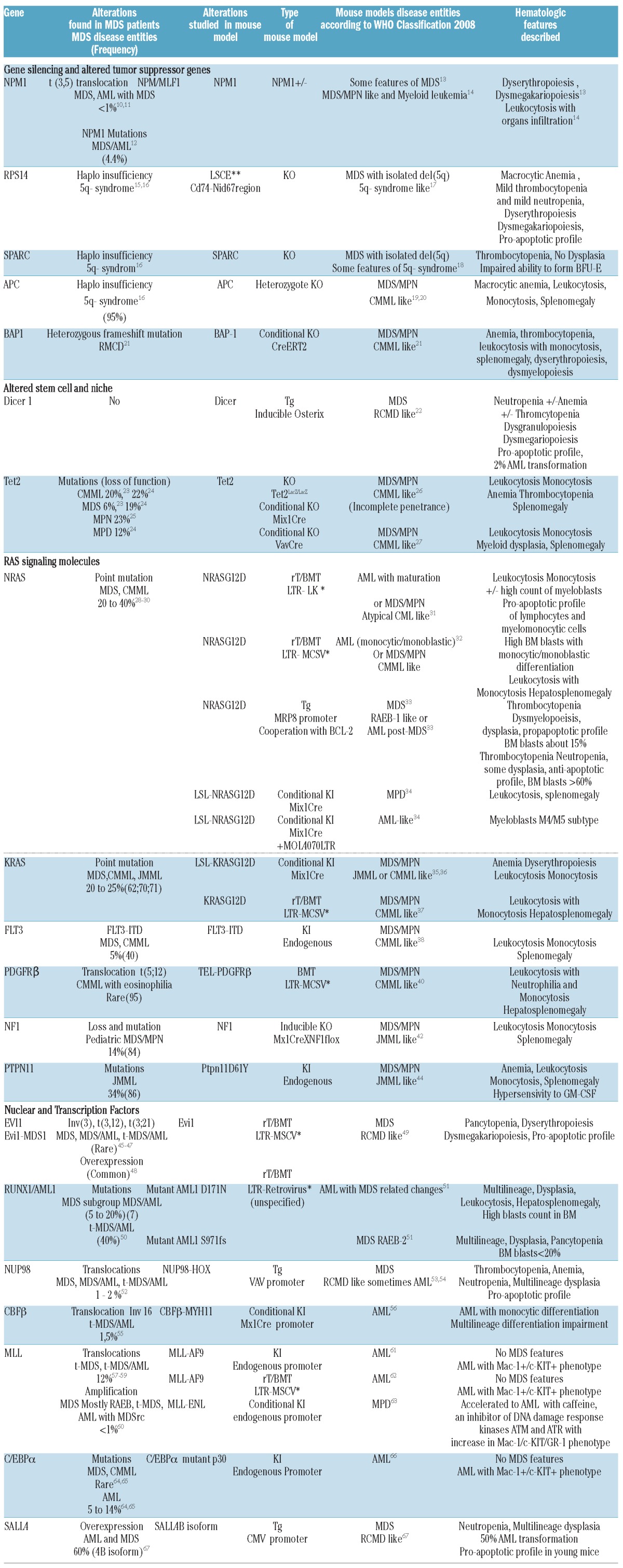
Figure 2.

Multi-step model of leukemogenesis. Starting with a stem cell, an initiating event occurs which gives rise to an abnormal clone with a growth advantage, which expands. Further genetic events drive the progression of the disease towards overt leukemia. Genomic instability and misrepair increase as disease progresses. Some of the events that may drive progression include increased methylation to down-regulate genes and mutations of the ribosome machinery to further deregulate genes.
Novel signaling pathways and cells involved in MDS/MPN
Recent insights into the pathogenesis of MDS have highlighted the importance of genomic instability (GI), epigenetic alterations and abnormalities of the spliceosome machinery. The MDS stem cell and its niche have also proved to be of significance (Figure 2).
Genomic instability (GI)
Karyotypic changes are indicative of increased GI. Increased GI can result in increased misrepair. Non-homologous end joining (NHEJ), the main pathway for double-strand break repair, gives rise to chromosomal deletions and translocations. It has been hypothesized that increased GI can drive initiation and progression of malignant disease.68 Aging mice display increased chromosome instability mediated by MDM2, a regulator of the p53 tumor suppressor and DNA repair.69 The expression of many oncogenes, such as mutant RAS, results in increased DNA damage-inducing reactive oxygen species (ROS). As MDS is essentially a disease associated with aging, it is not surprising that increased chromosome instability has been observed in a subgroup of MDS that is associated with a poor prognosis (20%), thereby suggesting that GI may be a driver of disease progression.70 GI in mouse models of MDS has yet to be investigated; although a mouse model of AML with MDS-related changes mediated by RAS and BCL-233 displayed increased ROS, DNA damage and misrepair which can be rescued by treatment with either RAC1 inhibitors or antioxidants.71 Furthermore, BM cells isolated from NPM1+/-MPN-like mice display multiple centrosomes, thus indicating that centrosome amplification is associated with NPM1 loss and may participate in leukemia pathogenesis in this mouse model.14 DNA damage and chromosomal instability is observed in Fanconi anemia (FA) patients, which is an important cause of childhood MDS.72 Unfortunately, mouse models harboring a disrupted mouse homolog of FANCC fail to develop MDS.73 However, the double mutant Fancc−/−, Fancg−/− mice develop spontaneous hematologic abnormalities including BM failure, AML, MDS and complex random chromosomal abnormalities that the single mutant mice do not display.74
Altered gene expression through epigenetic modifications and gene silencing
Dysregulation of the epigenetic machinery can lead to oncogenic transformation. Approximately 30% of MDS patients show inactivation, by hypermethylation, of the gene encoding p15INK4b.75 This strongly suggests that the INK4 family member plays a role in the regulation of cell cycle arrest at the early G1 phase as a myeloid tumor suppressor.76 Ink4b KO mice (Ink4b-/-) show defects in the differentiation of common myeloid progenitors, resulting in an imbalance between erythroid and myeloid potential, although follow-up analysis of these mice did not reveal any signs of MDS.77 Other genes modified by epigenetic mechanisms have been identified, such as DAPK1, CDH1 (E-Cadherins), and TSP1 (thrombospondin).78,79 Hypermethylation of DNA is known to contribute to progression of MDS to AML, thereby inhibiting different tumor suppressor genes. Mouse models designed to address this epigenetic progression have yet to be characterized. An exception is the EVI1 mouse model, in which EVI1 expression in the BM gives rise to an MDS phenotype and causes silencing of the miRNA124 by methylation. This epigenetic event results in perturbation of cell division and self-renewal in this mouse model.80
The TET2 gene (Ten-Eleven Translocation-2) was originally identified from an MDS patient with a rearrangement of 4q2424 and numerous groups have identified somatic inactivating mutations (frameshifts or misense) in MDS, MPD and CMML patients.23-25 As one of the most frequent mutations found in myeloid malignancies, TET2 has been proposed to be a tumor suppressor as loss of function tends to lead to disruption of progenitor myeloid differentiation leading to disease progression.81 Its function is to convert 5-methlycytosine to 5-hdroxymethyl cytosine (5-hmc) and it is, therefore, involved in the epigenetic control of gene regulation; 5-hmc has been found to be decreased in granulocyte DNA from MPD patients with TET2 mutations.81 Animal models of TET2 using conditional knock-out systems give rise to CMML-like diseases: Tet2LacZ mice, Tet2Lox mice26 and Tet2−/−VavCre+ mice27 develop a CMML-like phenotype with splenomegaly, leukocytosis with abnormal myelomonocytic differentiation, and expansion of the hematopoietic stem cell compartment.26,27
Altered stem cell and niche in MDS mouse models
Blood and BM samples from most MDS patients are technically difficult to engraft into NOD/SCID-β2mnull mice. When xenografts are obtained, they regenerate progeny that display phenotypic and genotypic abnormalities of the original neoplastic clone, such as trisomy 8 and del(5q). Interestingly, injecting BM cells isolated from MDS patients with human stroma-derived cells, via intramedullary injection, into NOD/SCID-β2mnull mice82 showed the engraftment of early precursors containing del(5q) with a loss of more mature cells that had additionally acquired trisomy 8. Therefore, the 5q deletion may be an earlier event than trisomy 8.82 In another study, the results of MDS sample engraftment suggested the presence of a relatively late type of ‘multilineage but myeloid-restricted’ neoplastic ‘stem’ cell with repopulating activity, limited self-renewal ability, and skewed differentiation potential.83 These findings also show that the repopulating cells present in MDS patients include residual normal or pre-neoplastic repopulating cells with normal features that outcompete the neoplastic clone if adequately stimulated. Recently, a transgenic approach has provided evidence that disturbance of the endosteal niche can result in MDS.22 Transgenic mice lacking Dicer1 expression in mesenchymal cells of the osteolineage were generated, OsxCre+/Dicerfl/fl.22 The OsxCre+/Dicerfl/fl mutant mice develop fatal neutropenia with hyperplastic BM and dysmyelopoiesis which is highly suggestive of MDS. Increased apoptosis of Lin-/c-KIT+ progenitor cells has also been observed in these mice. However, the MDS phenotype was not transplantable. Interestingly, when OsxCre+/Dicerfl/fl mice were transplanted with BM cells from wild-type mice they developed the same MDS phenotype. Although mutations of Dicer have yet to be discovered in MDS patients, gene expression analysis of Dicer-/- osteoprogenitors showed a downregulation of SDB, the gene found mutated in Schwachman-Bodian-Diamond syndrome, which induces BM failure and MDS in children. Recently, downregulation of Dicer has been found associated with poor prognosis in chronic lymphocytic leukemia (CLL) patients.84 Recent studies have highlighted the role of innate immune signaling molecules in the physiopathology of BM failure: in 5q- patients, deletion of chromosome 5 correlates with a loss of two miRs, miR145 and miR146a,85 which are abundant in hematopoietic progenitors and target two molecules implicated in innate immune signaling,86 Toll-interleukin-1 receptor domain-containing adaptor protein (TIRAP) and tumor necrosis factor receptor-associated factor-6 (TRAF6), respectively. In KO mouse models of miR145 and miR146a targeted specifically in hematopoietic stem and progenitor cells, animals develop thrombocytosis, mild neutropenia, and megakaryocytic dysplasia, which represent several clinical features of 5q-MDS.87
MDS mouse models of chromosomal deletions: candidate genes for 5q- syndrome
Deletion of chromosome 5 (5q-) is the most common cytogenetic abnormality found in MDS (10-15% of patients). It is associated with refractory anemia, thrombocytosis, hypolobulated megakaryocytes and low risk of evolving into AML.16 Because deletion of 5q represents the major cytogenetic abnormality associated with MDS, it has been hypothesized that this region may contain an important MDS suppressor gene.16,18 One such candidate is the NPM (Nucleophosmin) gene, located in the 5q region. Mutations of the NPM1 gene have also been found in 4.4% of MDS patients.12 NPM is a nuclear phosphoprotein, which has a putative function in ribosome assembly and DNA repair. While NPM1-/- mice lack definitive hematopoiesis, NPM1+/- mice develop features similar to the human 5q-syndrome, such as dyserythropoiesis and dysmegakaryopoiesis that is sometimes associated with thrombocytosis,13 albeit after a long latency period of 10-24 months.14 The commonly deleted region (CDR) associated with the 5q-syndrome has been narrowed down to a region of approximately 1.5 Mb located between 5q31 and 5q32.
The CDR is divided into two regions of synteny on mouse chromosomes 11 and 18. Partial deletion of this region in mice results in macrocytic anemia, prominent erythroid dysplasia, and monolobulated megakaryocytes, and is associated with moderate thrombocytopenia and neutropenia.17 The candidate genes included in this region are: Cd74, RPS14, NDST1, SYNPO, MYOZ3, RBM22, DCTN4 and NID67. In humans, the candidate genes of the CDR include FAT2 (FAT tumor suppressor homolog 2), SPARC (secreted protein, acidic and rich in cysteine), and RPS14 (40S ribosomal protein S14). The FAT2 candidate gene was evaluated in vivo but deficient mice failed to develop hematologic disease.17 RPS14 is a strong candidate as demonstrated in vitro, whereby silencing RPS14 expression by RNA interference in human BM cells reproduced a 5q-cell phenotype.15 Interestingly, Diamond-Blackfan anemia is also associated with expression of the ribosomal protein RPS1988 and transgenic mice expressing an inducible transgene of a mutant form of RPS19 (loss of function) develop anemia with dyserythropoiesis, reduced erythroid progenitors and a block in terminal erythroid maturation.89 RPS14 is thought to be a tumor suppressor.16
SPARC is a component protein of the extracellular matrix. SPARC-null mice develop mild thrombocytopenia and exhibit an impaired ability to form BFU-E colonies.18 SPARC is also a candidate tumor suppressor as methylation of the SPARC promoter has been observed in both lung and pancreatic cancers90,91 and is often associated with breast cancer cell invasion.92
The human APC (adenomatous polyposis coli) gene is located on chromosome 5q22, in close proximity to the region that is deleted in 5q-syndrome. APC is a well-characterized tumor suppressor associated with colon cancer. APC/min mice (carrying a truncated, non-functional allele of APC) develop an MDS/MPN hematologic disease with macrocytic anemia, leukocytosis, monocytosis and splenomegaly.19 The mice display altered stem cell function demonstrated by an impaired repopulating potential in secondary recipients.19 However, APC haploinsufficiency is observed in 95% of MDS patients with 5q-syndrome and APC+/- mice develop a disease that recapitulates several characteristic features of human MDS.20 Thus, haploinsufficiency is a mechanism of activating the oncogenic potential of a gene. However, other candidate genes located in the 5q-region either fail to confer hematopoietic disease or have not yet been studied.
Spliceosomal mutations
Recurrent mutations in a number of genes encoding core spliceosomal proteins have been identified in all subgroups of MDS. Heterozygous missense mutations in the U2AF1 and SF3B1 genes that encode spliceosome subunits have been described. SF3B1 mutation is prevalent in both low-risk MDS with ringed sideroblasts and chronic lymphocytic leukemia (CLL) and is associated with a good prognosis. U2AF1 and SRSF2 are frequently mutated in myeloid hematopoietic malignancies, especially in CMML and advanced forms of MDS, and are associated with shorter survival.93-96 SF3B1 null homozygous mice are not viable and heterozygous mutant mice are healthy with reduced levels of transcripts and protein exhibiting skeletal alterations with associated changes in ectopic homeobox transcription factor HOX gene expressions in the vertebrae of embryos, leading to the conclusion that SF3B1 mediates repression of HOX genes.97 HOX gene abnormalities have been implicated in fusions giving rise to myeloid malignancies, as described below. GEP of MDS patients show HOX deregulation with high risk of transformation to AML98 and that HOX upregulation is associated with poor prognosis AML,99 although no study has yet correlated the deregulation of HOX genes with SF3B1 mutations. However, the study in mouse97 shows that SF3B1 spliceosomal protein is required to repress HOX genes, so there may well be a link between these two genes in the context of human disease, as predicted by the mouse model.
An epigenetic regulator belonging to the polycomb group of proteins, ASXL1, is involved in the activation and silencing of HOX genes and ASXL1 mutations do occur in a proportion of MDS patients and are associated with poor prognosis.100-102 However, the ASXL1-deficient mice have a mild phenotype with defects in differentiation of progenitors and do not develop MDS, possibly due to redundancy of pathways.103
These abnormalities and deregulation of target genes identify a ubiquitous pathway that undoubtedly contributes to malignancy.
Tumor suppressors
The p53 protein, a tumor suppressor involved in many types of cancer, has been reported to be mutated in up to 10% of MDS patients with high-risk subtypes.28 Although p53 KO mice are highly susceptible to develop AML after benzene exposure,104 and mutant p53 KI mice are susceptible to develop cancers such as lymphoma, thymoma and sarcoma, no signs of MDS have been reported in association with p53 mutations in these animals. Many pathways converge to the p53 pathway leading to deregulation of apoptosis. Examples are the RPS14 and SPARC described above as potential candidates of the 5q- deletion have increased p53 expression.17,91,105 Reports describing the increased expression of the tumor suppressor gene p53 in 5q- syndrome patients, and the partial rescue of the phenotype observed when Cd74-Nid67 deficient mice are crossed with p53-/- mice, provide further insight into the development of the disease. Upregulation of p53 has been observed in a subset of patients. RPS19 deficiency also activates p53 and, like RPS14 deficiency, is associated with dyserythropoiesis.105
Likewise, although lack of the IRF-1 gene expression has been observed in 100% of MDS patients assessed in one study,106,107 IRF-1-/- mice show no sign of MDS development despite impaired granulocytic differentiation and decreased expression of C/EBPα and C/EBPɛ in CD11b+ cells.108 This tends to suggest that these abnormalities are not sufficient by themselves to confer disease.
Mutations of the deubiquitinating enzyme BAP1 is found in hereditary cancer syndrome and predisposes to various malignancies, particularly mesothelioma and uveal melanoma. In mice, the BAP1 knockout is lethal. However, when a conditional knockout is employed and deletion as an adult is induced, the mice develop features of human MDS.21 KI mice expressing BAP1 interact with the polycomb proteins ASXL1. These are frequently mutated in MDS101 and ASXL2. The authors postulate that a BAP1/ASXL1 complex may suppress the development of MDS. A heterozygous frameshift mutation of BAP1 was identified in an MDS patient with refractory cytopenias and multi-lineage dysplasia (RCMD, WHO classification),2 similar to the features observed in the mouse model. In GEP studies, BAP1 expression was found to be reduced in CD34+ cells from MDS patients compared to healthy controls,96 consistent with BAP1 being a tumor suppressor. In the mouse model, loss of only 1 copy of BAP1 (Bap1fl/+) was shown to have a mild phenotype but had progressive hematologic defects, as predicted by the heterozygous mutation found in the MDS patient. The identification of additional chromosomal deletions associated with MDS such as 7q- and 20q, suggest that a tumor suppressor gene may map to these regions. Mice with a 7q22 deletion have been created but no phenotype was found, and no cooperation was found with oncogenes. Interestingly, the death inducer-obliterator (Dido) gene, which maps to 20q, has been targeted in mice and when deleted gives rise to a transplantable hematologic disease with features consistent with MDS/MPN.109
Mouse models of activated signaling pathways in MDS, MDS/MPN and MDS/AML
Mutations of genes coding for proteins of the RAS signaling pathway have been reported to participate in the pathogenesis of MDS. The RAS pathway is complex and integrated within a network of pathways known to be implicated in cell proliferation, differentiation and apoptosis. Deregulation of the RAS pathway may result from genetic alterations of the genes coding for tyrosine kinase receptors or RAS/GTPase activating protein (GAP)-related proteins.
RAS mutations have been described in MDS and MDS/MPN patients correlating with prognosis in earlier studies;28-30 mutations have been identified in up to 20% of patients with MDS. Point mutations interfere with the intrinsic activity of RAS and result in constitutive signaling and subsequent activation of downstream components. NRAS mutations are more frequent in myeloid malignancies than KRAS mutations, while HRAS mutations are rare. The most common NRAS mutations are found at residue G12 in the P-loop and at the catalytic residue Q61. The glycine to valine mutation at residue 12 renders the RAS GTPase domain insensitive to inactivation by GAP, which causes the protein to remain in an ‘on’ state, whereas the glutamine to lysine mutation at residue 61 reduces the rate of RAS-GTP hydrolysis. NRAS mutations are associated with a poor prognosis in MDS patients and an increased rate of transformation to AML.28,30 Over the past ten years, mouse models have been established to study the effects of increased signaling through the RAS pathway. The results of a comparative analysis of the different models emphasize that the promoter determines the temporal and the spatial expression of the mutant gene as well as the strength of its expression. Activation of the RAS pathway may also be able to block differentiation110 and induce AML, either when expressed at high levels, by activation of the pathways downstream of RAS or with co-expression of other co-operating genes.
RAS mutation mouse models of MDS/MPN
Several transplantation assays using retrovirally-infected BM cells from donor mice to express mutant NRASD12 in murine hematopoietic cells give rise to an MDS/MPN (CMML)-like,32,37 and AML-like32 diseases. Transgenic mice expressing KRASG12D from its endogenous locus using an Mx1Cre system also develop a rapid and lethal MDS/MPN with leukocytosis, monocytosis, severe anemia and BM features of dyserythropoiesis resembling JMML and CMML.35,36,111,112 Furthermore, mice expressing NRASG12D from its endogenous locus (the Mx1Cre Lox-STP-Lox (LSL)-NRASG12D mice) develop an indolent MPD with a long latency, and eventually die of MPD, MDS-like lymphoproliferation found concomitant with myeloid disease and histiocytic sarcoma. Together with the MOL4070LTR retrovirus, these mice develop AML,34 stressing once again the requirement of genetic events working together for the onset of disease. Overexpression of EVI1 as a co-operating event for AML transformation was identified. A transgenic model expressing mutant NRASG12V via the VAV promoter113 and rt/BMT models expressing either MRASQ71L (a recently identified RAS isoform, also known as RRAS) or HRASG12V develop mast cell MPN-like diseases including mastocytosis, mast cell leukemia and malignant histiocytosis.114
Mutated RAS mouse models of MDS and MDS/AML
Kogan et al. have described a transgenic mouse model expressing mutant NRASD12 under the control of the myeloid promoter MRP8 with a phenotype associated with dysplastic features in the myeloid BM.115 Crossing NRASD12 and hBCL-2 transgenic mice yields two distinct models of MDS and AML with MDS-related changes depending on the promoter driving BCL-2 expression: when the expression of the MRP8-driven NRASD12 transgene is associated with the expression of the hBCL-2 gene driven by the MMTV-LTR promoter, mice develop a disease resembling human MDS with excess BM blasts or MDS-like and increased apoptosis. However, when both transgenes, NRAS and hBCL2, are driven by the same MRP8 promoter, the mice develop a disease with characteristics of human AML, high BM blast counts, and persistence of MDS dysplastic features in myeloid cells, and an apoptosis resistance profile. Expanded LSK (Lin-/Sca-1+/c-Kit+) populations and increased hBCL-2 expression in the RAS-GTP complex within the expanded Sca-1+ compartment are observed in both disease models. The diseases are transplantable, which underscores the view that these genetic alterations are functional at the stem cell level.33
In the MDS-like mouse model, most RAS and BCL-2 double-staining localizes to the plasma membrane where active pro-apoptotic RAS is normally located, whereas in the AML post-MDS disease, RAS and BCL-2 co-localize at the mitochondria, where anti-apoptotic BCL-2 is normally found. The co-localization of NRAS and BCL2 was also found in MDS patients.33 The apoptotic profile of these NRASD12/hBCL-2 mice has been characterized by terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end-labeling (TUNEL) of liver sections showing increased TUNEL in the single mutant NRASD12 mice and the MMTVtTA/TBCL-2/NRASD12 mice.33 Analysis of the AML post-MDS model shows a stepwise increase in double-strand breaks, increased misrepair and increased ROS production.71
Other RAS pathway mouse models of MPN/MDS
Approximately 30% of MDS/MPN JMML patients harbor an NF1 gene mutation, and approximately 14% of JMML patients are also diagnosed with neurofibromatosis 1.41 NF1 encodes neurofibromin, a tumor suppressor gene product that has a GAP function. Transgenic mice created using the inducible Mix1-cre system to express an inactive, mutant NF1 protein develop a progressive and fatal MDS/MPN with granulocytosis, monocytosis and splenomegaly, thus representing an appropriate model for JMML.42 Likewise, PTPN11 encodes the tyrosine phosphatase SHP2, which is frequently mutated in JMML patients (34%).43 Chan et al. have developed an inducible KI mouse model that expresses the inactive PTPN11D61Y mutation, these mice die of a fatal myeloproliferative disease (MPD). The selective hypersensitivity of BM cells to GM-CSF, the hallmark of JMML, confirms the JMML model.44
Abnormalities of the RAS signaling pathways tend to suggest that these mouse models are master regulators and in some cases, depending on the context, can alone give rise to rapid disease.
Tyrosine kinases
Amongst the genes coding for tyrosine kinase receptors, the FLT-3 Internal Tandem Duplication (ITD) mutation is found in 5% of MDS patients including CMML,116 while mutation incidence is 33% for patients who progress to AML.117 RAS mutations and FLT3 ITD have rarely been found together, suggesting that their pathways are convergent or that only one of these pathways needs to be altered to give rise to disease. Various methods have been used to express FLT3-ITD in hematopoietic cells in animal models, which mainly develop an MPN phenotype.118-120 In particular, two KI models closely resemble human CMML, exhibiting leukocytosis, monocytosis and splenomegaly.38,121
Translocations involving TEL, a member of the ETS transcription factor family, have been found to be associated with MDS and MDS/MPN. The fusion partners of TEL identified in association with MDS include SYK, MDS2, CDX2 and MN1, while the expression of the TEL-PDGFR fusion protein has been shown to be associated with CMML. The translocation t(5;12) (TEL-PGFRβ) rarely occurs in MPN and MDS.39 Murine transplantation of BM cells transfected with TEL-PDGFRβ results in the development of an MDS/MPN disease resembling CMML.40
MDS mouse models of altered transcription factor and nuclear factor expression in MDS/MPN
Unlike in AML, alterations in transcriptional factor activity resulting from chromosomal translocations are less frequent in MDS (13-16%).122,123 Nevertheless, several transcription factor genes have been reported to be mutated or truncated in MDS, including RUNX1 (AML1), CBFβ, C/EBPα, TEL, MLL, EVI1, RARα, P53, IRF-1, and SALL4.7 Both point mutations in the RUNT domain and truncation of the C-terminus domain of the RUNX1 (AML1) gene are found in 15-40% of MDS patients with excess blasts.7 Mice expressing the RUNX1 (AML1) S291fs mutation develop MDS with excess blasts and erythroid dysplasia.51 TEL has been discussed above.
Abnormalities of EVI1 (ectopic viral integration 1), such as Inv3, t(3;3), t(3;21), are found in up to 2% of MDS patients.45-47 EVI1 is a nuclear transcription factor that is required for normal hematopoiesis; EVI1 deficient mice die as embryos and it has been shown to be essential for the maintenance of HSCs.124-126 GATA-2, essential for proliferation of definitive HSCs, is one of the targets of EVI1 and is reduced in HSCs of EVI1 deficient mouse embryos. Expression of EVI1 is frequently increased in MDS48 and associated with poor prognosis in AML patients.127 Transplantation of lineage-negative BM cells infected with the MSCV retrovirus containing the EVI1 gene induces fatal MDS with pancytopenia, hypercellular BM, dyserythropoiesis and dysmegakaryopoiesis through molecular inhibition of GATA-1 by EVI1.49,128 Transgenic mice with EVI1 driven by the Sca1 promoter had impaired erythropoiesis129 and increased susceptibility to development of myeloid leukemia upon retroviral infection of newborn mice with the Cas-Br-M MuLV.
Translocations involving NUP98 have been identified in both MDS and therapy-related MDS (t-MDS) patients.52,130,131 Nucleoporins are molecules involved in the nuclear import and export of both proteins and RNA. NUP98 has been identified as a fusion partner of 19 various proteins classified into two groups: the homeobox genes (HOXA9, HOXD13 and PMX1) and non-homeobox genes (DDX10, RNA helicase; and TOP1, human DNA topoisomerase 1).52,132 NUP98-HOXD13 fusions under the control of the VAV promoter develop fatal MDS within 14 months.52-54,130,131 CBFβ-MYH11-mediated translocation (inversion 16) is not frequently found in MDS but is associated with t-MDS with eosinophilia.55 CBFβ-MYH11 transgenic mice expressing the fusion protein under the control of the MRP8 promoter have been reported to develop dysgranulopoiesis115 whereas the CBF-MYH11 KI mouse model develops AML.56 Chromosomal translocations involving MLL, such as t(11;16)(MLL-CBP)58,59 and t(11;19),57 as well as MLL gene tandem duplications (MLL-PTD),7 have been described in MDS and t-MDS, although frequencies are low. However, none of the MLL-AF9 mouse models, whether it be KI61 or retroviral BM transduction/transplantation assay (rt/BMT) models, appear to develop MDS, and overt AML rapidly develops.62,133 The conditional MLL-ENL mice develop MPD early (within 7 months) and later develop AML. This can be accelerated by treating with caffeine, an inhibitor of ATM and ATR kinases which are part of the fail-safe DNA damage response machinery,63 thus, demonstrating the importance of GI in the development of disease. Likewise, KI mice with CEBPα mutations develop AML.66
SALL4 overexpression has also been detected in MDS.67,134,135 Interestingly, transgenic mice expressing the SALL4B isoform under the control of the CMV promoter develop MDS-like features within 6-8 months, with dysplastic neutrophils, erythrocytes and megakaryocytes, as well as increased blast counts leading to AML transformation in 50% of mice.67
Growth factors
Myeloid differentiation-associated cytokine pathways have been implicated in MDS. Approximately 40% of patients with severe congenital neutropenia express truncated granulocyte colony-stimulating factor receptor (G-CSFR) protein, and those who develop secondary MDS typically harbor an acquired G-CSFR mutation.136 Alterations in erythropoietin (EPO), granulocyte-macrophage colony-stimulating factor (GM-CSF) and G-CSF signaling have been recapitulated in mice. Alone they do not induce MDS but instead confer maturation disturbances, proliferative advantage of an abnormal clone, and consequent effects on apoptosis.137-139 The macrophage colony-stimulating factor receptor (M-CSFR) encoded by FMS is mutated in approximately 12% of MDS patients and 20% of CMML patients and is associated in some cases with a poor outcome.140 CSF-1 deficient osteopetrotic Op/Op mice display a macrophage deficiency and monocytopenia as well as defective bone formation, but do not develop MDS.141-143
Conclusions
To engineer relevant animal models of MDS, we must ask what genetic alterations are important, how many genetic events are required, and in which stem cells should they be expressed for the development of a given MDS disease in patients. However, this review emphasizes that the expression or inactivation of one such candidate gene in mouse models can generate various disease profiles, MDS, MDS/MPN and AML, depending on the vector, the promoter or the mouse strain with a predominant description of the MDS/MPN or AML phenotypes. This suggests that, very often, the driver of the malignant clone is proliferation and/or that induction of dysmyelopoiesis with increased apoptosis is either rare or difficult to analyze. The Knudson 2-hit hypothesis of loss of one allele and mutation of the other in the case of p53 may not be appropriate for MDS as time is needed for disease to develop; the age of onset in the majority of cases is 65-75 years. This suggests that multiple steps may be required. It is also feasible that mutations in a gene that controls chromatin structure/methylation may hit several genes at once, and again such a mutation could initiate the disease but will still need other events for the disease to develop if it is to resemble the human condition. This has hampered the identification and establishment of true MDS mouse models. Nevertheless, this review shows that there are now a number of mouse models that may qualify as bona fide MDS models based on their pro-apoptotic features, and possibly more may qualify once their apoptotic status has been investigated. It is clear from the use of the emerging technologies of gene expression profiling, sequencing and epigenomics that the expression of one gene or transgene in human or mouse stem cells results from and in the deregulation of a number of networks and pathways. Increased genomic instability as a result of increased inflammation, irradiation, genotoxic stress, and mutagens, is one way of generating mutations, deletions, and translocations, and these alterations in turn enhance genomic instability. Thus mouse models may serve as tools that can be used to investigate the integrity of the genome and dissect the various altered pathways. The identification of the NRAS:BCL2 complex in MDS and MDS-AML patients from data observed in MDS and AML-post MDS mouse models is one such example.
Of the animal models for which apoptosis has been identified, the genes used include NRASD12 in co-operation with BCL-2,33 transcription factors of the Class II type EVI1,49 Sall4B,67 genes important in ribosome biogenesis NPM,13 RPS14,17 NUP98-HOXD13,53,54 and the microenvironment Dicer 1,22 which appear to confer MDS-like diseases.
However, despite the power of mouse model-based biology, there is no getting away from the criticism that mice are not men. The ongoing development of immunodeficient mice that can be xenografted with human MDS BM hematopoietic and/or stromal cells offers a further level of understanding of MDS-genesis and a patient-tailored study that is not limited to one or few transgenes.
Finally, the potential of these mice as pre-clinical models is unique as they allow us to test the drugs required to target a specific pathway (apoptosis, proliferation, differentiation), a disease status (MDS, MDS/MPD or AML), and hopefully in the near future a diseased stem cell (MDS, AML or mesenchymal). Furthermore, mouse models that resemble MDS transformation to AML further highlight the possibility of discovering drugs that may either prevent transformation or allow us to reverse an AML to an MDS phenotype. Recent clinical data have stressed the survival advantage obtained with such therapeutic approaches.
Acknowledgments
SB is a recipient of an INSERM fellowship. RAP and CC are members of the COST action BM0801 and the European Leukemia Network.
Footnotes
Authorship and Disclosures: Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Hebeda KM, Fend F. Changed concepts and definitions of myeloproliferative neoplasms (MPN), myelodysplastic syndromes (MDS) and myelodysplastic/myeloproliferative neoplasms (MDS/MPN) in the updated 2008 WHO classification. J Hematop. 2009;2(4):205-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114(5):937-51 [DOI] [PubMed] [Google Scholar]
- 3.Parker JE, Mufti GJ. Ineffective haemopoiesis and apoptosis in myelodysplastic syndromes. Br J Haematol. 1998;101(2):220-30 [DOI] [PubMed] [Google Scholar]
- 4.Rajapaksa R, Ginzton N, Rott LS, Greenberg PL. Altered oncoprotein expression and apoptosis in myelodysplastic syndrome marrow cells. Blood. 1996;88(11):4275-87 [PubMed] [Google Scholar]
- 5.Bennett JM. World Health Organization classification of the acute leukemias and myelodysplastic syndrome. Int J Hematol. 2000;72(2):131-3 [PubMed] [Google Scholar]
- 6.Harris NL, Jaffe ES, Diebold J, Flandrin G, Muller-Hermelink HK, Vardiman J, et al. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues: report of the Clinical Advisory Committee meeting-Airlie House, Virginia, November 1997. J Clin Oncol. 1999;17(12):3835-49 [DOI] [PubMed] [Google Scholar]
- 7.Dicker F, Haferlach C, Sundermann J, Wendland N, Weiss T, Kern W, et al. Mutation analysis for RUNX1, MLL-PTD, FLT3-ITD, NPM1 and NRAS in 269 patients with MDS or secondary AML. Leukemia. 2010;24(8):1528-32 [DOI] [PubMed] [Google Scholar]
- 8.Akagi T, Ogawa S, Dugas M, Kawamata N, Yamamoto G, Nannya Y, et al. Frequent genomic abnormalities in acute myeloid leukemia/myelodysplastic syndrome with normal karyotype. Haematologica. 2009; 94(2):213-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tormo M, Marugan I, Calabuig M. Myelodysplastic syndromes: an update on molecular pathology. Clin Transl Oncol. 2010;12(10):652-61 [DOI] [PubMed] [Google Scholar]
- 10.Arber DA, Chang KL, Lyda MH, Bedell V, Spielberger R, Slovak ML. Detection of NPM/MLF1 fusion in t(3;5)-positive acute myeloid leukemia and myelodysplasia. Hum Pathol. 2003;34(8):809-13 [DOI] [PubMed] [Google Scholar]
- 11.Yoneda-Kato N, Look AT, Kirstein MN, Valentine MB, Raimondi SC, Cohen KJ, et al. The t(3;5)(q25.1;q34) of myelodysplastic syndrome and acute myeloid leukemia produces a novel fusion gene, NPM-MLF1. Oncogene. 1996;12(2):265-75 [PubMed] [Google Scholar]
- 12.Bains A, Luthra R, Medeiros LJ, Zuo Z. FLT3 and NPM1 mutations in myelodysplastic syndromes: Frequency and potential value for predicting progression to acute myeloid leukemia. Am J Clin Pathol. 2011;135(1):62-9 [DOI] [PubMed] [Google Scholar]
- 13.Grisendi S, Bernardi R, Rossi M, Cheng K, Khandker L, Manova K, et al. Role of nucleophosmin in embryonic development and tumorigenesis. Nature. 2005;437(7055):147-53 [DOI] [PubMed] [Google Scholar]
- 14.Sportoletti P, Grisendi S, Majid SM, Cheng K, Clohessy JG, Viale A, et al. Npm1 is a haploinsufficient suppressor of myeloid and lymphoid malignancies in the mouse. Blood. 2008;111(7):3859-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ebert BL, Pretz J, Bosco J, Chang CY, Tamayo P, Galili N, et al. Identification of RPS14 as a 5q-syndrome gene by RNA interference screen. Nature. 2008;451(7176):335-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eisenmann KM, Dykema KJ, Matheson SF, Kent NF, Deward AD, West RA, et al. 5q-myelodysplastic syndromes: chromosome 5q genes direct a tumor-suppression network sensing actin dynamics. Oncogene. 2009;28(39):3429-41 [DOI] [PubMed] [Google Scholar]
- 17.Barlow JL, Drynan LF, Hewett DR, Holmes LR, Lorenzo-Abalde S, Lane AL, et al. A p53-dependent mechanism underlies macrocytic anemia in a mouse model of human 5q- syndrome. Nat Med. 2010;16(1):59-66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lehmann S, O'Kelly J, Raynaud S, Funk SE, Sage EH, Koeffler HP. Common deleted genes in the 5q- syndrome: thrombocytopenia and reduced erythroid colony formation in SPARC null mice. Leukemia. 2007;21(9):1931-6 [DOI] [PubMed] [Google Scholar]
- 19.Lane SW, Sykes SM, Al-Shahrour F, Shterental S, Paktinat M, Lo CC, et al. The Apc(min) mouse has altered hematopoietic stem cell function and provides a model for MPD/MDS. Blood. 2010;115(17):3489-97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang J, Fernald AA, Anastasi J, Le Beau MM, Qian Z. Haploinsufficiency of Apc leads to ineffective hematopoiesis. Blood. 2010;115(17):3481-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dey A, Seshasayee D, Noubade R, French DM, Liu J, Chaurushiya MS, et al. Loss of the tumor suppressor BAP1 causes myeloid transformation. Science. 2012;337(6101):1541-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raaijmakers MH, Mukherjee S, Guo S, Zhang S, Kobayashi T, Schoonmaker JA, et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature. 2010;464(7290):852-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tefferi A, Lim KH, Abdel-Wahab O, Lasho TL, Patel J, Patnaik MM, et al. Detection of mutant TET2 in myeloid malignancies other than myeloproliferative neoplasms: CMML, MDS, MDS/MPN and AML. Leukemia. 2009;23(7):1343-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Delhommeau F, Dupont S, Della VV, James C, Trannoy S, Masse A, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360(22):2289-301 [DOI] [PubMed] [Google Scholar]
- 25.Tefferi A, Pardanani A, Lim KH, Abdel-Wahab O, Lasho TL, Patel J, et al. TET2 mutations and their clinical correlates in polycythemia vera, essential thrombocythemia and myelofibrosis. Leukemia. 2009;23(5):905-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quivoron C, Couronné L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell. 2011;20(1):25-38 [DOI] [PubMed] [Google Scholar]
- 27.Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20(1):11-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Padua RA, Guinn BA, Al-Sabah AI, Smith M, Taylor C, Pettersson T, et al. RAS, FMS and p53 mutations and poor clinical outcome in myelodysplasias: a 10-year follow-up. Leukemia. 1998;12(6):887-92 [DOI] [PubMed] [Google Scholar]
- 29.Bowen DT, Frew ME, Hills R, Gale RE, Wheatley K, Groves MJ, et al. RAS mutation in acute myeloid leukemia is associated with distinct cytogenetic subgroups but does not influence outcome in patients younger than 60 years. Blood. 2005;106(6):2113-9 [DOI] [PubMed] [Google Scholar]
- 30.Paquette RL, Landaw EM, Pierre RV, Kahan J, Lubbert M, Lazcano O, et al. N-ras mutations are associated with poor prognosis and increased risk of leukemia in myelodysplastic syndrome. Blood. 1993;82(2):590-9 [PubMed] [Google Scholar]
- 31.MacKenzie KL, Dolnikov A, Millington M, Shounan Y, Symonds G. Mutant N-ras induces myeloproliferative disorders and apoptosis in bone marrow repopulated mice. Blood. 1999;93(6):2043-56 [PubMed] [Google Scholar]
- 32.Parikh C, Subrahmanyam R, Ren R. Oncogenic NRAS rapidly and efficiently induces CMML- and AML-like diseases in mice. Blood. 2006;108(7):2349-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Omidvar N, Kogan S, Beurlet S, le Pogam C, Janin A, West R, et al. BCL-2 and mutant NRAS interact physically and functionally in a mouse model of progressive myelodysplasia. Cancer Res. 2007;67(24):11657-67 [DOI] [PubMed] [Google Scholar]
- 34.Li Q, Haigis KM, McDaniel A, Harding-Theobald E, Kogan SC, Akagi K, et al. Hematopoiesis and leukemogenesis in mice expressing oncogenic NrasG12D from the endogenous locus. Blood. 2011;117(6):2022-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Braun BS, Tuveson DA, Kong N, Le DT, Kogan SC, Rozmus J, et al. Somatic activation of oncogenic Kras in hematopoietic cells initiates a rapidly fatal myeloproliferative disorder. Proc Natl Acad Sci USA. 2004;101(2):597-602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Van Meter ME, Diaz-Flores E, Archard JA, Passegue E, Irish JM, Kotecha N, et al. K-RasG12D expression induces hyperproliferation and aberrant signaling in primary hematopoietic stem/progenitor cells. Blood. 2007;109(9):3945-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parikh C, Subrahmanyam R, Ren R. Oncogenic NRAS, KRAS, and HRAS exhibit different leukemogenic potentials in mice. Cancer Res. 2007;67(15):7139-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee BH, Tothova Z, Levine RL, Anderson K, Buza-Vidas N, Cullen DE, et al. FLT3 mutations confer enhanced proliferation and survival properties to multipotent progenitors in a murine model of chronic myelomonocytic leukemia. Cancer Cell. 2007;12(4):367-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Greipp PT, Dewald GW, Tefferi A. Prevalence, breakpoint distribution, and clinical correlates of t(5;12). Cancer Genet Cytogenet. 2004;153(2):170-2 [DOI] [PubMed] [Google Scholar]
- 40.Stover EH, Chen J, Lee BH, Cools J, McDowell E, Adelsperger J, et al. The small molecule tyrosine kinase inhibitor AMN107 inhibits TEL-PDGFRbeta and FIP1L1-PDGFRalpha in vitro and in vivo. Blood. 2005;106(9):3206-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Side L, Taylor B, Cayouette M, Conner E, Thompson P, Luce M, et al. Homozygous inactivation of the NF1 gene in bone marrow cells from children with neurofibromatosis type 1 and malignant myeloid disorders. N Engl J Med. 1997;336(24):1713-20 [DOI] [PubMed] [Google Scholar]
- 42.Le DT, Kong N, Zhu Y, Lauchle JO, Aiyigari A, Braun BS, et al. Somatic inactivation of Nf1 in hematopoietic cells results in a progressive myeloproliferative disorder. Blood. 2004;103(11):4243-50 [DOI] [PubMed] [Google Scholar]
- 43.Tartaglia M, Niemeyer CM, Fragale A, Song X, Buechner J, Jung A, et al. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet. 2003;34(2):148-50 [DOI] [PubMed] [Google Scholar]
- 44.Chan G, Kalaitzidis D, Usenko T, Kutok JL, Yang W, Mohi MG, et al. Leukemogenic Ptpn11 causes fatal myeloproliferative disorder via cell-autonomous effects on multiple stages of hematopoiesis. Blood. 2009;113(18):4414-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mitani K, Ogawa S, Tanaka T, Miyoshi H, Kurokawa M, Mano H, et al. Generation of the AML1-EVI-1 fusion gene in the t(3;21)(q26;q22) causes blastic crisis in chronic myelocytic leukemia. EMBO J. 1994;13(3):504-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mochizuki N, Shimizu S, Nagasawa T, Tanaka H, Taniwaki M, Yokota J, et al. A novel gene, MEL1, mapped to 1p36.3 is highly homologous to the MDS1/EVI1 gene and is transcriptionally activated in t(1;3)(p36;q21)-positive leukemia cells. Blood. 2000;96(9):3209-14 [PubMed] [Google Scholar]
- 47.Suzukawa K, Parganas E, Gajjar A, Abe T, Takahashi S, Tani K, et al. Identification of a breakpoint cluster region 3′ of the ribophorin I gene at 3q21 associated with the transcriptional activation of the EVI1 gene in acute myelogenous leukemias with inv(3)(q21q26). Blood. 1994;84(8):2681-8 [PubMed] [Google Scholar]
- 48.Russell M, List A, Greenberg P, Woodward S, Glinsmann B, Parganas E, et al. Expression of EVI1 in myelodysplastic syndromes and other hematologic malignancies without 3q26 translocations. Blood. 1994;84(4):1243-8 [PubMed] [Google Scholar]
- 49.Laricchia-Robbio L, Fazzina R, Li D, Rinaldi CR, Sinha KK, Chakraborty S, et al. Point mutations in two EVI1 Zn fingers abolish EVI1-GATA1 interaction and allow erythroid differentiation of murine bone marrow cells. Mol Cell Biol. 2006;26(20):7658-66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harada Y, Harada H. Molecular pathways mediating MDS/AML with focus on AML1/RUNX1 point mutations. J Cell Physiol. 2009;220(1):16-20 [DOI] [PubMed] [Google Scholar]
- 51.Watanabe-Okochi N, Kitaura J, Ono R, Harada H, Harada Y, Komeno Y, et al. AML1 mutations induced MDS and MDS/AML in a mouse BMT model. Blood. 2008;111(8):4297-308 [DOI] [PubMed] [Google Scholar]
- 52.Lam DH, Aplan PD. NUP98 gene fusions in hematologic malignancies. Leukemia 2001;15(11):1689-95 [DOI] [PubMed] [Google Scholar]
- 53.Choi CW, Chung YJ, Slape C, Aplan PD. Impaired differentiation and apoptosis of hematopoietic precursors in a mouse model of myelodysplastic syndrome. Haematologica. 2008;93(9):1394-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Slape C, Lin YW, Hartung H, Zhang Z, Wolff L, Aplan PD. NUP98-HOX translocations lead to myelodysplastic syndrome in mice and men. J Natl Cancer Inst Monogr. 2008;(39):64-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leroy H, Roumier C, Grardel-Duflos N, Macintyre E, Lepelley P, Fenaux P, et al. Unlike AML1, CBFbeta gene is not deregulated by point mutations in acute myeloid leukemia and in myelodysplastic syndromes. Blood. 2002;99(10):3848-50 [DOI] [PubMed] [Google Scholar]
- 56.Kuo YH, Gerstein RM, Castilla LH. Cbfβ-SMMHC impairs differentiation of common lymphoid progenitors and reveals an essential role for RUNX in early B-cell development. Blood. 2008;111(3):1543-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Block AW, Carroll AJ, Hagemeijer A, Michaux L, van LK, Olney HJ, et al. Rare recurring balanced chromosome abnormalities in therapy-related myelodysplastic syndromes and acute leukemia: report from an international workshop. Genes Chromosomes Cancer. 2002;33(4):401-12 [DOI] [PubMed] [Google Scholar]
- 58.Rowley JD, Reshmi S, Sobulo O, Musvee T, Anastasi J, Raimondi S, et al. All patients with the T(11;16)(q23;p13.3) that involves MLL and CBP have treatment-related hematologic disorders. Blood. 1997;90(2):535-41 [PubMed] [Google Scholar]
- 59.Taki T, Sako M, Tsuchida M, Hayashi Y. The t(11;16)(q23;p13) translocation in myelodysplastic syndrome fuses the MLL gene to the CBP gene. Blood. 1997;89(11):3945-50 [PubMed] [Google Scholar]
- 60.Andersen MK, Christiansen DH, Kirchhoff M, Pedersen-Bjergaard J. Duplication or amplification of chromosome band 11q23, including the unrearranged MLL gene, is a recurrent abnormality in therapy-related MDS and AML, and is closely related to mutation of the TP53 gene and to previous therapy with alkylating agents. Genes Chromosomes Cancer. 2001;31(1):33-41 [DOI] [PubMed] [Google Scholar]
- 61.Johnson JJ, Chen W, Hudson W, Yao Q, Taylor M, Rabbitts TH, et al. Prenatal and postnatal myeloid cells demonstrate stepwise progression in the pathogenesis of MLL fusion gene leukemia. Blood. 2003;101(8):3229-35 [DOI] [PubMed] [Google Scholar]
- 62.Chen W, Kumar AR, Hudson WA, Li Q, Wu B, Staggs RA, et al. Malignant transformation initiated by Mll-AF9: gene dosage and critical target cells. Cancer Cell. 2008;13(5):432-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Takacova S, Slany R, Bartkova J, Stranecky V, Dolezel P, Luzna P, et al. DNA Damage Response and Inflammatory Signaling Limit the MLL-ENL-Induced Leukemogenesis In Vivo. Cancer Cell. 2012;21(4):517-31 [DOI] [PubMed] [Google Scholar]
- 64.Fuchs O, Provaznikova D, Kocova M, Kostecka A, Cvekova P, Neuwirtova R, et al. CEBPA polymorphisms and mutations in patients with acute myeloid leukemia, myelodysplastic syndrome, multiple myeloma and non-Hodgkin's lymphoma. Blood Cells Mol Dis. 2008;40(3):401-5 [DOI] [PubMed] [Google Scholar]
- 65.Gombart AF, Hofmann WK, Kawano S, Takeuchi S, Krug U, Kwok SH, et al. Mutations in the gene encoding the transcription factor CCAAT/enhancer binding protein alpha in myelodysplastic syndromes and acute myeloid leukemias. Blood. 2002;99(4):1332-40 [DOI] [PubMed] [Google Scholar]
- 66.Kirstetter P, Schuster MB, Bereshchenko O, Moore S, Dvinge H, Kurz E, et al. Modeling of C/EBPalpha mutant acute myeloid leukemia reveals a common expression signature of committed myeloid leukemia-initiating cells. Cancer Cell. 2008;13(4):299-310 [DOI] [PubMed] [Google Scholar]
- 67.Ma Y, Cui W, Yang J, Qu J, Di C, Amin HM, et al. SALL4, a novel oncogene, is constitutively expressed in human acute myeloid leukemia (AML) and induces AML in transgenic mice. Blood. 2006;108(8):2726-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sallmyr A, Fan J, Rassool FV. Genomic instability in myeloid malignancies: increased reactive oxygen species (ROS), DNA double strand breaks (DSBs) and error-prone repair. Cancer Lett. 2008;270(1):1-9 [DOI] [PubMed] [Google Scholar]
- 69.Lushnikova T, Bouska A, Odvody J, Dupont WD, Eischen CM. Aging mice have increased chromosome instability that is exacerbated by elevated Mdm2 expression. Oncogene. 2011;30(46):4622-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Heilig CE, Loffler H, Mahlknecht U, Janssen JW, Ho AD, Jauch A, et al. Chromosomal instability correlates with poor outcome in patients with myelodysplastic syndromes irrespectively of the cytogenetic risk group. J Cell Mol Med. 2010;14(4):895-902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rassool FV, Gaymes TJ, Omidvar N, Brady N, Beurlet S, Pla M, et al. Reactive oxygen species, DNA damage, and error-prone repair: a model for genomic instability with progression in myeloid leukemia? Cancer Res. 2007;67(18):8762-71 [DOI] [PubMed] [Google Scholar]
- 72.Soulier J. Fanconi anemia. Hematology Am Soc Hematol Educ Program. 2011;2011492-7 [DOI] [PubMed] [Google Scholar]
- 73.Aube M, Lafrance M, Charbonneau C, Goulet I, Carreau M. Hematopoietic stem cells from fancc(-/-) mice have lower growth and differentiation potential in response to growth factors. Stem Cells. 2002;20(5):438-47 [DOI] [PubMed] [Google Scholar]
- 74.Pulliam-Leath AC, Ciccone SL, Nalepa G, Li X, Si Y, Miravalle L, et al. Genetic disruption of both Fancc and Fancg in mice recapitulates the hematopoietic manifestations of Fanconi anemia. Blood. 2010;116(16):2915-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tien HF, Tang JH, Tsay W, Liu MC, Lee FY, Wang CH, et al. Methylation of the p15(INK4B) gene in myelodysplastic syndrome: it can be detected early at diagnosis or during disease progression and is highly associated with leukaemic transformation. Br J Haematol. 2001;112(1):148-54 [DOI] [PubMed] [Google Scholar]
- 76.Drexler HG. Review of alterations of the cyclin-dependent kinase inhibitor INK4 family genes p15, p16, p18 and p19 in human leukemia-lymphoma cells. Leukemia. 1998;12(6):845-59 [DOI] [PubMed] [Google Scholar]
- 77.Rosu-Myles M, Taylor BJ, Wolff L. Loss of the tumor suppressor p15Ink4b enhances myeloid progenitor formation from common myeloid progenitors. Exp Hematol. 2007;35(3):394-406 [DOI] [PubMed] [Google Scholar]
- 78.Greco M, D'Alo F, Scardocci A, Criscuolo M, Fabiani E, Guidi F, et al. Promoter methylation of DAPK1, E-cadherin and thrombospondin-1 in de novo and therapy-related myeloid neoplasms. Blood Cells Mol Dis. 2010;45(3):181-5 [DOI] [PubMed] [Google Scholar]
- 79.Issa JP. Epigenetic changes in the myelodysplastic syndrome. Hematol Oncol Clin North Am. 2010;24(2):317-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dickstein J, Senyuk V, Premanand K, Laricchia-Robbio L, Xu P, Cattaneo F, et al. Methylation and silencing of miRNA-124 by EVI1 and self-renewal exhaustion of hematopoietic stem cells in murine myelodysplastic syndrome. Proc Natl Acad Sci USA. 2010;107(21):9783-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pronier E, Almire C, Mokrani H, Vasanthakumar A, Simon A, da Costa Reis Monte Mor, et al. Inhibition of TET2-mediated conversion of 5-methylcytosine to 5-hydroxymethylcytosine disturbs erythroid and granulomonocytic differentiation of human hematopoietic progenitors. Blood. 2011;118(9):2551-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kerbauy DM, Lesnikov V, Torok-Storb B, Bryant E, Deeg HJ. Engraftment of distinct clonal MDS-derived hematopoietic precursors in NOD/SCID-beta2-microglobulin-deficient mice after intramedullary transplantation of hematopoietic and stromal cells. Blood. 2004;104(7):2202-3 [DOI] [PubMed] [Google Scholar]
- 83.Thanopoulou E, Cashman J, Kakagianne T, Eaves A, Zoumbos N, Eaves C. Engraftment of NOD/SCID-beta2 microglobulin null mice with multilineage neoplastic cells from patients with myelodysplastic syndrome. Blood. 2004;103(11):4285-93 [DOI] [PubMed] [Google Scholar]
- 84.Zhu DX, Fan L, Lu RN, Fang C, Shen WY, Zou ZJ, et al. Downregulated Dicer expression predicts poor prognosis in chronic lymphocytic leukemia. Cancer Sci. 2012;103(5):875-81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ebert BL. Genetic deletions in AML and MDS. Best Pract Res Clin Haematol. 2010;23(4):457-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Starczynowski DT, Kuchenbauer F, Argiropoulos B, Sung S, Morin R, Muranyi A, et al. Identification of miR-145 and miR-146a as mediators of the 5q- syndrome phenotype. Nat Med. 2010;16(1):49-58 [DOI] [PubMed] [Google Scholar]
- 87.Wegrzyn J, Lam JC, Karsan A. Mouse models of myelodysplastic syndromes. Leuk Res. 2011;35(7):853-62 [DOI] [PubMed] [Google Scholar]
- 88.Draptchinskaia N, Gustavsson P, Andersson B, Pettersson M, Willig TN, Dianzani I, et al. The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia. Nat Genet. 1999;21(2):169-75 [DOI] [PubMed] [Google Scholar]
- 89.Devlin EE, Dacosta L, Mohandas N, Elliott G, Bodine DM. A transgenic mouse model demonstrates a dominant negative effect of a point mutation in the RPS19 gene associated with Diamond-Blackfan anemia. Blood. 2010;116(15):2826-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gao J, Song J, Huang H, Li Z, Du Y, Cao J, et al. Methylation of the SPARC gene promoter and its clinical implication in pancreatic cancer. J Exp Clin Cancer Res. 2010;2928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Suzuki M, Hao C, Takahashi T, Shigematsu H, Shivapurkar N, Sathyanarayana UG, et al. Aberrant methylation of SPARC in human lung cancers. Br J Cancer. 2005;92(5):942-8 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 92.Helleman J, Jansen MP, Ruigrok-Ritstier K, van Staveren IL, Look MP, Meijer-van Gelder ME, et al. Association of an extracellular matrix gene cluster with breast cancer prognosis and endocrine therapy response. Clin.Cancer Res. 2008;14(17):5555-64 [DOI] [PubMed] [Google Scholar]
- 93.Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478(7367):64-9 [DOI] [PubMed] [Google Scholar]
- 94.Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011;365(15):1384-95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Makishima H, Visconte V, Sakaguchi H, Jankowska AM, Abu KS, Jerez A, et al. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood. 2012;119(14):3203-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Graubert TA, Shen D, Ding L, Okeyo-Owuor T, Lunn CL, Shao J, et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat Genet. 2012;44(1):53-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Isono K, Mizutani-Koseki Y, Komori T, Schmidt-Zachmann MS, Koseki H. Mammalian polycomb-mediated repression of Hox genes requires the essential spliceosomal protein Sf3b1. Genes Dev. 2005;19(5):536-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mills KI, Kohlmann A, Williams PM, Wieczorek L, Liu WM, Li R, et al. Microarray-based classifiers and prognosis models identify subgroups with distinct clinical outcomes and high risk of AML transformation of myelodysplastic syndrome. Blood. 2009;114(5):1063-72 [DOI] [PubMed] [Google Scholar]
- 99.Eklund E. The role of Hox proteins in leukemogenesis: insights into key regulatory events in hematopoiesis. Crit Rev Oncog. 2011;16(1-2):65-76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Devillier R, Gelsi-Boyer V, Brecqueville M, Carbuccia N, Murati A, Vey N, et al. Acute myeloid leukemia with myelodysplasia-related changes are characterized by a specific molecular pattern with high frequency of ASXL1 mutations. Am J Hematol. 2012;87(7):659-62 [DOI] [PubMed] [Google Scholar]
- 101.Gelsi-Boyer V, Brecqueville M, Devillier R, Murati A, Mozziconacci MJ, Birnbaum D. Mutations in ASXL1 are associated with poor prognosis across the spectrum of malignant myeloid diseases. J Hematol Oncol. 2012;512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bejar R, Stevenson KE, Caughey BA, Abdel-Wahab O, Steensma DP, Galili N, et al. Validation of a Prognostic Model and the Impact of Mutations in Patients With Lower-Risk Myelodysplastic Syndromes. J Clin Oncol. 2012;30(27):3376-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fisher CL, Pineault N, Brookes C, Helgason CD, Ohta H, Bodner C, et al. Loss-of-function Additional sex combs like 1 mutations disrupt hematopoiesis but do not cause severe myelodysplasia or leukemia. Blood. 2010;115(1):38-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hirabayashi Y, Yoon BI, Li GX, Kanno J, Inoue T. Mechanism of benzene-induced hematotoxicity and leukemogenicity: current review with implication of microarray analyses. Toxicol Pathol. 2004;32(Suppl):212-6 [DOI] [PubMed] [Google Scholar]
- 105.Dutt S, Narla A, Lin K, Mullally A, Abayasekara N, Megerdichian C, et al. Haploinsufficiency for ribosomal protein genes causes selective activation of p53 in human erythroid progenitor cells. Blood. 2011;117(9):2567-76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Harada H, Kondo T, Ogawa S, Tamura T, Kitagawa M, Tanaka N, et al. Accelerated exon skipping of IRF-1 mRNA in human myelodysplasia/leukemia; a possible mechanism of tumor suppressor inactivation. Oncogene. 1994;9(11):3313-20 [PubMed] [Google Scholar]
- 107.Maratheftis CI, Bolaraki PE, Giannouli S, Kapsogeorgou EK, Moutsopoulos HM, Voulgarelis M. Aberrant alternative splicing of interferon regulatory factor-1 (IRF-1) in myelodysplastic hematopoietic progenitor cells. Leuk Res. 2006;30(9):1177-86 [DOI] [PubMed] [Google Scholar]
- 108.Testa U, Stellacci E, Pelosi E, Sestili P, Venditti M, Orsatti R, et al. Impaired myelopoiesis in mice devoid of interferon regulatory factor 1. Leukemia. 2004;18(11):1864-71 [DOI] [PubMed] [Google Scholar]
- 109.Futterer A, Campanero MR, Leonardo E, Criado LM, Flores JM, Hernandez JM, et al. Dido gene expression alterations are implicated in the induction of hematological myeloid neoplasms. J Clin Invest. 2005; 115(9):2351-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Darley RL, Hoy TG, Baines P, Padua RA, Burnett AK. Mutant N-RAS induces erythroid lineage dysplasia in human CD34+ cells. J Exp Med. 1997;185(7):1337-47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Braun BS, Archard JA, Van Ziffle JA, Tuveson DA, Jacks TE, Shannon K. Somatic activation of a conditional KrasG12D allele causes ineffective erythropoiesis in vivo. Blood. 2006;108(6):2041-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chan IT, Kutok JL, Williams IR, Cohen S, Kelly L, Shigematsu H, et al. Conditional expression of oncogenic K-ras from its endogenous promoter induces a myeloproliferative disease. J Clin Invest. 2004;113(4):528-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wiesner SM, Jones JM, Hasz DE, Largaespada DA. Repressible transgenic model of NRAS oncogene-driven mast cell disease in the mouse. Blood. 2005;106(3):1054-62 [DOI] [PubMed] [Google Scholar]
- 114.Guo X, Schrader KA, Xu Y, Schrader JW. Expression of a constitutively active mutant of M-Ras in normal bone marrow is sufficient for induction of a malignant mastocytosis/mast cell leukemia, distinct from the histiocytosis/monocytic leukemia induced by expression of activated H-Ras. Oncogene. 2005;24(14):2330-42 [DOI] [PubMed] [Google Scholar]
- 115.Kogan SC, Lagasse E, Atwater S, Bae SC, Weissman I, Ito Y, et al. The PEBP2betaMYH11 fusion created by Inv(16)(p13;q22) in myeloid leukemia impairs neutrophil maturation and contributes to granulocytic dysplasia. Proc Natl Acad Sci USA. 1998;95(20):11863-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pinheiro RF, de Sa ME, Silva MR, Alberto FL, Chauffaille ML. FLT3 internal tandem duplication during myelodysplastic syndrome follow-up: a marker of transformation to acute myeloid leukemia. Cancer Genet Cytogenet. 2008;183(2):89-93 [DOI] [PubMed] [Google Scholar]
- 117.Shih LY, Huang CF, Wang PN, Wu JH, Lin TL, Dunn P, et al. Acquisition of FLT3 or N-ras mutations is frequently associated with progression of myelodysplastic syndrome to acute myeloid leukemia. Leukemia. 2004;18(3):466-75 [DOI] [PubMed] [Google Scholar]
- 118.Grundler R, Miething C, Thiede C, Peschel C, Duyster J. FLT3-ITD and tyrosine kinase domain mutants induce 2 distinct phenotypes in a murine bone marrow transplantation model. Blood. 2005;105(12):4792-9 [DOI] [PubMed] [Google Scholar]
- 119.Kelly LM, Liu Q, Kutok JL, Williams IR, Boulton CL, Gilliland DG. FLT3 internal tandem duplication mutations associated with human acute myeloid leukemias induce myeloproliferative disease in a murine bone marrow transplant model. Blood. 2002;99(1):310-8 [DOI] [PubMed] [Google Scholar]
- 120.Lee BH, Williams IR, Anastasiadou E, Boulton CL, Joseph SW, Amaral SM, et al. FLT3 internal tandem duplication mutations induce myeloproliferative or lymphoid disease in a transgenic mouse model. Oncogene. 2005;24(53):7882-92 [DOI] [PubMed] [Google Scholar]
- 121.Li L, Piloto O, Nguyen HB, Greenberg K, Takamiya K, Racke F, et al. Knock-in of an internal tandem duplication mutation into murine FLT3 confers myeloproliferative disease in a mouse model. Blood. 2008;111(7):3849-58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Breccia M, Mengarelli A, Mancini M, Biondo F, Gentilini F, Latagliata R, et al. Myelodysplastic syndromes in patients under 50 years old: a single institution experience. Leuk Res. 2005;29(7):749-54 [DOI] [PubMed] [Google Scholar]
- 123.Li L, Liu XP, Nie L, Yu MH, Zhang Y, Qin TJ, et al. Unique cytogenetic features of primary myelodysplastic syndromes in Chinese patients. Leuk Res. 2009;33(9):1194-8 [DOI] [PubMed] [Google Scholar]
- 124.Kataoka K, Sato T, Yoshimi A, Goyama S, Tsuruta T, Kobayashi H, et al. Evi1 is essential for hematopoietic stem cell self-renewal, and its expression marks hematopoietic cells with long-term multilineage repopulating activity. J Exp Med. 2011;208(12):2403-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yuasa H, Oike Y, Iwama A, Nishikata I, Sugiyama D, Perkins A, et al. Oncogenic transcription factor Evi1 regulates hematopoietic stem cell proliferation through GATA-2 expression. EMBO J. 2005;24(11):1976-87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Goyama S, Yamamoto G, Shimabe M, Sato T, Ichikawa M, Ogawa S, et al. Evi-1 is a critical regulator for hematopoietic stem cells and transformed leukemic cells. Cell Stem Cell. 2008;3(2):207-20 [DOI] [PubMed] [Google Scholar]
- 127.van Waalwijk van Doorn-Khosrovani Barjesteh, Erpelinck C, van Putten WL, Valk PJ, van der Poel-van de Luytgaarde, Hack R, et al. High EVI1 expression predicts poor survival in acute myeloid leukemia: a study of 319 de novo AML patients. Blood. 2003;101(3):837-45 [DOI] [PubMed] [Google Scholar]
- 128.Buonamici S, Li D, Chi Y, Zhao R, Wang X, Brace L, et al. EVI1 induces myelodysplastic syndrome in mice. J Clin Invest. 2004;114(5):713-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Louz D, van den Broek M, Verbakel S, Vankan Y, van Lom K, Joosten M, et al. Erythroid defects and increased retrovirally-induced tumor formation in Evi1 transgenic mice. Leukemia. 2000;14(11):1876-84 [DOI] [PubMed] [Google Scholar]
- 130.Beachy SH, Aplan PD. Mouse models of myelodysplastic syndromes. Hematol Oncol Clin North Am. 2010;24(2):361-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lin YW, Slape C, Zhang Z, Aplan PD. NUP98-HOXD13 transgenic mice develop a highly penetrant, severe myelodysplastic syndrome that progresses to acute leukemia. Blood. 2005;106(1):287-95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Romana SP, Radford-Weiss I, Ben AR, Schluth C, Petit A, Dastugue N, et al. NUP98 rearrangements in hematopoietic malignancies: a study of the Groupe Francophone de Cytogenetique Hematologique. Leukemia. 2006;20(4):696-706 [DOI] [PubMed] [Google Scholar]
- 133.Somervaille TC, Cleary ML. Identification and characterization of leukemia stem cells in murine MLL-AF9 acute myeloid leukemia. Cancer Cell. 2006;10(4):257-68 [DOI] [PubMed] [Google Scholar]
- 134.Shuai X, Zhou D, Shen T, Wu Y, Zhang J, Wang X, et al. Overexpression of the novel oncogene SALL4 and activation of the Wnt/beta-catenin pathway in myelodysplastic syndromes. Cancer Genet Cytogenet. 2009;194(2):119-24 [DOI] [PubMed] [Google Scholar]
- 135.Yang J, Chai L, Gao C, Fowles TC, Alipio Z, Dang H, et al. SALL4 is a key regulator of survival and apoptosis in human leukemic cells. Blood. 2008;112(3):805-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Tidow N, Kasper B, Welte K. Clinical implications of G-CSF receptor mutations. Crit Rev Oncol Hematol. 1998;28(1):1-6 [DOI] [PubMed] [Google Scholar]
- 137.Hermans MH, Antonissen C, Ward AC, Mayen AE, Ploemacher RE, Touw IP. Sustained receptor activation and hyperproliferation in response to granulocyte colony-stimulating factor (G-CSF) in mice with a severe congenital neutropenia/acute myeloid leukemia-derived mutation in the G-CSF receptor gene. J Exp Med. 1999; 189(4):683-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Liu F, Kunter G, Krem MM, Eades WC, Cain JA, Tomasson MH, et al. Csf3r mutations in mice confer a strong clonal HSC advantage via activation of Stat5. J Clin Invest. 2008;118(3):946-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Socolovsky M, Nam H, Fleming MD, Haase VH, Brugnara C, Lodish HF. Ineffective erythropoiesis in Stat5a(-/-)5b(-/-) mice due to decreased survival of early erythroblasts. Blood. 2001;98(12):3261-73 [DOI] [PubMed] [Google Scholar]
- 140.Ridge SA, Worwood M, Oscier D, Jacobs A, Padua RA. FMS mutations in myelodysplastic, leukemic, and normal subjects. Proc Natl Acad Sci.USA. 1990;87(4):1377-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lieschke GJ, Grail D, Hodgson G, Metcalf D, Stanley E, Cheers C, et al. Mice lacking granulocyte colony-stimulating factor have chronic neutropenia, granulocyte and macrophage progenitor cell deficiency, and impaired neutrophil mobilization. Blood. 1994;84(6):1737-46 [PubMed] [Google Scholar]
- 142.Wiktor-Jedrzejczak W, Bartocci A, Ferrante AW, Jr, Ahmed-Ansari A, Sell KW, Pollard JW, et al. Total absence of colony-stimulating factor 1 in the macrophage-deficient osteopetrotic (op/op) mouse. Proc Natl Acad Sci USA. 1990;87(12):4828-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Yoshida H, Hayashi S, Kunisada T, Ogawa M, Nishikawa S, Okamura H, et al. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature. 1990;345(6274):442-4 [DOI] [PubMed] [Google Scholar]