

Pediatric acute lymphoblastic leukemia (ALL) can now been cured in the vast majority of cases diagnosed in the developed world. However, the treatment regimes rely on good supportive care and multi-agent chemotherapy; and cannot be delivered in poor countries like Malawi. Therefore, many patients are left untreated. The difficulties in treating hematologic malignancies in sub-Saharan Africa include a lack of medical resources (e.g. chemotherapy agents, supportive care, laboratory support), the inability of families to access basic services, and endemic infections.1 In 2010, the first Malawi ALL protocol was developed jointly by oncology teams in Malawi and the UK. As this was the first attempt to treat ALL in Malawi, it was essential to set realistic goals and be mindful of the limited resources and infrastructure available. Therefore, the primary objective was to induce morphological remission, thereby allowing the children to go home clinically well and participate in normal life. The secondary objectives included delivering the protocol without excessive toxicity, monitoring outcome, and sustaining remission. Based on our experience of treating Burkitt's lymphoma, we knew the following would be critical for successful implementation of the protocol: i) length of hospital stay; ii) number of follow-up appointments; iii) toxicity, as measuring daily electrolyte levels and blood counts is not possible.2,3 Therefore, the protocol was simple, short and only used drugs with proven anti-leukemic effect4-7 (Figure 1). The children were all hyperhydrated (3 L/m 2/24 h) and given allopurinol at the initiation of treatment in an attempt to reduce the risk of tumor lysis. Children with presumed infection or pyrexia were treated with intravenous antibiotics (initially penicillin and genatamicin) after excluding malaria.
Figure 1.
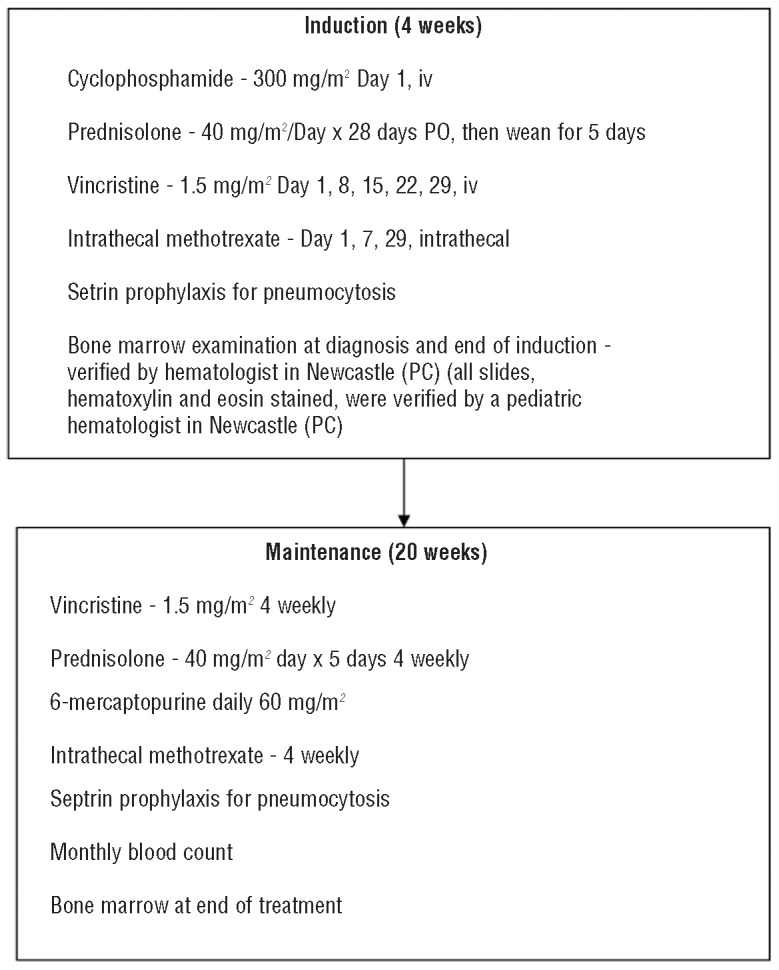
Details of treatment for the first Malawi ALL protocol.
All children aged one to 16 years with morphologically diagnosed ALL were eligible for the protocol unless they had a life-threatening infection or the family could not comply with the treatment and follow up. The study was approved by the local ethics committee in Blantyre and all families gave informed consent. Twenty patients (11 boys, 9 girls) with a median age of 7.3 years were treated between December 2009 and August 2011 (Table 1). All children were tested for HIV status; only one was positive. The median hemoglobin concentration at diagnosis was 4.9 g/dL, median white cell count (WCC) 41.6×109/L, and median platelet count 18×109/L. Presenting history included prolonged bleeding (45%), nutrition (good 10%, fair 65%, poor 20% and kwashiorkhor or ‘swollen belly’ 5%) and bone pain (35%). One patient had extensive lymphadenopathy. A total of 9 patients were screened by fluorescence in situ hybridization (FISH) for established chromosomal abnormalities; 2 were positive (ETV6-RUNX1 and BCR-ABL1) but none had a MYC, MLL or IGH@ translocation.
Table 1.
Characteristics of ALL patients treated on the first Malawi ALL protocol.

Two children, both with a high WCC, died before treatment could be administered. A further 7 patients died during induction therapy (Table 1). Among the 11 patients that completed induction therapy, all achieved a morphological remission by Day 28. Maintenance therapy was well tolerated and 2 children completed therapy. Five children had presumed relapses during maintenance (3 confirmed by bone marrow examination) and 2 after completion of treatment (one confirmed on bone marrow examination), all of whom died of their disease. One patient died of presumed bacterial meningitis during maintenance. There was no failure to complete treatment in those who did not relapse. The median time to death of those completing induction was 163 days (range 91-435). All but 2 patients relapsed during treatment. Three patients are still alive after 554, 232 and 93 days after treatment began. There was no relationship between outcome and white blood count, nutrition, overt bleeding, platelet count or bone pain.
We have demonstrated that it is possible to deliver ALL treatment in a low-resource setting. However, there is a period of intense learning as evidenced by the early death rate that was higher in the first 12 patients. Treatment efficacy may also be compromised by late presentation and relatively poor presenting condition. Our primary objective to deliver induction therapy and achieve a complete remission was achieved in 55% of patients, all of whom returned to their communities well and able to resume normal life, albeit for some with a limited time-frame. We are continuing to develop this ALL protocol in a realistic stepwise fashion. The second Malawi ALL protocol will have a 7-day prednisolone pre-phase (to combat the high initial death rate) and 9 months of treatment (to lengthen remission duration). We are mindful that future protocols that should include more drugs will need to be accompanied by advances in supportive care.
Taking the first step to treat pediatric ALL is a brave move but an important one, and our success should encourage others. Although the protocol was simple compared to those in the developed world, it is important that protocols evolve and develop in situ. Attempts to try and provide the same treatment as resource rich countries are likely to fail. While it can be disheartening in the beginning, many children who otherwise would die within days are now surviving for several months, and we are now aiming to achieve a small number of long-term survivors from the successive protocol.
Acknowledgments
The children and families in Malawi who bravely and with dignity face diseases such as ALL despite the many hardships they endure.
Footnotes
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Gopal S, Wood WA, Lee SJ, et al. Meeting the challenge of hematologic malignancies in sub-Saharan Africa. Blood. 2012;119(22):5078-87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hesseling P, Broadhead R, Mansvelt E, et al. The 2000 Burkitt lymphoma trial in Malawi. Pediatr Blood Cancer. 2005;44:245-50 [DOI] [PubMed] [Google Scholar]
- 3.Hesseling P, Molyneux E, Kamiza S, Israels T, Broadhead R. Endemic Burkitt lymphoma: a 28-day treatment schedule with cyclophosphamide and intrathecal methotrexate. Ann Trop Paediatr. 2009; 29:29-34 [DOI] [PubMed] [Google Scholar]
- 4.Wolff JA, Brubaker CA, Murphy ML, Pierce MI, Severo N. Prednisone therapy of acute childhood leukemia: prognosis and duration of response in 330 treated patients. J Pediatr. 1967;70:626-31 [DOI] [PubMed] [Google Scholar]
- 5.Elion GB, Hitchings GH. Antagonists of nucleic acid derivatives. V. Pteridines. J Biol Chem. 1951;188:611-21 [PubMed] [Google Scholar]
- 6.Elion GB, Hitchings GH, Van Der Werff H. Antagonists of nucleic acid derivatives. VI. Purines. J Biol Chem. 1951;192:505-18 [PubMed] [Google Scholar]
- 7.Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med. 2006;354:166-78 [DOI] [PubMed] [Google Scholar]
- 8.Moorman AV. The clinical relevance of chromosomal and genomic abnormalities in B-cell precursor acute lymphoblastic leukaemia. Blood Rev. 2012;26(3):123-35 [DOI] [PubMed] [Google Scholar]