

Abstract
Amyloidosis of the gastrointestinal tract, with biopsy-proven disease, is rare. We reviewed a series of patients who presented with biopsy-proven gastrointestinal amyloidosis and report their clinical characteristics, treatments, and survival. This is a retrospective review of data prospectively collected from January 1998 to December 2011 in a tertiary referral center; 2,334 patients with all types of amyloidosis were evaluated during this period. Seventy-six patients (3.2%) had biopsy-proven amyloid involvement of the gastrointestinal tract. Their median age was 61 years (range, 34-79). Systemic amyloidosis with dominant gastrointestinal involvement was present in 60 (79%) patients, whereas the other 16 (21%) patients had amyloidosis localized to the gastrointestinal tract without evidence of an associated plasma cell dyscrasia or other organ involvement. Of the 60 systemic cases, 50 (83%) had immunoglobulin light-chain, five (8%) had familial lysozyme, three (5%) had wild-type transthyretin, and two (3%) had mutant transthyretin amyloidosis. The most frequent symptoms for all patients were weight loss in 33 (45%) and gastrointestinal bleeding in 27 (36%). Incidental identification of amyloidosis on routine endoscopic surveillance played a role in the diagnosis of seven patients with systemic immunoglobulin light-chain, and four patients with immunoglobulin light-chain localized to the gastrointestinal tract. Amyloid protein subtyping was performed in 12 of the cases of localized disease, and all had lambda light chain disease. Of the 50 patients with systemic immunoglobulin light-chain amyloidosis, 45 were treated with anti-plasma cell therapy. The median survival has not been reached for this group. For the 16 patients with localized gastrointestinal amyloidosis, supportive care was the mainstay of treatment; none received anti-plasma cell therapy. All 16 are alive at a median follow-up of 36 months (range, 1-143). Patients with biopsy-proven gastrointestinal amyloidosis often present with weight loss and bleeding. In localized cases, all that underwent typing were due to lambda light chain amyloidosis and none progressed to systemic disease during the period of follow-up. Most patients with systemic disease had immunoglobulin light-chain, and their tolerance of therapy and median survival were excellent. Although a rare manifestation of amyloidosis, staining for amyloid should be considered in patients undergoing gastrointestinal biopsy who have unexplained chronic gastrointestinal symptoms.
Introduction
The amyloidoses are a heterogeneous group of diseases, characterized by the misfolding of extracellular proteins, generating insoluble fibrils resulting in disruption of tissue structure and function.1,2 Fibrils are comprised of protofilaments, made up of beta-sheet rich polypeptides. By electron microscopy, amyloid fibrils are ∼10 nm in diameter; by polarized light microscopy, the regular structure of the fibris leads to apple green-birefringence following staining with Congo red dye.1 At least 27 different human proteins have been identified to date as amyloidogenic, causing both systemic and localized disease.3 Systemic amyloidosis, the most common type of amyloid disease, is characterized by production of amyloidogenic precursor proteins at a site remote from the organs of amyloid deposition. Localized amyloidosis, in comparison, is defined by the production of precursor proteins in the same location as amyloid deposition.4,5
Systemic amyloidosis can be acquired, as in immunoglobulin light chain (AL) amyloidosis caused by an underlying clonal plasma cell dyscrasia, or hereditary, as most commonly occurs by modification of the transthyretin gene (mutant ATTR). Wild-type transthyretin can also form amyloid disease in older patients, in a process termed senile systemic amyloidosis. Both AL and ATTR amyloidosis are characterized by multi-system disease, typically involving the heart and kidneys in AL, whereas ATTR preferentially affects the heart and nervous system; often with gastrointestinal (GI) symptoms such as diarrhea and weight loss due to autonomic neuropathy.6 Familial or hereditary amyloidosis encompasses multiple amyloidogenic precursor proteins in addition to ATTR, including apolipoprotein AI, apolipoprotein AII, fibrinogen Aα chain, and lysozyme (ALys).3 ALys is a non-neuropathic hereditary disease that mimics AL amyloidosis, predominantly affecting the GI tract, liver, and kidneys.7 Localized amyloidosis is less common, typically occurring in the respiratory tract, bladder, breast, skin, or GI tract.2,8
GI involvement in AL amyloidosis is defined as the presence of GI symptoms with direct biopsy verification.9 Although standardized guidelines for the diagnosis and staging of non-AL forms of systemic amyloidosis and localized amyloidosis have not been developed, clinicians often define organ involvement as is done for AL amyloidosis. While many patients with systemic AL disease have amyloid deposits in blood vessels from GI tract biopsies, GI tract involvement with both symptoms and biopsy proof of involvement of the intestinal tissue itself is less common. In one series of 445 patients with AL amyloidosis, GI involvement was identified in 7% of the patients.1
Commonly reported symptoms of GI amyloidosis include esophageal reflux, constipation, nausea, and abdominal pain. Other symptoms such as diarrhea, weight loss, and early satiety, which are commonly attributed to GI amyloidosis, may be caused by other mechanisms such as autonomic neuropathy, small bowel bacterial overgrowth, or cardiac cachexia.2,10,11 Autonomic neuropathy plays a major role in the GI symptoms described in ATTR amyloidosis, contributing to overall morbidity in disease progression, and may also complicate liver transplantation.12,13 GI amyloid deposits identified by endoscopic biopsy occur most often in the duodenum, followed by the stomach, colo-rectum, and esophagus.14 Reports of amyloidosis localized to the GI tract are limited to case reports and small series of patients.15
This retrospective study details clinical characteristics, pathological findings, risk of progression to systemic amyloidosis, indications for chemotherapy, and associated toxicities as well as survival in a series of 76 patients.
Design and Methods
Patients
Data were collected retrospectively from January 1, 1998 to December 31, 2011, using the Boston University Amyloid Treatment and Research Program database. A total of 2,334 consecutive patients with all types of amyloidosis were evaluated during this period. Patients with documented GI amyloidosis were included in this series; GI amyloidosis was diagnosed when patients presented with GI symptoms and had biopsy proof of involvement of the GI tract. Histologically, vascular amyloid deposition only was not considered sufficient for a diagnosis; amyloid deposition had to occur in the extravascular space, e.g. within the muscularis mucosa, submucosal, and muscularis propria, as proof of GI tract involvement. Biopsies from outside institutions were reviewed, with repeat Congo red staining if necessary for confirmation. Patients with evidence of an underlying clonal plasma cell or lymphoplasmacytic disorder and AL amyloidosis were considered as having systemic AL type, including patients with co-existent multiple myeloma and Waldenström's macroglobulinemia as well as those with primary AL. Localized amyloidosis was diagnosed when GI amyloid deposition was present without evidence of systemic disease or an associated plasma cell dyscrasia. Immunohistochemical staining, mass spectrometry, or immunogold electron microscopy was used to type the amyloid deposits. When proteomics failed to identify the amyloid precursor, gene sequencing was used to identify age-related amyloidosis (wild-type TTR) or one of the non-TTR familial amyloidoses.
Patients with GI symptoms without biopsy confirmation, or those who presented with an initial manifestation of hepatic involvement or isolated macroglossia were excluded from analysis.
Data collection
The Institutional Review Board of Boston University Medical Campus approved data collection, and written informed consent was obtained from each patient, in accordance with the Declaration of Helsinki. GI symptoms were recorded for each patient. Time from symptom onset to diagnosis and diagnosis to amyloid treatment and research program evaluation were determined. Other organ involvement, including cardiac, pulmonary, renal, neurological, soft tissue, and hepatic involvement was reviewed. For patients with AL amyloidosis, data included results of bone marrow biopsy, serum free light chain (FLC) analysis, and serum and urine immunofixation electrophoresis. The difference between the involved FLC and the uninvolved FLC (dFLC) at presentation was calculated, when applicable. Serum creatinine, B-type natriuretic peptide, serum albumin, and alkaline phosphatase levels were collected for all patients; troponin-I data were not available until June 2008, thus this measurement is not available for all patients. The median overall survival and associated Kaplan-Meier survival curves were calculated for patients with systemic AL amyloidosis. For those patients diagnosed with systemic AL who were never treated, charts were reviewed to confirm the diagnosis.
Follow-up information was obtained for all of these patients at the time of this analysis by telephone or e-mail.
Results
Patients' characteristics
A total of 76 patients, 3% of all referred patients during the study period, had dominant GI amyloidosis (Table 1). Women accounted for less than half of the cases: 29 of 76 (38%). The median age at the time of diagnosis was 61 years (range, 34-79). Sixty patients (79%) had evidence of systemic amyloidosis, while 16 patients (21%) had localized GI amyloidosis (Figure 1). Symptoms for all patients included weight loss in 33 patients (43%), GI bleeding in 27 (36%), heartburn in 25 (33%), early satiety in 25 (33%), diarrhea in 22 (29%), and abdominal pain in 21 (28%). The median follow-up after initial evaluation was 35 months (range, 1-159).
Table 1.
Patients' characteristics and demographics.

Figure 1.
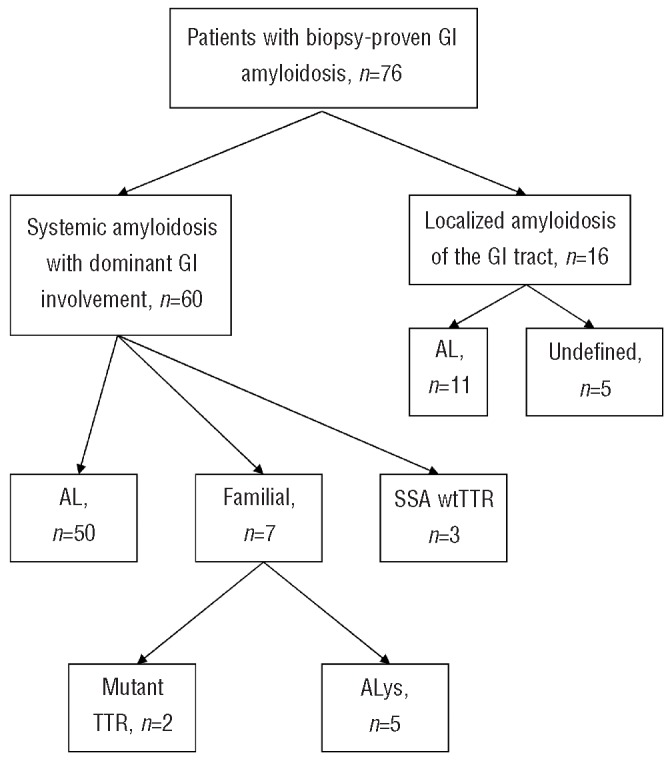
Gastrointestinal amyloidosis: distribution of patients. SSA: senile systemic amyloidosis.
Systemic amyloidosis with dominant gastrointestinal involvement
Among the patients with systemic amyloidosis, 50 (83%) had AL, five (8%) had ALys, three (5%) had senile systemic amyloidosis, and two (3%) had mutant ATTR (Figure 1).
Fifty patients presented with systemic AL amyloidosis and dominant GI involvement; the median age at the time of diagnosis was 63 years (range, 46-79) (Tables 2 and 3). Women accounted for 18 (36%) of these patients. The median time from symptoms to diagnosis was 5 months (range, 0-220). The median time from amyloid diagnosis to evaluation was 2 months (range, 0-80). For patients with AL, the most common biopsy site leading to a diagnosis of amyloidosis was the small bowel in 25 patients (50%) followed by stomach in 22 (44%) and colon in 16 (32%). The diagnosis was made during routine surveillance gastrointestinal endoscopy in seven (14%) patients; this included colorectal cancer screening and surveillance endoscopy for Barrett's esophagus.
Table 2.
Systemic and localized amyloidosis of the GI tract.
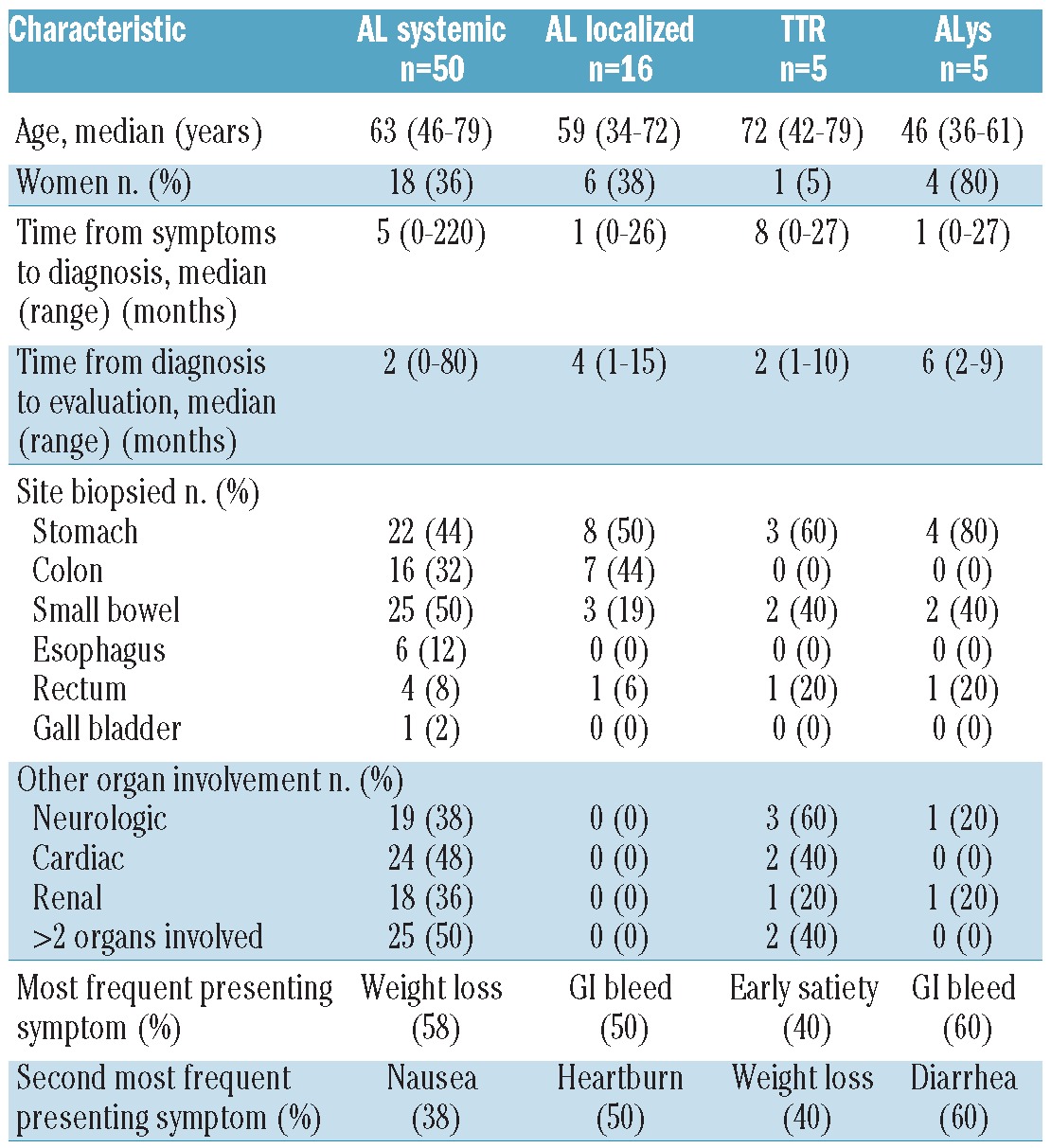
Table 3.
AL amyloidosis of the GI tract.
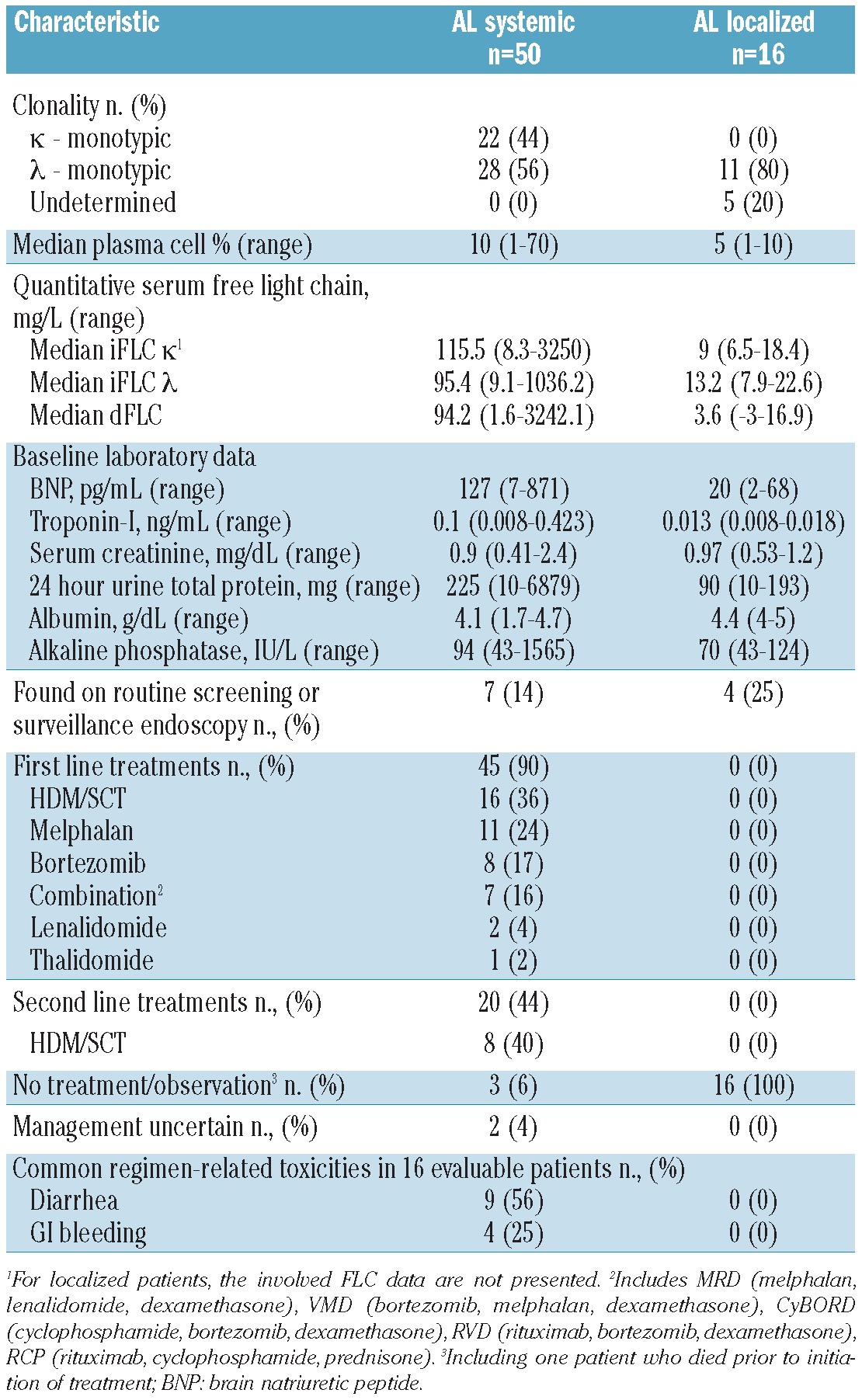
Other organs/systems involved included the neurological system in 19 (38%) patients, the heart in 24 (48%), and the kidneys in 18 (36%); 50% of the patients had more than two organs/systems involved. Of the seven patients with an initial diagnosis made by surveillance endoscopy, four had more than one organ/system involved; three of these patients had systemic amyloidosis affecting the GI tract only. Clonality of the underlying plasma cell dyscrasia was λ in 28 (56%) and κ in 22 (44%). Of the 50 patients with systemic AL, seven (14%) had coexistent multiple myeloma and two (4%) had coexistent Waldenström's macroglobulinemia. The median troponin-I level was 0.1 ng/mL (range, 0.008-0.423) and the median B-type natriuretic peptide concentration was 127 pg/mL (range, 7-871). The median serum albumin concentration was 4.1 g/dL (range, 1.7-4.7) while the median serum creatinine was 0.9 mg/dL (range, 0.41-2.4). The median 24-hour urine total protein excretion was 225 mg (range, 10-6879). At the time of diagnosis, the median plasma cell burden on bone marrow biopsy was 10% (range, 1-70). Among the patients for whom FLC analysis was available, the median involved κ FLC was 115.5 mg/L (range, 8.3-3250), and the median involved λ FLC was 95.4 mg/L (range, 9.1-1036). The median dFLC was 94.2 mg/L (range, 1.6-3242.1).
Forty-five of the 50 patients with systemic AL received systemic therapy; the first-line treatments included high-dose melphalan/stem cell transplantation (HDM/SCT) in 16 (36%), melphalan and dexamethasone in 11 (24%), bortezomib in 8 (18%), multidrug regimens in seven (16%), lenalidomide in two (4%), and thalidomide in one (2%). Multidrug regimens included bortezomib, melphalan and dexamethasone (VMD); cyclophosphamide, bortezomib, and dexamethasone (CyBorD); rituximab, cyclophosphamide, and prednisone (R-CP); rituximab, bortezomib and dexamethasone (RVD); and melphalan, lenalidomide and dexamethasone (MRD). Two of the 50 patients were observed without chemotherapy and one patient died prior to the initiation of treatment. In two patients, management of the disease was uncertain after the initial visit to this referral center (4%). Of the seven patients with systemic AL diagnosed by surveillance endoscopy, four underwent treatment with HDM/SCT, one received MRD, one received bortezomib, and in one patient treatment was not known. Of the 45 patients who received first-line treatments, 20 (44%) required additional treatment. Eight of these patients underwent treatment with HDM/SCT (40%).
For the 16 patients for whom toxicity data were available, the most frequent treatment-related toxicities (NCI CTCAEv3.0 grade 1-4) were diarrhea in nine (56%), nausea in eight (50%), constipation in ten (62%), GI bleeding in four (25%), and anorexia in seven (44%). Only one patient (6%) experienced severe GI hemorrhage – defined as requiring transfusion, operative intervention, or interventional radiology. One patient (6%) had life-threatening dehydration; no other life-threatening GI toxicities were documented. The survival rate at 2 and 4 years was 84% and 72%, respectively. With a median follow-up of 25 months after evaluation, the median overall survival for patients with systemic AL amyloidosis of the GI tract had not been reached (Figure 2).
Figure 2.
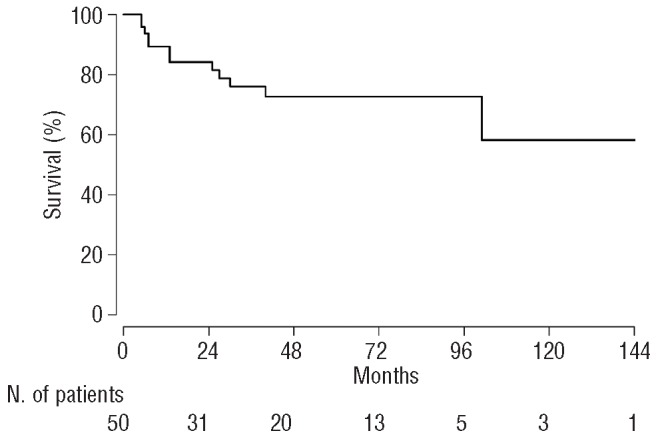
Kaplan-Meier curve showing overall survival in patients with systemic AL amyloidosis and GI involvement. The median overall survival was not.
Five patients with positive GI biopsies for amyloid were found to have ATTR amyloidosis, either mutant (2 patients) or wild-type (3 patients). The median age at the time of diagnosis was 72 years (range, 42-79). One ATTR patient had T60A, and the other had V30M. The median time from symptom onset to diagnosis was 8 months (range, 0-27), and the median time from diagnosis to evaluation was 2 months (range, 1-10). ATTR amyloidosis was found in the stomach in three patients (60%), small bowel in two (40%), and rectum in one (20%). The most frequent presenting symptoms were early satiety in two patients (40%) and weight loss in two (40%). The median serum albumin concentration was 4.3 g/dL. Both patients with mutant TTR also had cardiac and neurological involvement. All three patients with wild-type TTR had isolated GI tract involvement, with no coexistent cardiac involvement. The two patients with mutant TTR participated in a clinical trial evaluating diflunisal in familial amyloidosis (ClinicalTrials.gov identifier NCT00294671), and one of them is currently listed for a liver transplant. All three patients with wild-type ATTR were treated with supportive care only.
Five patients had ALys (7%). The median age at the time of diagnosis was 46 years (range, 36-61). Unusually for systemic amyloid diseases, women were in the majority (4 of the 5, 80%). The median time from symptom onset to diagnosis was 1 month (range, 0-27) and the median time from diagnosis to evaluation was 6 months (range, 2-9). The most frequent positive biopsies were stomach in four (80%) patients and small bowel in two (40%). Lysozyme gene sequencing demonstrated that four patients, all from different pedigrees, had a W64R substitution, and one patient had Y54N. GI bleeding and diarrhea were the presenting symptoms in three patients (60%) and heartburn in two (40%). Isolated GI tract involvement was documented in three patients (60%), and one patient had renal involvement (20%); the liver was not involved in any of these patients. Treatment for all ALys patients was supportive management and observation only, no patient is known to have undergone liver or kidney transplantation.
Localized amyloidosis of the gastrointestinal tract
Sixteen patients had localized amyloidosis of the GI tract, without any evidence of an underlying plasma cell dyscrasia or other organ involvement. These patients had normal bone marrow biopsies without evidence of clonality by immunohistochemistry or in situ hybridization, and normal serum FLC levels and ratio. Two patients had free λ on serum immunofixation electrophoresis, and two patients had free λ on urine immunofixation electrophoresis, but none had any other evidence of an underlying clonal plasma cell dyscrasia. They also lacked clinical evidence of involvement of other organs, and had no sign of amyloid deposition in a fat aspirate. The patients' characteristics are summarized in Tables 2 and 3. There were six women (38%). The median age at the time of diagnosis was 59 years (range, 34-72). Serendipitous discovery on routine endoscopic surveillance screening occurred in four (25%). The time from symptom onset to diagnosis was 1 month (range, 0-26). After initial diagnosis, the median time to evaluation was 4 months (range, 1-15). With a median follow-up after evaluation of 39 months (range, 1-121), all 16 patients (100%) were alive; 15 out of 16 (94%) had no evidence suggestive of progression to systemic disease, while this information was not available for one patient.
The most frequent symptoms for patients with localized GI involvement were GI bleeding in eight (50%), heartburn in eight (50%), and constipation in three.19 The most frequent biopsy sites were stomach in eight (50%) patients, colon in seven (44%), and small bowel in three (19%). In 11 of the 16 cases, the amyloid type was determined using immunohistochemistry, immunoelectron microscopy, or mass spectrometry, and all of these proved to be comprised of λ light chains. Supportive care without systemic treatment was recommended for these patients.
Discussion
Of 2,334 patients evaluated at our center in a 13-year period, 76 (3.3%) were diagnosed with biopsy-proven involvement of the GI tract. For these patients, weight loss, diarrhea, and early satiety were among the most common symptoms reported, in keeping with prior reports.10,11 These symptoms can also occur with autonomic neuropathy due to amyloidosis, and are not diagnostic of GI involvement in the absence of biopsy-proof.
The median time from diagnosis to treatment in patients with systemic disease was surprisingly short, being only 5 months. This contrasts, for example, with a median time of 48 months for patients presenting with peripheral neuropathy.16 Several factors might explain the more expedient time to diagnosis in patients with GI amyloidosis. The GI tract is more accessible to biopsy, so endoscopy may be employed sooner in the course of the workup for symptoms. However, early diagnosis might also reflect a selection bias for those patients who are successfully diagnosed and referred. Many patients with GI amyloidosis might not have been diagnosed, if Congo red staining of biopsied material was not performed.
AL amyloidosis was the most common cause of GI amyloid involvement, occurring in 50 patients as systemic disease and in 11 patients with localized disease. Thus, overall, among 76 patients with biopsy-proven GI amyloidosis, 61 (80%) cases were due to light chain deposition. In all 11 patients with localized AL amyloidosis the amyloid deposits were caused by a λ monoclonal light chain, in keeping with prior reports of localized disease, in which the majority had documented λ light chain disease.17 None of the patients with localized AL was found to progress to systemic disease, consistent with the hypothesis that localized AL is due to local production of light chains by a clone of plasma cells residing in the target organ.18
Patients with systemic AL amyloidosis were offered anti-plasma cell chemotherapy. HDM/SCT was offered as first-line therapy, as we and others have observed a high rate of hematologic complete responses and durable remissions in patients who achieve a complete response; we found an overall 6.3-year median survival and 43% complete response rate in our recent update.19 However, patients with GI amyloidosis and evidence of active GI bleeding or ulceration were not offered myeloablative chemotherapy, because of the risk of catastrophic GI hemorrhage (Figures 3 and 4).20 In the series we report here, severe GI bleeding occurred in only 6% of patients treated with HDM/SCT, which is comparable to rates of GI hemorrhage in other reported transplant series.21,22 None of our selected patients died due to GI bleeding. Patients who were not candidates for HDM/SCT were treated in clinical trials with novel agents or with melphalan and dexamethasone.
Figure 3.
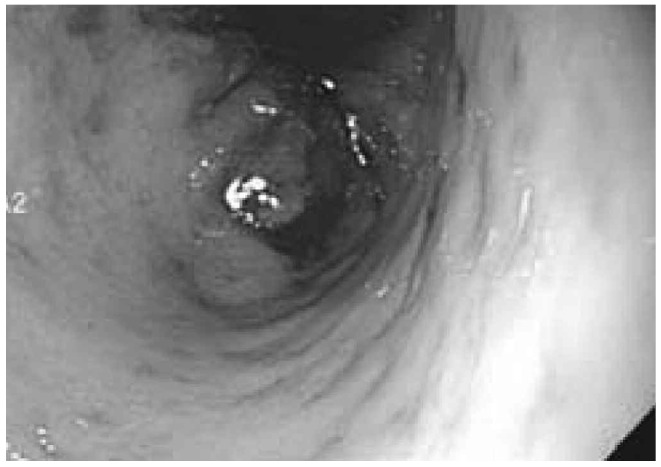
Endoscopic appearance of gastric ulceration in a patient with AL amyloidosis and severe gastrointestinal hemorrhage.
Figure 4.
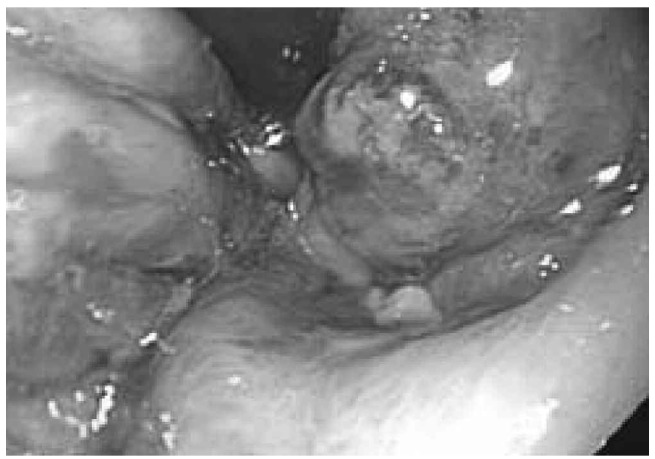
Endoscopic appearance of gastric submucosal hematoma in a patient with AL amyloidosis and severe gastrointestinal hemorrhage.
Ten patients (13%) with biopsy-proven GI amyloidosis had non-AL disease. Thus, accurate typing is critical to avoid the use of chemotherapy in these patients. Of these ten cases, five were due to TTR and five were due to ALys. Three of the five cases due to TTR were associated with senile systemic amyloidosis rather than a mutant TTR. Although wild-type TTR predominantly involves the heart, an autopsy series, reported by Matsutani et al., identified GI involvement in 7.8% of cases.23
In keeping with prior reports, all of the patients with ALys amyloidosis in our series presented with symptomatic GI amyloidosis proven by biopsy. Their presentation was very similar to that described in another report;7 however, in contrast to that study, none of our patients is known to have undergone either liver or renal transplantation, and all are still alive.
Patients with GI amyloidosis require supportive care for nausea with ondansetron, granisetron, or prochlorperazine, and for diarrhea with loperamide, lomotil, or deodorized tincture of opium.2 For dysmotility and early satiety, metoclopromide has been tried with limited success. Erythromycin, a pro-kinetic medication, was shown to have some benefit in improving delayed gastric emptying and early satiety in an ATTR patient.24
Amyloid protein typing was not performed in five patients (31%) of the group of 16 patients with biopsy-proven GI amyloidosis without evidence of systemic disease because sufficient tissue was not available. All five of these patients had a clinical course consistent with those patients who had λ light chain localized disease, with none receiving systemic treatment, and all still being alive today. Given these findings, these patients likely have localized AL disease, despite the lack of confirmatory amyloid protein typing.
In summary, we report the characteristics of a large series of 76 patients with biopsy-proven GI amyloidosis. Serendipitous biopsies appeared to play a role in the initial diagnosis of some patients with amyloidosis, and endoscopy with blind biopsies stained with Congo red should be considered for any patient with unexplained weight loss, GI bleeding, or early satiety, particularly in association with a monoclonal gammopathy or symptoms or signs of other organ disease (neuropathy, nephropathy, and cardiomyopathy). It is crucial to distinguish the type of amyloid, in order that patients with non-AL amyloidosis are not subjected to cytotoxic chemotherapy. While systemic AL amyloidosis is the most common cause of GI amyloid involvement, only carefully selected patients with systemic AL and involvement of the GI tract can be treated safely with HDM/SCT.
Localized GI amyloidosis without evidence of systemic involvement appears to be a distinct entity that, in keeping with other forms of localized amyloidosis, is due to a local population of plasma cells producing amyloidogenic immunoglobulin light chains. Patients with this form of amyloidosis should be monitored and treated symptomatically, but the rate of progression to systemic disease among them is low and their survival is excellent without systemic therapy, as none of these patients died from their disease during the follow-up period.
Acknowledgments
The authors acknowledge the participation of colleagues in the Amyloid Treatment and Research Program at Boston University School of Medicine and the Stem Cell Transplant Program at Boston Medical Center, as well as other clinical colleagues involved in the evaluation and management of the patients described in this article.
Funding: The development of the Amyloid Repository and Database was supported by P01 HL68705 and by grants from the Gruss and Wildflower Foundations. Most of all, the authors thank the patients who participated in our research programs.
Footnotes
Authorship and Disclosures: Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med. 2003; 349(6):583-96 [DOI] [PubMed] [Google Scholar]
- 2.Sattianayagam PT, Hawkins PN, Gillmore JD. Systemic amyloidosis and the gastrointestinal tract. Nat Rev Gastroenterol Hepatol. 2009;6(10):608-17 [DOI] [PubMed] [Google Scholar]
- 3.Sipe JD, Benson MD, Buxbaum JN, Ikeda S, Merlini G, Saraiva MJ, et al. Amyloid fibril protein nomenclature: 2010 recommendations from the nomenclature committee of the International Society of Amyloidosis. Amyloid. 2010;17(3-4):101-4 [DOI] [PubMed] [Google Scholar]
- 4.Dember LM. Emerging treatment approaches for the systemic amyloidoses. Kidney Int. 2005;68(3):1377-90 [DOI] [PubMed] [Google Scholar]
- 5.Westermark P, Benson MD, Buxbaum JN, Cohen AS, Frangione B, Ikeda S, et al. A primer of amyloid nomenclature. Amyloid. 2007;14(3):179-83 [DOI] [PubMed] [Google Scholar]
- 6.Falk RH, Comenzo RL, Skinner M. The systemic amyloidoses. N Engl J Med. 1997;337(13):898-909 [DOI] [PubMed] [Google Scholar]
- 7.Sattianayagam PT, Gibbs SD, Rowczenio D, Pinney JH, Wechalekar AD, Gilbertson JA, et al. Hereditary lysozyme amyloidosis - phenotypic heterogeneity and the role of solid organ transplantation. J Intern Med. 2012;272(1):36-44 [DOI] [PubMed] [Google Scholar]
- 8.Charlot M, Seldin DC, O'Hara C, Skinner M, Sanchorawala V. Localized amyloidosis of the breast: a case series. Amyloid. 2011; 18(2):72-5 [DOI] [PubMed] [Google Scholar]
- 9.Gertz MA, Comenzo R, Falk RH, Fermand JP, Hazenberg BP, Hawkins PN, et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis, Tours, France, 18-22 April 2004. Am J Hematol. 2005;79(4):319-28 [DOI] [PubMed] [Google Scholar]
- 10.Hayman SR, Lacy MQ, Kyle RA, Gertz MA. Primary systemic amyloidosis: a cause of malabsorption syndrome. Am J Med. 2001;111(7):535-40 [DOI] [PubMed] [Google Scholar]
- 11.Madsen LG, Gimsing P, Schiodt FV. Primary (AL) amyloidosis with gastrointestinal involvement. Scand J Gastroenterol. 2009; 44(6):708-11 [DOI] [PubMed] [Google Scholar]
- 12.Suhr O, Danielsson A, Holmgren G, Steen L. Malnutrition and gastrointestinal dysfunction as prognostic factors for survival in familial amyloidotic polyneuropathy. J Intern Med. 1994;235(5):479-85 [DOI] [PubMed] [Google Scholar]
- 13.Suhr OB, Anan I, Ahlstrom KR, Rydh A. Gastric emptying before and after liver transplantation for familial amyloidotic polyneuropathy, Portuguese type (Val30Met). Amyloid. 2003;10(2):121-6 [DOI] [PubMed] [Google Scholar]
- 14.Tada S, Iida M, Iwashita A, Matsui T, Fuchigami T, Yamamoto T, et al. Endoscopic and biopsy findings of the upper digestive tract in patients with amyloidosis. Gastrointest Endosc. 1990;36(1):10-4 [DOI] [PubMed] [Google Scholar]
- 15.Ebert EC, Nagar M. Gastrointestinal manifestations of amyloidosis. Am J Gastroenterol. 2008;103(3):776-87 [DOI] [PubMed] [Google Scholar]
- 16.Duston MA, Skinner M, Anderson J, Cohen AS. Peripheral neuropathy as an early marker of AL amyloidosis. Arch Intern Med. 1989;149(2):358-60 [PubMed] [Google Scholar]
- 17.Biewend ML, Menke DM, Calamia KT. The spectrum of localized amyloidosis: a case series of 20 patients and review of the literature. Amyloid. 2006;13(3):135-42 [DOI] [PubMed] [Google Scholar]
- 18.Hamidi Asl K, Liepnieks JJ, Nakamura M, Benson MD. Organ-specific (localized) synthesis of Ig light chain amyloid. J Immunol. 1999;162(9):5556-60 [PubMed] [Google Scholar]
- 19.Cibeira MT, Sanchorawala V, Seldin DC, Quillen K, Berk JL, Dember LM, et al. Outcome of AL amyloidosis after high-dose melphalan and autologous stem cell transplantation: long-term results in a series of 421 patients. Blood. 2011;118(16):4346-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seldin DC, Andrea N, Berenbaum I, Berk JL, Connors L, Dember LM, et al. High-dose melphalan and autologous stem cell transplantation for AL amyloidosis: recent trends in treatment-related mortality and 1-year survival at a single institution. Amyloid. 2011;18(Suppl 1):122-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Comenzo RL, Gertz MA. Autologous stem cell transplantation for primary systemic amyloidosis. Blood. 2002;99(12):4276-82 [DOI] [PubMed] [Google Scholar]
- 22.Skinner M, Sanchorawala V, Seldin DC, Dember LM, Falk RH, Berk JL, et al. High-dose melphalan and autologous stem-cell transplantation in patients with AL amyloidosis: an 8-year study. Ann Intern Med. 2004;140(2):85-93 [DOI] [PubMed] [Google Scholar]
- 23.Matsutani H, Hoshii Y, Setoguchi M, Kawano H, Gondo T, Takahashi M, et al. Vascular amyloid of unknown origin and senile transthyretin amyloid in the lung and gastrointestinal tract of old age: histological and immunohistochemical studies. Pathol Int. 2001;51(5):326-32 [DOI] [PubMed] [Google Scholar]
- 24.Saglam F, Celik A, Cavdar C, Sifil A, Atila K, Kaya GC, et al. A renal transplant recipient with delayed gastric emptying in amyloidosis due to familial Mediterranean fever improved with erythromycin: a case report. Transplant Proc. 2008;40(1):308-9 [DOI] [PubMed] [Google Scholar]