

Abstract
RecG is a DNA translocase that helps to maintain genomic integrity. Initial studies suggested a role in promoting recombination, a possibility consistent with synergism between recG and ruv null alleles and reinforced when the protein was shown to unwind Holliday junctions. In this article we describe novel suppressors of recG and show that the pathology seen without RecG is suppressed on reducing or eliminating PriB, a component of the PriA system for replisome assembly and replication restart. Suppression is conditional, depending on additional mutations that modify ribosomal subunit S6 or one of three subunits of RNA polymerase. The latter suppress phenotypes associated with deletion of priB, enabling the deletion to suppress recG. They include alleles likely to disrupt interactions with transcription anti-terminator, NusA. Deleting priB has a different effect in ruv strains. It provokes abortive recombination and compromises DNA repair in a manner consistent with PriB being required to limit exposure of recombinogenic ssDNA. This synergism is reduced by the RNA polymerase mutations identified. Taken together, the results reveal that RecG curbs a potentially negative effect of proteins that direct replication fork assembly at sites removed from the normal origin, a facility needed to resolve conflicts between replication and transcription.
Introduction
The assembly of replication fork complexes at sites removed from the normal chromosomal origin plays a vital role in maintaining the integrity of the bacterial genome and in securing its duplication (Gabbai and Marians, 2010). In Escherichia coli, it relies on the PriA and PriC proteins to load the DnaB replicative helicase. Transfer of DnaB from a complex with DnaC to what becomes the template for lagging strand synthesis is a key step in fork assembly. Once loaded, DnaB recruits DnaG primase and PolIII holoenzymes, thus establishing a fully fledged fork complex, or replisome (Tougu et al., 1994; Kim et al., 1996a,b). Promiscuous loading of DnaB is prevented by prior binding of SSB protein to any exposed ssDNA (LeBowitz and McMacken, 1986). DnaA protein overcomes this barrier at oriC by opening the DNA in a sequence directed manner that excludes SSB (Messer, 2002). PriA and PriC achieve the same end, but in a sequence-independent manner at branched DNA structures.
The PriA system relies on PriA itself plus PriB and DnaT (Sandler and Marians, 2000; Gabbai and Marians, 2010). PriA is a DNA helicase with a 3′–5′ polarity of strand translocation. It has a strong affinity for three-strand junctions, enabling it to target a D-loop intermediate in recombination, or a fork structure, with high specificity (McGlynn et al., 1997; Nurse et al., 1999). PriB is related to SSB and binds with high affinity to ssDNA. It stabilizes a PriA–DNA complex, stimulates PriA helicase activity and facilitates binding of DnaT. The tripartite PriA–PriB–DnaT complex enables DnaB loading, thus nucleating replisome assembly (Cadman et al., 2005; Lopper et al., 2007; Gabbai and Marians, 2010). The PriC system appears to be directed at stalled forks, especially forks with a gap between the branch point and the 3′ leading strand hydroxyl (Heller and Marians, 2005). As with the PriA system, PriC facilitates DnaB loading in the presence of SSB. It can do so in vitro without the aid of other proteins (Heller and Marians, 2005), but may require the 3′–5′ helicase activity of either Rep or PriA to do so efficiently in vivo (Sandler, 2000; Mahdi et al., 2006; Gabbai and Marians, 2010).
Null mutations in priA reduce cell viability, compromise recombination and DNA repair, and block DnaA-independent, stable DNA replication (SDR). This pleiotropic phenotype is suppressed by missense mutations in dnaC (Sandler et al., 1996; 1999; Gregg et al., 2002). In the case of dnaC810, the altered DnaC protein overcomes the SSB barrier to load DnaB without the aid of PriA (Liu et al., 1999). A partial deletion of DnaT behaves much like a priA null (McCool et al., 2004). Surprisingly, a strain deleted for priB shows little loss of viability and is reasonably proficient in recombination and DNA repair. The same is true of a strain deleted for priC. However, a strain deleted for both priB and priC is barely viable (Sandler, 2000). Viability is improved by dnaC809, which encodes the same amino acid substitution as dnaC810 (Sandler et al., 1996), and is restored to almost wild-type levels by dnaC809,820, which encodes an additional substitution (Sandler et al., 1999). On the basis of these and other observations demonstrating that priA priC and priA rep double mutants are inviable, Sandler (2000) concluded that there is cross-talk between the PriA and PriC systems, and proposed the existence of PriA–PriB, PriA–PriC and PriC–Rep pathways.
Although these pathways have evolved to promote cell survival, they establish a potential for replication to initiate when doing so offers no obvious advantage and might even be detrimental. Indeed, two proteins appear capable of curbing such activity, namely RNase HI and RecG. They reduce spurious initiations at R-loops, either by digesting the invading RNA strand or by unwinding the structure respectively (Horiuchi et al., 1984; Ogawa et al., 1984; Vincent et al., 1996; Fukuoh et al., 1997). Loss of either protein is associated with a substantial increase in DnaA-independent DNA synthesis. The loss of both is lethal (von Meyenburg et al., 1987; Asai and Kogoma, 1994a,b; Masai et al., 1994; Hong et al., 1995; Rudolph et al., 2009a,b).
Many features of the recG null phenotype are suppressed by mutations (e.g. priA300, srgA1) that reduce or eliminate the helicase activity of PriA (Al-Deib et al., 1996; Jaktaji and Lloyd, 2003; Rudolph et al., 2009a; Zhang et al., 2010). Unlike a priA null allele, these mutations do not reduce viability and retain the ability to promote DNA repair and recombination (Kogoma et al., 1996; Sandler et al., 1996; Jaktaji and Lloyd, 2003). The srgA1 allele of priA is especially informative. The mutant protein unwinds a three-way branched structure mimicking a replication fork. However, it has lost the ability to unwind a 3′ flap structure mimicking a fork with no leading strand at the branch point (Gregg et al., 2002), a structure RecG unwinds with high efficiency (McGlynn and Lloyd, 2001; Tanaka and Masai, 2006). This has led to the idea that 3′ flaps are generated accidentally during replication, but are eliminated via the combined actions of RecG and ssDNA exonucleases. Without RecG to unwind the structure, PriA is more likely to target the flap, thus triggering replisome assembly and re-replication of the already replicated DNA, with pathological consequences (Rudolph et al., 2009b; 2010a).
In this work, we describe how reducing or abolishing PriB can also lead to suppression of the recG null phenotype. However, the suppression requires additional mutations that alter 30S ribosomal subunit S6, or one of three major subunits of RNA polymerase, namely RpoA, RpoB or RpoC. These RNA polymerase mutations suppress a negative feature of the deletion priB phenotype that masks the ability to suppress recG. They also reduce a synergism between priB and ruv null alleles that we attribute to abortive recombination provoked by the exposure of ssDNA. We conclude that RecG is needed to curb a potential danger of replisome assembly directed at sites removed from oriC by the PriA system, a facility required to resolve conflicts between DNA replication and transcription.
Results
Recent studies exploiting priA and ssb suppressors of the recG null phenotype revealed how RecG protein might limit pathological events that disrupt the normal course of chromosome duplication (Rudolph et al., 2009a,b; 2010a,b; Zhang et al., 2010). In a new screen of ΔrecG derivatives selected for increased resistance to mitomycin C we isolated a novel clone that proved wild type for both priA and ssb. It carries instead a mutation in the rpsF gene encoding 30S ribosomal subunit S6 (Supplementary results). The G to T transversion identified and labelled rpsF292 converts the GAA codon for Glu98 to a TAA stop codon (Fig. 1A). This nonsense allele confers no obvious phenotype on its own, but is an effective and general suppressor of recG. Thus, it restores resistance to mitomycin C (Fig. 1B), alleviates the slight sensitivity to UV light (Fig. 1B and 2A, panels i and ii), and reduces the extended delay in replication of those cells surviving irradiation (Fig. 2B). It also overcomes the requirement for both Pol I and Dam proteins to maintain robust growth on LB agar (Fig. 2C), and improves the recovery of recombinants in conjugational and transductional crosses (Table 1). Its ability to do so depends on the presence of the RuvABC Holliday junction resolvase (Fig. 1B and 2A, panels i and ii; Table 1).
Fig. 1.

Suppression of recG by rpsF292.
A. Chromosomal location of rpsF and of downstream genes expressed from the same promoter (P). The position of the rpsF292 mutation and flanking markers exploited is also shown.
B. Effect of rpsF292 on the sensitivity of recG and ruv strains to mitomycin C and UV light. The strains examined are identified by genotype, followed in each case by the strain number in parentheses.
C. Expression of wild-type RpsF or PriB in trans reduces rpsF292 suppression of recG. Except for the presence of the indicated plasmid, the strains examined are identified by genotype, followed in each case by the strain number in parentheses.
Fig. 2.

Effect of rpsF292 and ΔpriB on the recG and ruv mutant phenotypes.
A. Sensitivity to UV light. The strains examined are identified by genotype, with the strain number in parentheses below the genotype.
B. Cell replication following UV irradiation. Strain genotypes are as identified, with strain numbers in parentheses. Data are means (± SE) of three independent experiments for irradiated and two for unirradiated cells. Data for MG1655 (wt) and its recG derivative, N4560, are reproduced for comparison from Rudolph et al. (2007b) and Rudolph et al. (2009a) respectively.
C. Synthetic lethality assays showing how rpsF292 overcomes the inviability of recG polA and recG dam cells. The plate assay exploited here and in subsequent figures is described in detail in Experimental procedures. The relevant genotype of the construct used is shown above the section of the plate photograph displayed. In each case the relevant plasmid genotype/relevant chromosome genotype (e.g. recG+/ΔrecG) is indicated, along with the strain number in parentheses. The fraction of white (Lac−) colonies is shown below with the number of white colonies/total colonies analysed in parentheses. White colonies arise from cells that lost the plasmid before plating whereas blue (Lac+) colonies or blue/white, sectored colonies arise from those that retained the plasmid.
Table 1.
Effect of rpsF292 on conjugational DNA transfer and recombination
| Relative number of transconjugants or P1 transductantsb | |||||||
|---|---|---|---|---|---|---|---|
| x KL548 | Hfr GY2200 | Hfr KL226 | |||||
| Strain number | Relevant genotype | Relative viabilitya | (F′ Pro+) | (λ)c | (Thr+Leu+) | (Pro+) | P1 transductants (Leu+) |
| AB1157 | rps+ rec+ ruv+ | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 |
| N7962 | rpsF292 | 0.93 | 1.18 | 1.22 | 0.99 | 1.17 | 0.82 |
| AM2123 | ΔrecG | 0.82 | 0.7 | 0.89 | 0.35 | 0.25 | 0.14 |
| N7985 | rpsF292 ΔrecG | 0.78 | 1.29 | 1.27 | 0.73 | 0.91 | 0.43 |
| N4454 | ΔruvABC | 0.62 | 0.62 | 0.84 | 0.42 | 0.43 | 0.21 |
| N7986 | rpsF292 ΔruvABC | 0.60 | 1.13 | 1.15 | 0.51 | 0.52 | 0.15 |
| AM2124 | ΔrecG ΔruvABC | 0.28 | 0.21 | 0.67 | 0.0018 | 0.0014 | 0.0011 |
| N7987 | rpsF292 ΔrecG ΔruvABC | 0.23 | 0.28 | 1.06 | 0.0024 | 0.0020 | 0.0048 |
Values for cell viability are based on the recipient cultures used in conjugational crosses. Those based on cultures of the same recipients used in P1 transductions are shown in Table S2. Although the culture conditions are not the same, the two estimates are generally very close.
Mating was for 30 (KL548), 40 (KL226) or 60 (GY2200) min and the transconjugant class selected is indicated. The phage P1 donor was strain W3110. Values for wild-type control strain AB1157 are set at 1. The actual mean values ± SE are shown in Table S2. Mutant strains were tested in parallel with AB1157 and the values shown are mean yields relative to AB1157 in each of three or more experiments. Numbers of experiments and standard errors are provided in Table S2.
λ plaques arise from zygotic induction of the λ prophage transferred by the Hfr.
The stop codon introduced by rpsF292 would be expected to eliminate the final 35 amino acids from the C-terminus of RpsF, the final two glutamic acids of which are needed for post-translational addition of a further four glutamates (Reeh and Pedersen, 1979; Kang et al., 1989). It might also cause premature termination of transcription and thus reduce expression of the downstream genes transcribed from the rpsF promoter. Significantly, these genes include priB, which is associated with the PriA system of replication restart. Previous studies revealed that mutations affecting the helicase activity of PriA suppress the sensitivity of recG cells to mitomycin-C (Al-Deib et al., 1996; Jaktaji and Lloyd, 2003). To determine which of these effects of rpsF292 might account for the suppression of recG, we introduced plasmids encoding the downstream genes into an rpsF292 ΔrecG double mutant. A priB+ construct makes the strain almost as sensitive to a combination of mitomycin C and UV light as a ΔrecG single mutant (Fig. 1C). In contrast, a plasmid encoding rpsR+ and rplI+ behaves like the vector. Thus it seems that reduced expression of PriB might be a substantial factor. However, a plasmid encoding rpsF+ also reduces resistance (Fig. 1C). The effect is not as great as seen with the priB+ plasmid, but the fact that there is any reduction in sensitivity at all does suggest that the truncation of RpsF contributes to the strength of the suppression.
ΔrpsF and ΔpriB are weak suppressors of the recG mutant phenotype
We made in-frame deletions of rpsF and priB to examine directly whether loss of either would suppress recG. Neither is essential for growth (Sandler et al., 1999; Bubunenko et al., 2007). The ΔrpsF allele clearly alleviates sensitivity to mitomycin C, although it is not as effective as rpsF292 (Fig. 3A). The resistance conferred is reversed by expressing rpsF+ from a plasmid (Fig. 3B). Given any polar effect of the rpsF deletion on downstream genes would persist in the presence of the rpsF+ plasmid, these data support the notion that inactivation of rpsF contributes substantially to the observed suppression of recG.
Fig. 3.
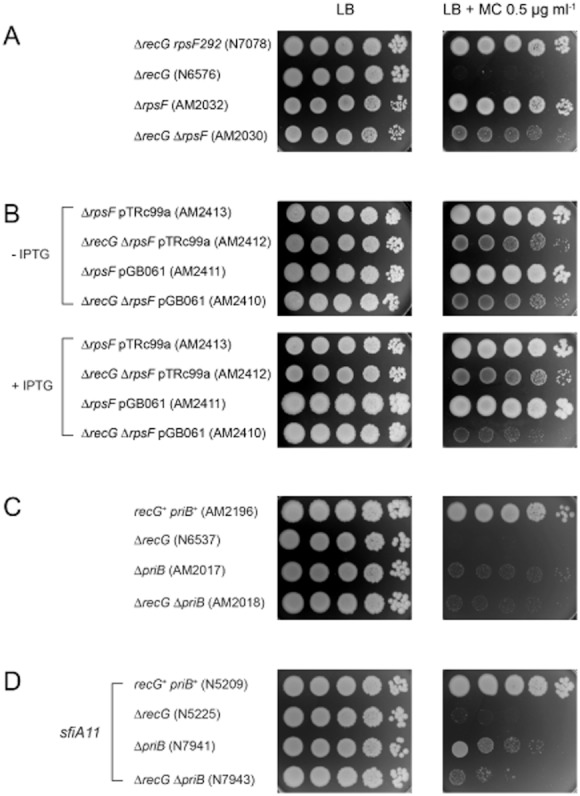
Effect of RpsF, PriB and SfiA depletion on sensitivity to mitomycin C.
A, C and D. Effect of rpsF, priB and sfiA null alleles, respectively, in the presence and absence of RecG.
B. Expression of rpsF+ in trans improves growth of ΔrpsF cells and reduces the suppression of ΔrecG.
The strains examined are identified by genotype, followed in each case by the strain number in parentheses.
The ΔpriB::dhfr allele we made confers slight sensitivity to UV light and moderate sensitivity to mitomycin C (Fig. 2A, panel iii; Fig. 3C). Another deletion, ΔpriB202 (Sandler et al., 1999), made without a resistance tag confers similar sensitivity to mitomycin C (data not shown). Neither allele is able to confer wild-type resistance to mitomycin C on a recG strain (Figs 3C and S2A). The recG priB double-deletion strain also remains slightly sensitive to UV light (Fig. 2A, panel iii). However, a side-by-side comparison reveals that a priB single mutant is not quite as sensitive to mitomycin C as a recG strain, and that a recG priB double mutant behaves like a priB strain (Fig. 3C and data not shown), indicating that there is some weak suppression of recG.
We investigated whether the sensitivity of a priB strain to mitomycin C might be due to increased expression of the SOS-induced division inhibitor encoded by sfiA (sulA). Previous studies had shown that sfiA inactivation enhances the viability of priA null cells (Nurse et al., 1991). We observed that it also improves the growth of both priB and priB recG strains in the presence of mitomycin C. However, the improvement is quite modest (Fig. 3D). There is no improvement with a recG strain. Taken together, these observations confirm that the recG phenotype is partially suppressed by the elimination of either RpsF or PriB. They are consistent with the notion that the strong suppression observed with rpsF292 is due to the combined effect of mutating RpsF and reducing the expression of PriB.
RNA polymerase mutations suppress ΔpriB and enable ΔpriB to suppress ΔrecG
Despite both ΔrecG and ΔpriB conferring sensitivity to mitomycin C, cultures of the double mutant readily accumulate resistant derivatives, suggesting that a single additional mutation might suffice to suppress sensitivity. We isolated 18 resistant clones of the recG priB strain AM2055 (Fig. S1), and established by DNA sequencing and genetic reconstruction that mutation of a single gene is responsible for the alleviation of sensitivity in at least 14 of these cases.
In no case was the suppressor an allele of priA. Instead, the mutations identified were located to genes encoding one of three major subunits of RNA polymerase. Several were found in rpoA and rpoB, and one in rpoC, with some alleles appearing more than once (Table 2). The rpoA[P293L] allele confers a requirement for methionine or cysteine for growth. The same requirement was previously associated with a K271E substitution (Thomas and Glass, 1991). It enabled us to identify rpoA[P293L] repeatedly in a further screen of ΔrecG ΔpriB strains selected for resistance to mitomycin C (Table S1 and strains not listed). The same screen also identified two independent rpoA isolates encoding a K298N substitution.
Table 2.
Properties of rpo suppressors of ΔpriB and ΔpriB ΔrecG
| Suppressorisolatea | Gene affected | DNA sequence change(s)b | Allele designation | RNAP feature affected | Rifampicin resistancec | Stringent phenotyped | rpo* activitye |
|---|---|---|---|---|---|---|---|
| AM2064/2066 | rpoA | CCT (Pro293) to CTT (Leu) | rpoA[P293L] | Alpha C-terminal domain | < 5 | ND | Weak negative |
| AM2072/2075 | |||||||
| AM2067 | rpoA | CTG (Leu253) to CGG (Arg) | rpoA[L253R] | Alpha C-terminal domain | < 5 | ND | Weak negative |
| AM2074 | rpoA | GAA (Glu273) to GAT (Asp) | rpoA[E273D] | Alpha C-terminal domain | < 5 | None | Neutral |
| AM2174 | rpoA | AAA (Lys298) to AAT (Asn) | rpoA[K298N] | Alpha C-terminal domain | < 5 | ND | Weak negative |
| AM2071 | rpoA | TCA (Ser49) to ACA (Thr) | rpoA[S49T,S309P] | Alpha C-terminal domain (S309P) | < 5 | ND | Negative |
| TCC (Ser309) to CCC (Pro) | |||||||
| AM2070 | rpoB | CGT (Arg452) to CTT (Leu) | rpoB[R452L] | Non-transcribed ssDNA channel | 10 | Very weak | Weak negative |
| AM2073 | rpoB | GGT (Gly1260) to GAT (Asp) | rpoB[G1260D] | RNA exit channel | 10 | Strong | Positive |
| AM2060/2069 | rpoB | TCG (Ser1332) to TTG (Leu) | rpoB[S1332L] | RpoB:RpoC interface; RNA exit? | < 5 | Strong | Weak positive |
| AM2063 | rpoB | Δ(G1336-C1344) | rpoB[ΔD446-L448] | Point of template DNA re-annealing | 5 | Very weak | Positive |
| AM2059 | rpoC | Δ(A643-T660) | rpoC[ΔK215-R220] | β′B rudder in the DNA channel? | < 5 | Strong | Negative |
Except for AM2174, the suppressor isolates are derivatives of strain AM2055 (ΔlacIZYA ΔrecG::apra zjf920::Tn10 ΔpriB202) selected for their resistance to mitomycin C. AM2064 and AM2066 came from the same culture of AM2055 and therefore may be siblings. AM2072 and AM2075 could also be siblings, but are independent of AM2064 and AM2066. AM2174 is a mitomycin C-resistant derivative of AM2167 (ΔlacIZYA ΔrecG::apra zjf920::Tn10 ΔpriB202 yheR::kan). The rpoA[P293L] allele was also identified in two other independent isolates, namely AM2173 and AM2191 (Table S1).
As defined in parentheses by the amino acid substitution(s) or deletion.
Strains were tested for growth on LB agar supplemented with rifampicin to a final concentration of 5, 10, 15, 20 or 50 μg ml−1. The parent strains show no resistance to rifampicin at 5 μg ml−1. The maximum concentration of rifampicin allowing growth to single colonies is indicated.
As determined by the ability of the rpo allele to allow a relA spoT strain to grow on minimal agar, i.e. to confer prototrophy (Cashel et al., 1996).
As determined from the survival of a ΔruvABC derivative irradiated with UV light at doses ranging from 5 to 60 J per m2 (McGlynn and Lloyd, 2000). Neutral: no effect; positive: improves survival; negative: reduces survival.
We transferred the rpo alleles to wild-type strain MG1655 and examined the sensitivity to mitomycin C of the rpo single mutant constructs and of derivatives carrying ΔrecG, ΔpriB or both. The priB and priB recG derivatives all proved quite resistant, as did the rpo single mutants. However, the recG derivative remained sensitive in every case, although slightly increased resistance was observed in a few instances, notably with rpoA[L253R], rpoA[E273D] and rpoB[ΔD446-L448] (Figs 4 and S2). These data demonstrate that the rpo mutations are suppressors of ΔpriB and when present enable ΔpriB to strongly suppress ΔrecG.
Fig. 4.

Effect of RNA polymerase mutations on sensitivity to DNA-damaging agents. Suppression of the sensitivity of priB and priB recG cells to mitomycin C by mutation of RpoA, RpoB or RpoC. The strains examined are identified by genotype, followed in each case by the strain number in parentheses.
The rpoB[G1260D] allele was identified previously among a subclass of stringent RNAP mutations that improve survival of UV-irradiated strains lacking the RuvABC Holliday junction resolvase (McGlynn and Lloyd, 2000; Trautinger and Lloyd, 2002). We considered whether suppression of priB might be a general property of these so-called rpo* mutations (McGlynn and Lloyd, 2000). We tested rpoB*35, which encodes an H1244Q substitution in the β-subunit that appears to destabilize transcription elongation complexes (McGlynn and Lloyd, 2000; Trautinger et al., 2005). This allele clearly increases the resistance of a priB strain to mitomycin C, but has little or no effect on a recG strain unless priB is deleted (Fig. S2J). However, with the exception of rpoB[G1260D], the rpo alleles identified here seem distinct from the rpo* class. Only one (rpoB[R452L]) confers the modest resistance to rifampicin characteristic of both rpoB*35 and rpoB[G1260D], and only two (rpoB[S1332L] and rpoC[ΔK215-R220]) confer a stringent phenotype (Table 2). The ability to affect the survival of UV-irradiated ΔruvABC cells also varies. Again, apart from rpoB[G1260D], which has a strong positive effect, only rpoB[ΔD446-L448] shows an ability to improve survival. Indeed, several have a substantial negative effect (Table 2; Fig. S3). No rpoA alleles were identified among the rpo* class of ruv suppressors described previously. It is also significant that the rpoA alleles identified here encode substitutions in RpoA that are unlikely to impinge on the DNA channel through RNA polymerase, a notable feature of the rpo* class (Trautinger and Lloyd, 2002). They appear instead to affect a C-terminal domain of the RpoA subunit that interacts with the transcription anti-terminator, NusA (Mah et al., 2000).
From these data it is clear that eliminating PriB has itself a significant negative effect on the ability of cells to withstand damage to their DNA. We probed ΔpriB strains in more detail to see if we could shed light on how the absence of PriB is able nevertheless to mask the recG phenotype and explain why its ability to do so is conditional on some alteration of RNA polymerase. We focused initially on cells lacking the RuvABC resolvase since previous studies demonstrated that the priA300 suppressor of recG has a negative effect on DNA repair in such cells (Jaktaji and Lloyd, 2003).
The absence of PriB provokes recombination
Our studies revealed that eliminating PriB increases the sensitivity of ΔruvABC cells to killing by UV light and reduces their ability to foster recombinants in genetic crosses. The increase in UV sensitivity approaches the synergism between ruv and recG null alleles (Fig. 2A, panels i and iii). Yields of haploid recombinants in genetic crosses are some 10-fold lower than with the ruv control (Tables 3A and S2). Inactivation of PriB alone has little or no effect on recombination, as reported (Sandler et al., 1999). The recovery of F-prime transconjugants with the priB ruv double mutant is reduced to an even greater extent (> 100-fold; Table 3A). Efficient zygotic induction of phage λ in the cross with Hfr GY2200 indicates that this latter defect is not due to reduced DNA transfer. Significantly, activation of the normally quiescent RusA Holliday junction resolvase via rus-1 or rus-2 insertions restores efficient recovery of both F-prime transconjugants and haploid recombinants (Tables 3A and S2). It also increases resistance to UV irradiation (Fig. 5A).
Table 3.
Effect of PriB on conjugational DNA transfer and recombination
| Relative numbers of transconjugants or P1 transductantsa | ||||||||
|---|---|---|---|---|---|---|---|---|
| KL548 | Hfr GY2200 | Hfr KL226d | ||||||
| Strain number | Relevantgenotype | Relative viabilitya | (F′ Pro+) | (λ)c | (Thr+Leu+) | (Pro+) | P1 transductants (Leu+) | |
| A | AM2077 | priB | 0.98 | 0.89 | 1.06 | 0.83 | 1.05 | 0.4 |
| N4454 | ruvABC | 0.62 | 0.62 | 0.84 | 0.42 | 0.43 | 0.18 | |
| AM2078 | priB ruvABC | 0.28 | 0.0017 | 0.72 | 0.026 | 0.034 | 0.011 | |
| N7946 | priB ruvABC rus-2 | 0.76 | 1.07 | 1.33 | 0.56 | 0.46 | 0.23 | |
| B | AM2089 | priB recG | 0.77 | 0.85 | 0.97 | 0.69 | 0.69 | 0.49 |
| AM2142 | priB recB | 0.25 | 0.15 | 0.56 | 0.00088 | 0.0005 | 0.0042 | |
| C | AM2096 | priB ruvABC recA | 0.48 | 0.59 | 0.84 | 0.000024 | 0.000023 | ND |
| N7938 | priB ruvABC lexA3 | 0.46 | 0.58 | 0.76 | 0.09 | 0.15 | ND | |
| N7940 | priB ruvABC sfiA | 0.29 | 0.006 | 0.70 | 0.05 | 0.016 | 0.0099 | |
| N8035 | priB ruvABC recB | 0.25 | 0.00008 | 0.93 | 0.00027 | 0.00061 | ND | |
| AM2097 | priB ruvABC recF | 0.57 | 0.64 | 0.87 | 0.37 | 0.37 | 0.14 | |
| AM2133 | priB ruvABC recJ | 0.42 | 0.42 | 0.86 | 0.088 | 0.14 | 0.11 | |
| AM2134 | priB ruvABC recQ | 0.51 | 0.55 | 0.65 | 0.08 | 0.10 | 0.08 | |
| D | N7915 | priB ruvABC dnaC809,820 | 0.61 | 0.85 | 1.16 | 0.25 | 0.56 | 0.19 |
| N7926 | priB ruvABC dnaC809,820 priC | 0.2 | 0.0028 | 0.93 | 0.036 | 0.29e | 0.008 | |
| N7918 | priB dnaC809,820 priC | 0.85 | 0.99 | 1.30 | 0.35 | 1.56 | 0.35 | |
| N7934 | ruvABC priC | 0.52 | 0.68 | 0.58 | 0.27 | 0.47 | 0.14 | |
| E | N7964 | priB ruvABC rpoB[G1260D] | 1.32 | 0.70 | 0.72 | 0.17 | 0.16 | 0.17 |
| N7948 | ruvABC rpoB[G1260D] | 1.41 | 1.06 | 0.47 | 0.26 | 0.28 | 0.24 | |
Values for cell viability are based on the recipient cultures used in conjugational crosses. Those based on cultures of the same recipients used in P1 transductions are shown in Table S2. Although the culture conditions are not the same, the two estimates are generally very close.
Mating was for 30 (KL548), 40 (KL226) or 60 (GY2200) min and the transconjugant class selected is indicated. The phage P1 donor was W3110. Values for wild-type control strain AB1157 are set at 1. The actual values ± SE are shown in Table S2. Mutant strains were tested in parallel with AB1157 and the values shown are mean yields relative to AB1157 in each of three or more experiments. Numbers of experiments, control mutant strains and standard errors are provided in Table S2. ND, not determined.
λ plaque forming units arising from zygotic induction of the λ prophage transferred by the Hfr.
Very similar values were obtained using N7610 as the Hfr donor, a ΔpriB::dhfr derivative of Hfr KL226.
The Hfr transfers priC+ proximal to the selected marker, hence the increased recovery of recombinant relative to the cross with Hfr GY2200, which transfers priC+ distal to the selected marker such that fewer of the selected transconjugants receive this allele.
Fig. 5.
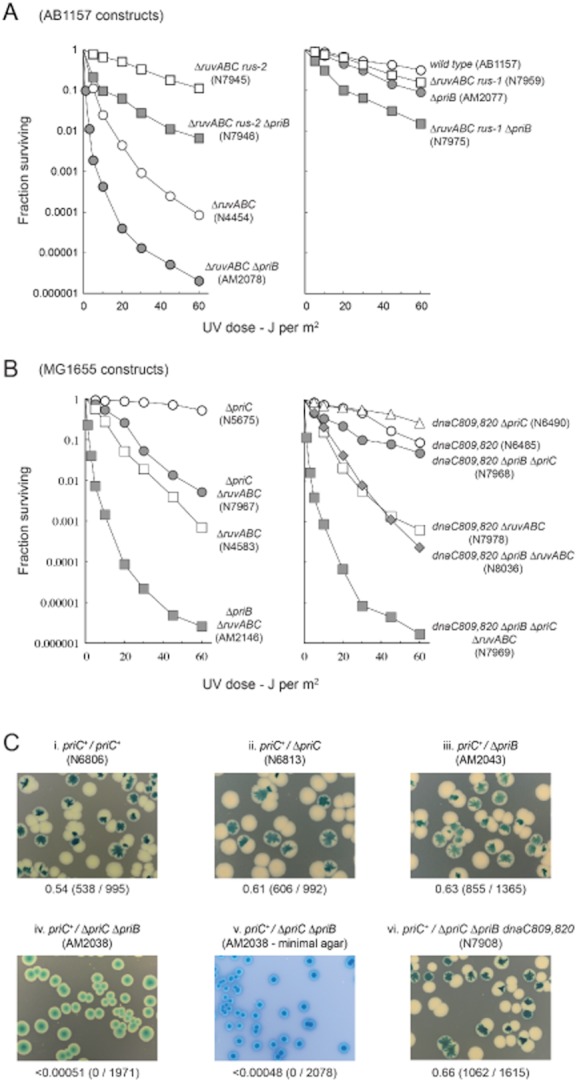
Suppression of the priB mutant phenotype.
A and B. Suppression of the synergism between priB and ruv (A) by rus-1 and rus-2 activation of the RusA resolvase and (B) by dnaC809,820. The strains examined are identified by genotype, and by the strain number in parentheses.
C. Synthetic lethality assays demonstrating the inviability of priB priC cells and the restoration of viability by dnaC809,820. Each image is labelled as described in the legend to Fig. 2C.
A notable feature of ruv mutant cells is that they foster the recovery of recombinants in genetic crosses with Hfr donors with a frequency only some two- to threefold lower than with a ruv+ control despite the lack of any other known activity capable of cleaving Holliday junctions (Table 3A) (Lloyd et al., 1984; Lloyd, 1991; Mandal et al., 1993; Mahdi et al., 1996). However, the viability of ruv cells is much reduced if the incidence of recombination is increased by exposure to UV light or other agents that damage DNA (Lloyd et al., 1984), or by mutations that compromise DNA macromolecular metabolism (Magner et al., 2007; Zhang et al., 2010). Viability is maintained in these circumstances if the RusA resolvase is expressed, demonstrating that the lethality observed without either resolvase is due to the accumulation of unresolved Holliday junctions (Mandal et al., 1993; Mahdi et al., 1996; Zhang et al., 2010). Thus, from the data presented it seems clear that PriB normally limits the incidence of recombination in conjugational crosses and during repair of UV-irradiated cells. Without PriB, recombination occurs more frequently in these situations, generating Holliday junctions. With no RuvABC available, these junctions persist, compromising viability. There is no evidence that recombination is essential in the absence of PriB. This is evident from the viability of priB derivatives lacking various combinations of the major activities linked with promoting recombination (Tables 3 and S2).
Eliminating PriB from recG cells has little effect on recombination (Table 3B). This is consistent with RuvABC acting independently of RecG (Lloyd, 1991). Importantly, a recB mutation reduces recombinant yields by some 200-fold or more (Table 3B), establishing that the vast majority of progeny recovered in crosses with ΔpriB recipients are still formed via a RecBCD-dependent mechanism, as in wild-type cells.
Homologous recombination prevents the recovery of F-prime transconjugants
RecA is essential for conjugational recombination in E. coli, but not for the recovery of F-prime transconjugants (Clark and Margulies, 1965). We exploited this fact to investigate whether the reduced recovery of F-prime transconjugants with priB ruv cells is due to abortive recombination between a newly transferred F-prime element and the recipient chromosome. We discovered that eliminating RecA restores the ability to recover F-prime transconjugants with high efficiency (Table 3C). Introducing a lexA3 mutation, which reduces expression of RecA and prevents induction of the SOS response (Sassanfar and Roberts, 1990), also restores efficient recovery of F-prime transconjugants. However, eliminating the SOS-induced SfiA division inhibitor does not (Table 3C), from which we conclude that the failure to recover these transconjugants is not due to lethal, SOS-induced cell filamentation. Taken together, the data indicate instead that in the absence of PriB, recombination between a newly transferred F-prime and the chromosome occurs in the vast majority (≥ 99%) of transconjugants and leads to the formation of at least one Holliday junction that physically links the two DNA elements. Without RuvABC or RusA to resolve the junction, the transconjugant is inviable.
Eliminating RecF, RecO or RecR also rescues F-prime transconjugants whereas the inactivation of RecBCD enzyme does not (Tables 3C and S2). The RecFOR proteins facilitate loading of RecA on single-stranded DNA (ssDNA) bound by SSB protein. They enable RecA to displace the SSB and form a stable nucleoprotein filament that promotes homologous DNA pairing and strand exchange (Cox, 2007). Thus, the recombination provoked in the absence of PriB is most likely initiated at one or more ssDNA gaps. This would fit with the fact that during conjugation a single strand of DNA is transferred to the recipient with a 5′–3′ polarity, where it is then made duplex by lagging strand synthesis (Willetts and Wilkins, 1984; Lloyd and Buckman, 1995). The transferred donor DNA is likely therefore to contain transient ssDNA gaps that provide potential templates for the binding of PriB, SSB or both. PriB resembles SSB in several respects and is known to bind ssDNA. Our results may be explained if gaps are more common, persist for longer or are simply more recombinogenic when there is no PriB present. This would fit with our observation that inactivating RecJ or RecQ also restores a robust recovery of F-prime transconjugants (Table 3C). Without PriB to bind the transferred F-prime strand, any newly synthesized lagging strand may be targeted by a combination of the helicase activity of RecQ and the 5′–3′ ssDNA exonuclease activity of RecJ, thus delaying gap closure.
Eliminating RecFOR, RecJ or RecQ also improves slightly the recovery of haploid recombinants in Hfr crosses (Tables 3 and S2). In such crosses, it is thought that RecBCD enzyme facilitates initiation of two recombination events, one at either end of the linear Hfr DNA fragment transferred to the recipient (Smith, 1991). If true, and if single-strand gaps do persist in the transferred Hfr DNA, then it would seem that additional exchanges initiated at these gaps might be detrimental to the recovery of recombinants when the RuvABC resolvase is missing. However, we note that eliminating RecFOR, RecJ or RecQ also improves the recovery of transductants in crosses with phage P1 (Tables 3 and S2). We are unaware of any evidence to suggest that the linear fragment of duplex donor DNA in transducing particles contains single-strand interruptions that might trigger recombination.
dnaC809,820 promotes recovery of F-prime transconjugants, but only if PriC is present
We exploited dnaC809,820 to examine the possibility that F-prime DNA strand transferred to a priB cell provokes recombination because of delayed or incomplete synthesis of the complementary (lagging) strand. The mutant DnaC protein is believed to load DnaB without the aid of PriA or PriC (Sandler, 2000). It might therefore compensate for the absence of PriB, and thus eliminate the observed synergism between priB and ruv. This proved to be the case. However, its ability to do so depends on PriC (Tables 3D and S2; Fig. 5B). The need for PriC is unexpected as dnaC809,820 has been reported to act as a very effective suppressor of the near inviability of a priB priC double mutant (Sandler, 2000). A synthetic lethality assay confirmed that it does so under our experimental conditions (Fig. 5C). Deletion of priC alone does not reduce the recovery of either F-prime transconjugants or haploid recombinants, nor does it increase sensitivity to UV light. Unlike ΔpriB it also does not enhance the ruv phenotype (Tables 3D and S2; Fig. 5B). So, while the mutant DnaC protein encoded by dnaC809,820 is able to overcome the synergism between priB and ruv null alleles, it can do so only with the aid of PriC. We assume PriC is needed to help direct DnaB loading. With RuvABC available, priB dnaC809,820 cells show little or no such requirement (Table 3D; Fig. 5B). From these data, we conclude that the newly transferred F-prime DNA strand provokes recombination in the absence of PriB because of a failure to initiate or complete synthesis of the complementary strand, thus increasing the likelihood of loading RecA.
RNA polymerase mutations reduce the synergism between priB and ruv
We tested the rpo alleles identified as suppressors of priB and priB recG cells to see if they too might alleviate the synergism observed between priB and ruv. We found that they do. All tested alleles restore efficient recovery of F-prime transconjugants, improve the yield of haploid recombinants and reduce killing by UV light (Tables 3E and 4; Fig. S3). The improved ability to survive UV irradiation varies according to how the rpo allele affects the survival of ruv (priB+) cells, although the data reveal an imperfect correlation (Fig. S3). Nevertheless, they do indicate that the rpo suppressors are somehow able to reduce the incidence of recombination events that require processing by RuvABC.
Table 4.
Effect of rpo suppressors of priB on the recovery of F-prime transconjugants in crosses with a ΔpriB ΔruvABC recipient
| Strain | Suppressor | Relative yield of F-prime transconjugantsa |
|---|---|---|
| AM2078 | None | 0.0017 |
| N8174 | rpoA[S49T, S309P] | 0.28 ± 0.07 |
| N8175 | rpoA[E273D] | 0.30 ± 0.02 |
| N8185 | rpoA[K298N] | 0.17 ± 0.03 |
| N8187 | rpoA[L253R] | 0.14 ± 0.06 |
| N8004 | rpoB*35[H1244Q] | 0.4 ± 0.03 |
| N8179 | rpoB[ΔD446-L448] | 0.34 ± 0.04 |
| N8180 | rpoB[S1332L] | 0.37 ± 0.05 |
| N8181 | rpoB[R452L] | 0.21 ± 0.09 |
| N8178 | rpoC[ΔK215-R220] | 0.28 ± 0.03 |
Values are relative to the yield with the wild-type (pri+ ruv+) control strain, AB1157, and are the means (± SE) of from three to five independent experiments.
The rpoA, rpoB and rpoC mutations improve the viability of priB polA cells
Our analysis of priB cells revealed that PriB is required to help maintain viability in the absence of DNA polymerase I, at least under conditions supporting rapid growth. Without it, these cells plate with high efficiency on minimal salts agar, but are able to establish many fewer and rather sickly colonies on LB agar (Fig. 6A and B). This finding is not that surprising given that these cells have been shown to require the PriA-dependent pathway of replication restart to maintain viability (Lee and Kornberg, 1991). The rpo suppressors of priB we have identified allow robust growth of priB polA cells on LB agar (Fig. 6B and C. This observation provides further support for the conclusion that the suppression of priB by the rpo alleles described is not limited to the elimination of sensitivity to mitomycin C, reinforcing the conclusion that the latter effect is not some consequence of changes in gene expression that reduce the uptake of mitomycin C or which increase its efflux.
Fig. 6.
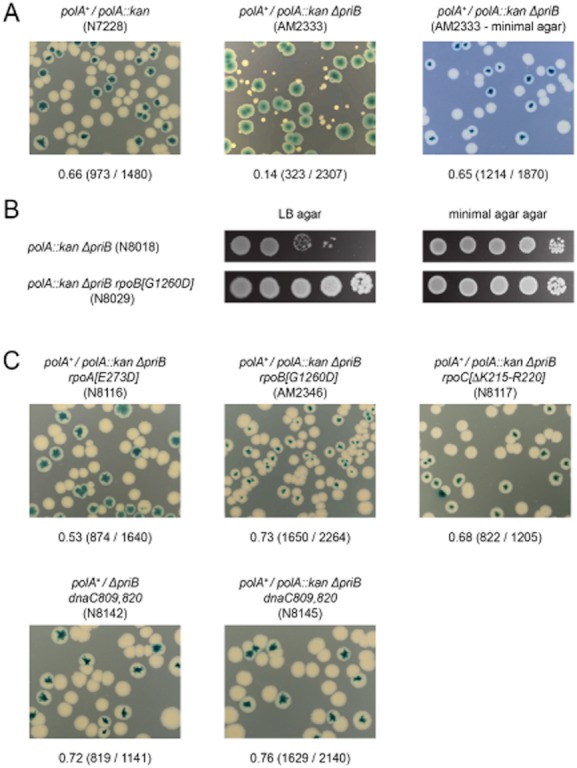
RNA polymerase and DnaC mutations improve the viability of polA priB cells.
A. Synthetic lethality of polA priB cells. Each assay is labelled as described in the legend to Fig. 2C.
B. Relative plating efficiency of polA priB cells on LB and minimal salts agar. The strains indicated were derived using 56/2 minimal salts agar media. Single colonies were grown in liquid 56/2 salts to an A650 of 0.4, serially diluted in 10-fold steps from 10−1 to 10−5 and 10 μl of samples of each dilution were spotted on LB or minimal salts agar as indicated. Plates were photographed after 48 h incubation.
C. Synthetic lethality assays demonstrating robust growth on LB agar of polA priB cells carrying the indicated rpoA alleles or dnaC809,820. Each image is labelled as described in the legend to Fig. 2C.
Discussion
We identified a novel suppressor of recG as a nonsense mutation in the rpsF gene encoding ribosomal subunit S6 (rpsF292; Fig. 1A). Because of its location upstream of priB, we thought it might act by exerting a polar effect, reducing synthesis of PriB and thus compromising DnaB loading. In other words, we suspected it might have an effect similar to previously identified priA suppressors that reduce the helicase activity of PriA (Al-Deib et al., 1996; Gregg et al., 2002; Jaktaji and Lloyd, 2003; Zhang et al., 2010). We dissected the contributions of rpsF and priB and demonstrated that a reduction in PriB synthesis might indeed be a substantial factor. However, the analysis revealed that the mutation of RpsF itself also makes a contribution (Fig. 1C). Indeed, we showed that an in-frame deletion of rpsF has suppressor activity (Fig. 3A and B).
The conclusion that reduced expression of priB is not by itself sufficient to explain the effect of rpsF292 is re-enforced by finding that a priB deletion is a weak suppressor of the mitomycin C sensitivity conferred by recG. However, this is not surprising as the deletion itself confers some sensitivity, and has other debilitating effects (see below). Intriguingly, the priB deletion becomes much more effective in the presence of an additional mutation in one of three major subunits of RNA polymerase. The mutations identified alleviate every aspect of the deletion priB phenotype we have tested, including the sensitivity to mitomycin C (Fig. 4A), the synergism with ruv (Table 3; Fig. 2) and the inviability with polA (Fig. 6). Although conditional, the fact that the absence of PriB can be a very effective suppressor is consistent with the view that much of the recG pathology is due to over-replication of the chromosome following PriA-mediated replisome assembly (Rudolph et al., 2009b; 2010a,2010b).
Analysis of the synergism with ruv revealed a strong tendency in cells lacking PriB for recombination to be provoked. However, this recombination is not essential, as is clear from the viability of deletion priB cells lacking RecA, RecBCD or RuvABC (Table 3). Analysis of the factors that eliminate the synergism with ruv indicated that recombination is provoked because one or more regions of ssDNA become exposed to RecA (Table 3, Fig. 5). We assume this occurs when the PriA–PriB–DnaT system is recruited to facilitate replisome assembly. PriB normally limits exposure of ssDNA by binding to the ssDNA exposed by PriA before transferring it via DnaT to the DnaC:DnaB complex (Lopper et al., 2007).
Our finding that mutations in RNA polymerase suppress the deletion priB phenotype would be consistent with the idea that PriB plays an important part in resolving conflicts between DNA replication and transcription. But if true, how could a deficiency in this activity be reconciled with the ability of deletion priB to suppress recG. Transcription complexes are substantial barriers to replication fork progression (Mirkin and Mirkin, 2007; Rudolph et al., 2007a; Merrikh et al., 2011), and may be particularly troublesome if they stall or backtrack (Trautinger et al., 2005; Dutta et al., 2011). Several recent studies indicate that recruitment of a second helicase motor helps drive forks through these barriers and that viability is compromised if the primary candidates are not available, as for example in rep uvrD strains (Guy et al., 2009; Baharoglu et al., 2010; Boubakri et al., 2010; Atkinson et al., 2011). Viability is improved by rpoB and rpoC mutations that destabilize transcribing RNA polymerases. Therefore, our finding that many features of the priB null phenotype are suppressed by some of the very same rpo mutations is highly significant, and especially so given dnaC809,820 is also a suppressor. It suggests that replication forks not only stall when they run into RNA polymerase, but also frequently require the re-loading of DnaB before replication can resume.
A need to re-load DnaB explains how a combination of priB and rpo mutations strongly suppresses recG. Assuming the recG phenotype is a consequence of PriA-dependent chromosome over-replication, as the results presented would suggest, it would be reasonable to suppose that this replication increases conflicts with transcription, especially if it were to initiate in the terminus area and proceed towards oriC, as suggested (Rudolph et al., 2010b). Eliminating PriB would prevent this over-replication by disrupting the replisome assembly needed for its initiation, while destabilizing RNA polymerase would itself reduce the need for PriB to rescue those forks assembled initially at oriC that subsequently ran into trouble. With PriB present in rpo recG cells, the over-replication triggered in the absence of RecG would negate any advantage gained from destabilizing RNA polymerase, thereby explaining the failure of the rpo mutation itself to suppress recG.
The RNA polymerase mutations implicated in reducing conflicts between replication and transcription most probably do so by reducing the stability of transcription complexes, thereby reducing the barrier to replication fork progression. In the case of rpoC[ΔK215-R220] and rpoB*35, destabilization has been demonstrated experimentally, and most likely reflects the disruption of important stabilizing interactions in the DNA channel (Bartlett et al., 1998; Trautinger et al., 2005). However, the rpoA alleles identified seem unlikely to compromise the intrinsic stability of RNA polymerase. With the exception of the S49T substitution encoded by rpoA[S49T,S309P], all affect the mobile C-terminal domain of the RpoA (alpha) subunit that interacts with NusA and with the emerging mRNA (Mah et al., 2000). The E273D, P293L and K298N substitutions may directly affect binding to NusA. The L253R and S309P substitutions are distant to the NusA binding interface, but might affect the total mobility of the domain and thus indirectly affect the interaction. NusA binding to RNA polymerase affects the β-flap domain of the RNA exit channel, exerting an allosteric effect on the trigger loop/bridge helix interaction required for translocation of the elongation complex, thus reducing elongation and increasing pausing (Bar-Nahum et al., 2005; Nudler, 2009). If the rpoA alleles reduce NusA binding, they might therefore destabilize transcription complexes indirectly by reducing pausing and uncoupling transcription from translation, enabling Rho to unwind the untranslated RNA (Epshtein et al., 2010; Dutta et al., 2011; Washburn and Gottesman, 2011). The idea that Rho might be a critical factor in reducing conflicts between replication and transcription is consistent with the reported synthetic lethality of recG rho double mutant cells (Harinarayanan and Gowrishankar, 2003), and with the identification here of ribosomal subunit S6 mutations as suppressors of recG. It may also be significant that the conditional rho-15 allele confers methionine auxotrophy (Guterman and Howitt, 1979), a property shared with rpoA[P293L], the most frequent suppressor in our screens for priB recG derivatives resistant to mitomycin C. If our interpretation is correct, it would follow that by coupling transcription with translation, and thus reducing Rho-mediated termination, the presence of NusA actually increases conflicts with replication. We assume that premature termination of transcription is a more immediate threat to growth and viability than is presented by blocking replication fork progression.
To conclude, we have identified novel suppressors of the recG mutant phenotype that combine a deficiency in the PriB component of the PriA–PriB–DnaT system of replisome assembly with modifications either to the ribosome or to RNA polymerase. By dissecting the properties of these suppressors and probing their modes of action, we have confirmed that the pathology resulting from loss of RecG is largely a consequence of unscheduled chromosome replication mediated by the PriA–PriB–DnaT system of replisome assembly. We have also presented evidence that this replication most likely increases conflicts with transcription and that PriB is needed to help resolve such conflicts. Eliminating PriB suppresses recG, presumably by reducing unscheduled replication, but only in the presence of an additional mutation to RNA polymerase that is itself likely to reduce conflicts between replication and transcription. The RNA polymerase mutations identified include rpoA alleles likely to disrupt interactions with NusA, leading us to suspect that factors controlling the coupling of transcription and translation may play a significant role in balancing the different pressures on replication and transcription.
Experimental procedures
Bacterial strains
The strains used are listed in Table S1. Chromosomal genes were inactivated using Tn10 or kan insertions conferring resistance to tetracycline (Tcr) and kanamycin (Kmr), respectively, or with deletions tagged with insertions conferring resistance to chloramphenicol (cat; Cmr), kanamycin (kan; Kmr), trimethoprim (dhfr; Tmr) or apramycin (apra; Aprar). The ΔpriB202 allele is an in-frame deletion of the priB-coding sequence (Sandler et al., 1999). It was introduced by co-transduction with zjf920::Tn10. A new in-frame deletion (ΔpriB::dhfr) was made using the one-step gene inactivation method of Datsenko and Wanner (2000). The entire priB sequence from start to stop codon was replaced with a dhfr sequence. The same method was used to make an in-frame deletion of rpsF (ΔrpsF::cat) and internal deletions of dam (Δdam::dhfr) and recR (ΔrecR::kan). The dam deletion leaves 42 bp of coding sequence at the 5′ end and 48 bp at the 3′ end while the recR deletion leaves 96 bp 5′ and 51 bp 3′. The yheB::kan and yheR::kan insertion alleles linked to rpoA, and the mutL::kan allele linked to rpsF, were identified using a library of random kan insertions in strain MG1655 generated using the EZ-Tn5 <kan-2> Tnp Transposome system (Epicentre Technologies). Neither of the yhe insertions has any obvious effect on growth or sensitivity to genotoxic agents (R.G. Lloyd, unpubl. work).
Plasmids
pRC7 is a low-copy-number, mini-F derivative of the lac+ construct pFZY1 (Bernhardt and de Boer, 2004). pJJ100 and pAM475 are derivatives of pRC7 carrying recG+ and polA+ respectively (Zhang et al., 2010). A priC+ derivative was made by PCR amplification of the coding region for priC from strain MG1655, plus some 100 bp of upstream promoter sequences, using 5′ and 3′ primers that incorporated flanking ApaI restriction sites. The amplified DNA was cut with ApaI and the priC+ fragment inserted into the ApaI site within the lacIq gene of pRC7, generating pAM421. This plasmid maintains robust growth of a ΔpriC ΔpriB strain, demonstrating that it expresses priC+. pT7 cloning vectors have been described (Tabor and Richardson, 1985). pAM494 is a derivative of pT7-7 carrying the adjacent rpsR+ and rplI+ genes inserted between the vector NdeI and HindIII sites. pAM496 and pAM499 are equivalent constructs carrying priB+ and rpsF+ respectively. pGB061 is an rpsF+ derivative of the expression vector pTRc99a (Amann et al., 1988). Expression of rpsF in strains harbouring pGB061 was induced by growth in LB media containing 0.15 mM IPTG. Media were supplemented with ampicillin for plasmid maintenance, except as specified in synthetic lethality assays with strains carrying pRC7 and its derivatives.
Media and general methods
LB broth and 56/2 minimal salts media, and methods for monitoring cell growth and for strain construction by P1vir-mediated transduction have been cited (Al-Deib et al., 1996; McGlynn and Lloyd, 2000; Trautinger et al., 2005). Resistance to rifampicin was measured by streaking culture samples on LB agar plates supplemented with rifampicin at a final concentration of 5, 10, 15, 20 and 50 μg ml−1 and scoring growth after overnight incubation.
Isolating mitomycin C-resistant suppressors of ΔrecG and ΔrecG ΔpriB strains
E. coli strains lacking RecG, or both RecG and PriB, are sensitive to mitomycin C. Several independent cultures of these strains were set up from single colonies and grown to mid-exponential phase in LB broth before plating 50–100 μl of samples on LB agar plates supplemented with mitomycin C at a final concentration of 0.5 μg ml−1. Resistant mutants establishing robust colonies appear within 24–36 h at 37°C. They arise at a frequency of approximately 0.1–1 per 106 colony-forming units (cfu) plated.
Measuring sensitivity to DNA damage
Sensitivity to UV light was measured using exponential phase cells grown to an A650 of 0.4 (1–2 × 108 cells ml−1 for strain MG1655). Samples of appropriate dilutions were irradiated on the surface of LB agar plates and survivors scored after 18–24 h incubation. Survival data are means from at least two, usually 3–6, independent experiments. Errors (SE) range between 5% and 15% of the mean. Sensitivity to mitomycin C (MC) was determined by growing cultures to an A650 of 0.4 and spotting 10 μl of serial 10-fold dilutions from 10−1 to 10−5 (from left to right in the images shown) on LB agar with or without mitomycin C at a final concentration of 0.5 μg ml−1 and incubating at 37°C, with or without prior exposure to UV light, as indicated. Plates were photographed after 24 h incubation, unless stated otherwise. Media contained ampicillin at a final concentration of 50 μg ml−1 in the case of strains harbouring Apr plasmids.
Multiplication of cells surviving UV irradiation
Cultures of each strain were grown in LB both to an A650 of 0.2, the cells pelleted, UV-irradiated or mock-irradiated on the surface of LB agar and resuspended in the original, but filter-sterilized supernatant and diluted 10 000-fold in conditioned medium prepared by growing the wild-type strain in fresh LB broth to an A650 of 0.2 with subsequent filter sterilization. The diluted cells were incubated with vigorous aeration at 37°C and samples removed at intervals were mixed with 2.5 ml of molten 0.6% top agar and plated on LB agar. Colonies were scored after 18–24 h at 37°C.
Genetic crosses and measures of recombination
F-prime and Hfr donors were mated with F− recipient strains in high-salt LB broth at 37°C as described (Lloyd et al., 1987; 1988). Measurements of cell viability relate to the number of cfu in the recipient culture at an A650 of 0.4, as determined with plating on non-selective 56/2 agar. All recipients were derivatives of the multi-auxotrophic, streptomycin-resistant strain, AB1157 (Table S1). Transconjugants were selected using 56/2 or LB agar, as appropriate, supplemented with 100 μg ml−1 streptomycin to counterselect donor cells. Transductions were conducted using phage P1 vir, following the recipes and protocols described (Miller, 1972).
Synthetic lethality assays
The rationale for synthetic lethality assays has been described (Bernhardt and de Boer, 2004; Mahdi et al., 2006). Essentially, a wild-type gene of interest is cloned in pRC7, a lac+, Apr mini-F plasmid that is rapidly lost, and used to cover a null mutation in the chromosome, in a Δlac background. A mutation in another gene of interest is then introduced into the chromosome. If the double mutant is viable, the plasmid-free cells segregated during culture will form white (Lac−) colonies or sectors of colonies on agar plates supplemented with X-gal and IPTG. If synthetically lethal, they will fail to grow and only solid blue (Lac+) colonies formed by cells retaining the plasmid will be observed. The segregation of white colonies that are significantly smaller than blue colonies is generally an indicator of reduced viability without the covering plasmid. Cultures of the constructs tested were grown in LB broth without ampicillin selection to an A650 of 0.4 before assaying for growth of plasmid-free cells on indicator plates. Plates were photographed after incubation for 48 h (LB agar) or 72 h (glucose minimal salts agar). Photographs were cropped to show a 3 cm × 2 cm section of the plate agar. Unless stated otherwise, images are from LB indicator plates.
Acknowledgments
We thank Adam Elenany and Mariam Malik for assistance with initial studies leading to the identification of rpsF and rpo suppressors, Christian Rudolph for helpful discussions, and Carol Buckman and Lynda Harris for excellent technical support. This work was supported by programme Grant G0800970 from the UK Medical Research Council.
Supporting information
Additional supporting information may be found in the online version of this article.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Al-Deib AA, Mahdi AA, Lloyd RG. Modulation of recombination and DNA repair by the RecG and PriA helicases of Escherichia coli K-12. J Bacteriol. 1996;178:6782–6789. doi: 10.1128/jb.178.23.6782-6789.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amann E, Ochs B, Abel KJ. Tightly regulated tac promoter vectors useful for the expression of unfused and fused proteins in Escherichia coli. Gene. 1988;69:301–315. doi: 10.1016/0378-1119(88)90440-4. [DOI] [PubMed] [Google Scholar]
- Asai T, Kogoma T. D-loops and R-loops: alternative mechanisms for the initiation of chromosome replication in Escherichia coli. J Bacteriol. 1994a;176:1807–1812. doi: 10.1128/jb.176.7.1807-1812.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asai T, Kogoma T. Roles of ruvA, ruvC and recG gene functions in normal and DNA damage-inducible replication of the Escherichia coli chromosome. Genetics. 1994b;137:895–902. doi: 10.1093/genetics/137.4.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson J, Gupta MK, Rudolph CJ, Bell H, Lloyd RG, McGlynn P. Localization of an accessory helicase at the replisome is critical in sustaining efficient genome duplication. Nucleic Acids Res. 2011;39:949–957. doi: 10.1093/nar/gkq889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baharoglu Z, Lestini R, Duigou S, Michel B. RNA polymerase mutations that facilitate replication progression in the rep uvrD recF mutant lacking two accessory replicative helicases. Mol Microbiol. 2010;77:324–336. doi: 10.1111/j.1365-2958.2010.07208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Nahum G, Epshtein V, Ruckenstein AE, Rafikov R, Mustaev A, Nudler E. A ratchet mechanism of transcription elongation and its control. Cell. 2005;120:183–193. doi: 10.1016/j.cell.2004.11.045. [DOI] [PubMed] [Google Scholar]
- Bartlett MS, Gaal T, Ross W, Gourse RL. RNA polymerase mutants that destabilize RNA polymerase-promoter complexes alter NTP-sensing by rrn P1 promoters. J Mol Biol. 1998;279:331–345. doi: 10.1006/jmbi.1998.1779. [DOI] [PubMed] [Google Scholar]
- Bernhardt TG, de Boer PA. Screening for synthetic lethal mutants in Escherichia coli and identification of EnvC (YibP) as a periplasmic septal ring factor with murein hydrolase activity. Mol Microbiol. 2004;52:1255–1269. doi: 10.1111/j.1365-2958.2004.04063.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boubakri H, de Septenville AL, Viguera E, Michel B. The helicases DinG, Rep and UvrD cooperate to promote replication across transcription units in vivo. EMBO J. 2010;29:145–157. doi: 10.1038/emboj.2009.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bubunenko M, Baker T, Court DL. Essentiality of ribosomal and transcription antitermination proteins analyzed by systematic gene replacement in Escherichia coli. J Bacteriol. 2007;189:2844–2853. doi: 10.1128/JB.01713-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadman CJ, Lopper M, Moon PB, Keck JL, McGlynn P. PriB stimulates PriA helicase via an interaction with single-stranded DNA. J Biol Chem. 2005;280:39693–39700. doi: 10.1074/jbc.M508521200. [DOI] [PubMed] [Google Scholar]
- Cashel M, Gentry DR, Hernandez VJ, Vinella D. In: The Stringent Response. In: Escherichia coli and Salmonella Cellular and Molecular Biology. 2nd edn. Neidhardt FC, Curtiss R III, Ingraham JL, Lin ECC, Low KB, Magasanik B, et al., editors. Washington, DC: ASM Press; 1996. pp. 1458–1496. [Google Scholar]
- Clark AJ, Margulies AD. Isolation and characterization of recombination deficient mutants of Escherichia coli K12. Proc Natl Acad Sci USA. 1965;53:451–459. doi: 10.1073/pnas.53.2.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox MM. Regulation of bacterial RecA protein function. Crit Rev Biochem Mol Biol. 2007;42:41–63. doi: 10.1080/10409230701260258. [DOI] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta D, Shatalin K, Epshtein V, Gottesman ME, Nudler E. Linking RNA polymerase backtracking to genome instability in E. coli. Cell. 2011;146:533–543. doi: 10.1016/j.cell.2011.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epshtein V, Dutta D, Wade J, Nudler E. An allosteric mechanism of Rho-dependent transcription termination. Nature. 2010;463:245–249. doi: 10.1038/nature08669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuoh A, Iwasaki H, Ishioka K, Shinagawa H. ATP-dependent resolution of R-loops at the ColE1 replication origin by Escherichia coli RecG protein, a Holliday junction-specific helicase. EMBO J. 1997;16:203–209. doi: 10.1093/emboj/16.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbai CB, Marians KJ. Recruitment to stalled replication forks of the PriA DNA helicase and replisome-loading activities is essential for survival. DNA Repair (Amst) 2010;9:202–209. doi: 10.1016/j.dnarep.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg AV, McGlynn P, Jaktaji RP, Lloyd RG. Direct rescue of stalled DNA replication forks via the combined action of PriA and RecG helicase activities. Mol Cell. 2002;9:241–251. doi: 10.1016/s1097-2765(02)00455-0. [DOI] [PubMed] [Google Scholar]
- Guterman SK, Howitt CL. Rho and ribosome mutation interaction: lethality of rho-15 in rpsL or rpsE strains, and rho-15 methionine auxotrophy in rps+ strains of Escherichia coli. Genetics. 1979;93:353–360. doi: 10.1093/genetics/93.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy CP, Atkinson J, Gupta MK, Mahdi AA, Gwynn EJ, Rudolph CJ, et al. Rep provides a second motor at the replisome to promote duplication of protein-bound DNA. Mol Cell. 2009;36:654–666. doi: 10.1016/j.molcel.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harinarayanan R, Gowrishankar J. Host factor titration by chromosomal R-loops as a mechanism for runaway plasmid replication in transcription termination-defective mutants of Escherichia coli. J Mol Biol. 2003;332:31–46. doi: 10.1016/s0022-2836(03)00753-8. [DOI] [PubMed] [Google Scholar]
- Heller RC, Marians KJ. The disposition of nascent strands at stalled replication forks dictates the pathway of replisome loading during restart. Mol Cell. 2005;17:733–743. doi: 10.1016/j.molcel.2005.01.019. [DOI] [PubMed] [Google Scholar]
- Hong X, Cadell GW, Kogoma T. Escherichia coli RecG and RecA proteins in R-loop formation. EMBO J. 1995;14:2385–2392. doi: 10.1002/j.1460-2075.1995.tb07233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi T, Maki H, Sekiguchi M. RNase H-defective mutants of Escherichia coli: a possible discriminatory role of RNase H in initiation of DNA replication. Mol Gen Genet. 1984;195:17–22. doi: 10.1007/BF00332717. [DOI] [PubMed] [Google Scholar]
- Jaktaji RP, Lloyd RG. PriA supports two distinct pathways for replication restart in UV-irradiated Escherichia coli cells. Mol Microbiol. 2003;47:1091–1100. doi: 10.1046/j.1365-2958.2003.03357.x. [DOI] [PubMed] [Google Scholar]
- Kang WK, Icho T, Isono S, Kitakawa M, Isono K. Characterization of the gene rimK responsible for the addition of glutamic acid residues to the C-terminus of ribosomal protein S6 in Escherichia coli K12. Mol Gen Genet. 1989;217:281–288. doi: 10.1007/BF02464894. [DOI] [PubMed] [Google Scholar]
- Kim S, Dallmann HG, McHenry CS, Marians KJ. Coupling of a replicative polymerase and helicase: a tau–DnaB interaction mediates rapid replication fork movement. Cell. 1996a;84:643–650. doi: 10.1016/s0092-8674(00)81039-9. [DOI] [PubMed] [Google Scholar]
- Kim S, Dallmann HG, McHenry CS, Marians KJ. tau couples the leading- and lagging-strand polymerases at the Escherichia coli DNA replication fork. J Biol Chem. 1996b;271:21406–21412. doi: 10.1074/jbc.271.35.21406. [DOI] [PubMed] [Google Scholar]
- Kogoma T, Cadwell GW, Barnard KG, Asai T. The DNA replication priming protein, PriA, is required for homologous recombination and double-strand break repair. J Bacteriol. 1996;178:1258–1264. doi: 10.1128/jb.178.5.1258-1264.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBowitz JH, McMacken R. The Escherichia coli dnaB replication protein is a DNA helicase. J Biol Chem. 1986;261:4738–4748. [PubMed] [Google Scholar]
- Lee EH, Kornberg A. Replication deficiencies in priA mutants of Escherichia coli lacking the primosomal replication n′ protein. Proc Natl Acad Sci USA. 1991;88:3029–3032. doi: 10.1073/pnas.88.8.3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Xu L, Sandler SJ, Marians KJ. Replication fork assembly at recombination intermediates is required for bacterial growth. Proc Natl Acad Sci USA. 1999;96:3552–3555. doi: 10.1073/pnas.96.7.3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd RG. Conjugational recombination in resolvase-deficient ruvC mutants of Escherichia coli K-12 depends on recG. J Bacteriol. 1991;173:5414–5418. doi: 10.1128/jb.173.17.5414-5418.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd RG, Buckman C. Conjugational recombination in Escherichia coli: genetic analysis of recombinant formation in Hfr x F− crosses. Genetics. 1995;139:1123–1148. doi: 10.1093/genetics/139.3.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd RG, Benson FE, Shurvinton CE. Effect of ruv mutations on recombination and DNA repair in Escherichia coli K12. Mol Gen Genet. 1984;194:303–309. doi: 10.1007/BF00383532. [DOI] [PubMed] [Google Scholar]
- Lloyd RG, Evans NP, Buckman C. Formation of recombinant lacZ+ DNA in conjugational crosses with a recB mutant of Escherichia coli K12 depends on recF, recJ, and recO. Mol Gen Genet. 1987;209:135–141. doi: 10.1007/BF00329848. [DOI] [PubMed] [Google Scholar]
- Lloyd RG, Porton MC, Buckman C. Effect of recF, recJ, recN, recO and ruv mutations on ultraviolet survival and genetic recombination in a recD strain of Escherichia coli K-12. Mol Gen Genet. 1988;212:317–324. doi: 10.1007/BF00334702. [DOI] [PubMed] [Google Scholar]
- Lopper M, Boonsombat R, Sandler SJ, Keck JL. A hand-off mechanism for primosome assembly in replication restart. Mol Cell. 2007;26:781–793. doi: 10.1016/j.molcel.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCool JD, Ford CC, Sandler SJ. A dnaT mutant with phenotypes similar to those of a priA2:kan mutant in Escherichia coli K-12. Genetics. 2004;167:569–578. doi: 10.1534/genetics.103.025296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlynn P, Lloyd RG. Modulation of RNA polymerase by (p)ppGpp reveals a RecG-dependent mechanism for replication fork progression. Cell. 2000;101:35–45. doi: 10.1016/S0092-8674(00)80621-2. [DOI] [PubMed] [Google Scholar]
- McGlynn P, Lloyd RG. Rescue of stalled replication forks by RecG: simultaneous translocation on the leading and lagging strand templates supports an active DNA unwinding model of fork reversal and Holliday junction formation. Proc Natl Acad Sci USA. 2001;98:8227–8234. doi: 10.1073/pnas.111008698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlynn P, Al-Deib AA, Liu J, Marians KJ, Lloyd RG. The DNA replication protein PriA and the recombination protein RecG bind D-loops. J Mol Biol. 1997;270:212–221. doi: 10.1006/jmbi.1997.1120. [DOI] [PubMed] [Google Scholar]
- Magner DB, Blankschien MD, Lee JA, Pennington JM, Lupski JR, Rosenberg SM. RecQ promotes toxic recombination in cells lacking recombination intermediate-removal proteins. Mol Cell. 2007;26:273–286. doi: 10.1016/j.molcel.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mah TF, Kuznedelov K, Mushegian A, Severinov K, Greenblatt J. The alpha subunit of E. coli RNA polymerase activates RNA binding by NusA. Genes Dev. 2000;14:2664–2675. doi: 10.1101/gad.822900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahdi AA, Sharples GJ, Mandal TN, Lloyd RG. Holliday junction resolvases encoded by homologous rusA genes in Escherichia coli K-12 and phage 82. J Mol Biol. 1996;257:561–573. doi: 10.1006/jmbi.1996.0185. [DOI] [PubMed] [Google Scholar]
- Mahdi AA, Buckman C, Harris L, Lloyd RG. Rep and PriA helicase activities prevent RecA from provoking unnecessary recombination during replication fork repair. Genes Dev. 2006;20:2135–2147. doi: 10.1101/gad.382306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal TN, Mahdi AA, Sharples GJ, Lloyd RG. Resolution of Holliday intermediates in recombination and DNA repair: indirect suppression of ruvA, ruvB and ruvC mutations. J Bacteriol. 1993;175:4325–4334. doi: 10.1128/jb.175.14.4325-4334.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masai H, Asai T, Kubota Y, Arai K, Kogoma T. Escherichia coli PriA protein is essential for inducible and constitutive stable DNA replication. EMBO J. 1994;13:5338–5345. doi: 10.1002/j.1460-2075.1994.tb06868.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrikh H, Machon C, Grainger WH, Grossman AD, Soultanas P. Co-directional replication–transcription conflicts lead to replication restart. Nature. 2011;470:554–557. doi: 10.1038/nature09758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messer W. The bacterial replication initiator DnaA. DnaA and oriC, the bacterial mode to initiate DNA replication. FEMS Microbiol Rev. 2002;26:355–374. doi: 10.1111/j.1574-6976.2002.tb00620.x. [DOI] [PubMed] [Google Scholar]
- von Meyenburg K, Boye E, Skarstad K, Koppes L, Kogoma T. Mode of initiation of constitutive stable DNA replication in RNase H-defective mutants of Escherichia coli K-12. J Bacteriol. 1987;169:2650–2658. doi: 10.1128/jb.169.6.2650-2658.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JH. Experiments in Molecular Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1972. [Google Scholar]
- Mirkin EV, Mirkin SM. Replication fork stalling at natural impediments. Microbiol Mol Biol Rev. 2007;71:13–35. doi: 10.1128/MMBR.00030-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nudler E. RNA polymerase active center: the molecular engine of transcription. Annu Rev Biochem. 2009;78:335–361. doi: 10.1146/annurev.biochem.76.052705.164655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurse P, Zavitz KH, Marians KJ. Inactivation of the Escherichia coli PriA DNA replication protein induces the SOS response. J Bacteriol. 1991;173:6686–6693. doi: 10.1128/jb.173.21.6686-6693.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurse P, Liu J, Marians KJ. Two modes of PriA binding to DNA. J Biol Chem. 1999;274:25026–25032. doi: 10.1074/jbc.274.35.25026. [DOI] [PubMed] [Google Scholar]
- Ogawa T, Pickett GG, Kogoma T, Kornberg A. RNase H confers specificity in the dnaA-dependent initiation of replication at the unique origin of the Escherichia coli chromosome in vivo and in vitro. Proc Natl Acad Sci USA. 1984;81:1040–1044. doi: 10.1073/pnas.81.4.1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeh S, Pedersen S. Post-translational modification of Escherichia coli ribosomal protein S6. Mol Gen Genet. 1979;173:183–187. doi: 10.1007/BF00330309. [DOI] [PubMed] [Google Scholar]
- Rudolph CJ, Dhillon P, Moore T, Lloyd RG. Avoiding and resolving conflicts between DNA replication and transcription. DNA Repair (Amst) 2007a;6:981–993. doi: 10.1016/j.dnarep.2007.02.017. [DOI] [PubMed] [Google Scholar]
- Rudolph CJ, Upton AL, Lloyd RG. Replication fork stalling and cell cycle arrest in UV-irradiated Escherichia coli. Genes Dev. 2007b;21:668–681. doi: 10.1101/gad.417607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph CJ, Upton AL, Harris L, Lloyd RG. Pathological replication in cells lacking RecG DNA translocase. Mol Microbiol. 2009a;73:352–366. doi: 10.1111/j.1365-2958.2009.06773.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph CJ, Upton AL, Lloyd RG. Replication fork collisions cause pathological chromosomal amplification in cells lacking RecG DNA translocase. Mol Microbiol. 2009b;74:940–955. doi: 10.1111/j.1365-2958.2009.06909.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph CJ, Mahdi AA, Upton AL, Lloyd RG. RecG protein and single-strand DNA exonucleases avoid cell lethality associated with PriA helicase activity in Escherichia coli. Genetics. 2010a;186:473–492. doi: 10.1534/genetics.110.120691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph CJ, Upton AL, Briggs GS, Lloyd RG. Is RecG a general guardian of the bacterial genome? DNA Repair (Amst) 2010b;9:210–223. doi: 10.1016/j.dnarep.2009.12.014. [DOI] [PubMed] [Google Scholar]
- Sandler SJ. Multiple genetic pathways for restarting DNA replication forks in Escherichia coli K-12. Genetics. 2000;155:487–497. doi: 10.1093/genetics/155.2.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandler SJ, Marians KJ. Role of PriA in replication fork reactivation in Escherichia coli. J Bacteriol. 2000;182:9–13. doi: 10.1128/jb.182.1.9-13.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandler SJ, Samra HS, Clark AJ. Differential suppression of priA2:kan phenotypes in Escherichia coli K-12 by mutations in priA, lexA, and dnaC. Genetics. 1996;143:5–13. doi: 10.1093/genetics/143.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandler SJ, Marians KJ, Zavitz KH, Coutu J, Parent MA, Clark AJ. dnaC mutations suppress defects in DNA replication- and recombination-associated functions in priB and priC double mutants in Escherichia coli K-12. Mol Microbiol. 1999;34:91–101. doi: 10.1046/j.1365-2958.1999.01576.x. [DOI] [PubMed] [Google Scholar]
- Sassanfar M, Roberts JW. Nature of the SOS-inducing signal in Escherichia coli. The involvement of DNA replication. J Mol Biol. 1990;212:79–96. doi: 10.1016/0022-2836(90)90306-7. [DOI] [PubMed] [Google Scholar]
- Smith GR. Conjugational recombination in E. coli: Myths and mechanisms. Cell. 1991;64:19–27. doi: 10.1016/0092-8674(91)90205-d. [DOI] [PubMed] [Google Scholar]
- Tabor S, Richardson CC. A bacteriophage T7 RNA polymerase/promoter system for controlled exclusive expression of specific genes. Proc Natl Acad Sci USA. 1985;82:1074–1078. doi: 10.1073/pnas.82.4.1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Masai H. Stabilization of a stalled replication fork by concerted actions of two helicases. J Biol Chem. 2006;281:3484–3493. doi: 10.1074/jbc.M510979200. [DOI] [PubMed] [Google Scholar]
- Thomas MS, Glass RE. Escherichia coli rpoA mutation which impairs transcription of positively regulated systems. Mol Microbiol. 1991;5:2719–2725. doi: 10.1111/j.1365-2958.1991.tb01980.x. [DOI] [PubMed] [Google Scholar]
- Tougu K, Peng H, Marians KJ. Identification of a domain of Escherichia coli primase required for functional interaction with the DnaB helicase at the replication fork. J Biol Chem. 1994;269:4675–4682. [PubMed] [Google Scholar]
- Trautinger BW, Lloyd RG. Modulation of DNA repair by mutations flanking the DNA channel through RNA polymerase. EMBO J. 2002;21:6944–6953. doi: 10.1093/emboj/cdf654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trautinger BW, Jaktaji RP, Rusakova E, Lloyd RG. RNA polymerase modulators and DNA repair activities resolve conflicts between DNA replication and transcription. Mol Cell. 2005;19:247–258. doi: 10.1016/j.molcel.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Vincent SD, Mahdi AA, Lloyd RG. The RecG branch migration protein of Escherichia coli dissociates R-loops. J Mol Biol. 1996;264:713–721. doi: 10.1006/jmbi.1996.0671. [DOI] [PubMed] [Google Scholar]
- Washburn RS, Gottesman ME. Transcription termination maintains chromosome integrity. Proc Natl Acad Sci USA. 2011;108:792–797. doi: 10.1073/pnas.1009564108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willetts N, Wilkins B. Processing of plasmid DNA during bacterial conjugation. Microbiol Rev. 1984;48:24–41. doi: 10.1128/mr.48.1.24-41.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Mahdi AA, Briggs GS, Lloyd RG. Promoting and avoiding recombination: contrasting activities of the Escherichia coli RuvABC Holliday junction resolvase and RecG DNA translocase. Genetics. 2010;185:23–37. doi: 10.1534/genetics.110.114413. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.