

Abstract
Filamentous fungi are a common cause of blindness and visual impairment worldwide. Using both murine model systems and in vitro human neutrophils, we found that NADPH oxidase produced by neutrophils was essential to control the growth of Aspergillus and Fusarium fungi in the cornea. We demonstrated that neutrophil oxidant production and antifungal activity are dependent on CD18, but not on the β-glucan receptor dectin-1. We used mutant A. fumigatus strains to show that the reactive oxygen species–sensing transcription factor Yap1, superoxide dismutases, and the Yap1-regulated thioredoxin antioxidant pathway are each required for protection against neutrophil-mediated oxidation of hyphae as well as optimal survival of fungal hyphae in vivo. We also demonstrated that thioredoxin inhibition using the anticancer drug PX-12 increased the sensitivity of fungal hyphae to both H2O2- and neutrophil-mediated killing in vitro. Additionally, topical application of PX-12 significantly enhanced neutrophil-mediated fungal killing in infected mouse corneas. Cumulatively, our data reveal critical host oxidative and fungal anti-oxidative mediators that regulate hyphal survival during infection. Further, these findings also indicate that targeting fungal anti-oxidative defenses via PX-12 may represent an efficacious strategy for treating fungal infections.
Introduction
Pathogenic fungi, such as Aspergillus and Fusarium species, can cause lethal pulmonary and systemic disease in immune-suppressed individuals, including those with HIV infection (1, 2). These organisms are also a major cause of infectious blindness and corneal ulcers in immunocompetent individuals, and in contrast to individuals with systemic and pulmonary fungal infections, there is no indication that fungal keratitis patients are other than fully immunocompetent (3, 4). In the hot and humid southeastern United States, fungal infections of the cornea account for up to 35% of all corneal ulcers (5, 6). Globally, fungal infections of the cornea account for up to 65% of corneal ulcers, with estimates of 80,000 total cases and 10,000 cornea transplants per year due to fungal infections in India alone (7–12). Other risk factors for disease in the USA, Britain, and Europe include contact lens wear, as illustrated by a 2005–2006 fungal keratitis outbreak associated with a lens care product (13). A. flavus, A. fumigatus, F. solani, and F. oxysporum are the main etiologic agents of fungal keratitis (14). These organisms are prevalent in vegetative matter and suspended in air, and are inoculated into the corneal stroma via traumatic injury associated with agricultural work (14). Current treatment with topical antimycotics is often ineffective, with up to 60% of cases requiring corneal transplantation (3, 14, 15).
Neutrophils are the predominant cell type infiltrating fungus-infected lungs and corneas, and contribute to tissue destruction by release of proteolytic enzymes and reactive oxygen and nitrogen species (13–17). Our recent studies characterizing fungus-infected human corneas in India showed that neutrophils constitute greater than 90% of cellular infiltrates in corneal ulcers in patients infected for less than 7 days and more than 70% total infiltrate at later stages of infection (18). Similarly, neutrophils are the first cells recruited to the corneal stroma in murine models of Aspergillus and Fusarium keratitis (19, 20), indicating that neutrophils are the main effector cells required for killing fungal hyphae. A role for neutrophils in control of fungal infection is also suggested by the increased incidence of systemic and pulmonary fungal infections in patients with neutropenia (2).
Neutrophils produce NADPH oxidase (NOX), which catalyzes the conversion of molecular O2 to superoxide anion (O2–) with the release of ROS and protons into the extracellular space (17, 21). Individuals with inherited defects in NOX such as in chronic granulomatous disease (CGD) exhibit an increased incidence of bacterial and fungal infections, supporting the concept that the specific expression of NOX by neutrophils is required for killing of fungi (22). However, even though it is the hyphal stage of these organisms that is invasive, most studies on CGD patients and transgenic mice with mutations in NOX genes have focused only on the role of NOX in killing conidia (23–26). Infected human corneas and lungs exhibit primarily hyphal stages of fungal growth, and conidia are rarely detected. Given that hyphae are significantly larger in size, and are not readily phagocytosed, they are likely killed through distinct mechanisms not required for anti-conidial defenses, and a recent study suggests that NOX is not required to control the growth of all filamentous fungi (27).
In the current study, we examined the role of ROS in killing Aspergillus and Fusarium hyphae by human neutrophils and in a murine model of fungal keratitis. We show that hyphae activate neutrophil NOX through CD18 and that NOX activation is essential for killing hyphae. In addition, utilizing mutant A. fumigatus strains, we show that the ROS-sensing transcription factor Yap1, the ROS-detoxifying enzyme superoxide dismutase, and the Yap1-regulated thioredoxin antioxidant pathway, but not catalases or fungal secondary metabolites such as gliotoxin are required for resistance to oxidation by neutrophils. Last, using pharmacologic inhibitors of thioredoxin, we provide proof of concept that targeting fungal anti-oxidative stress responses can enhance fungal clearance from infected tissues and may represent a new avenue for treatment of fungal infections.
Results
Neutrophils have an essential role in regulating fungal growth in the cornea.
To examine the role of neutrophils in fungal keratitis, we used two complementary approaches: systemic depletion of neutrophils from immune-competent C57BL/6 mice and adoptive transfer of neutrophils into Cxcr2–/– and Cd18–/– mice. In the first approach, neutrophils were depleted from transgenic C57BL/6 mice expressing eGFP downstream of the promoter LysM (LysM-eGFP mice; ref. 28) by i.p. injection of a neutrophil-specific monoclonal antibody (NIMPR-14), while control mice were given rat isotype antibody. Injection of 400 μg NIMP antibody on day 1 resulted in significantly decreased neutrophils in peripheral blood smears at 0, 24, and 48 hours after infection (S.M. Leal Jr., unpublished observations). After 24 hours, corneas were infected with conidia (40,000 in 2 μl) isolated from the A. fumigatus strain Af-dsRed, which constitutively expresses the red fluorescent protein dsRed under control of the glyceraldehyde dehydrogenase promoter. Subsequently, RFP+ fungal growth and eGFP+ neutrophil infiltration were assessed in live corneas. We also examined the effect of neutrophils on corneal opacity.
Figure 1, A and B, show-significantly increased eGFP+ neutrophils at 24 and 48 hours after infection in control, isotype-treated mice, but not in neutrophil-depleted (NIMP) mice, thus confirming neutrophil depletion in NIMP-treated mice. Conversely, Figure 1, A and C, shows significantly decreased dsRed-expressing fungal hyphae at 24 and 48 hours in isotype controls compared with NIMP-treated mice, which is consistent with increased CFU in NIMP-treated mice (Figure 1D). We also found significantly lower corneal opacity in neutrophil-depleted mice as measured by both area of opacity and total corneal opacity at 24 hours but not 48 hours after infection. Corneal opacity at 48 hours was likely due to fungus-mediated tissue pathology (image analysis methods described in Supplemental Figure 1; supplemental material available online with this article; doi: 10.1172/JCI63239DS1) (Figure 1, E and F). Figure 1A also shows a representative PAS-hematoxylin (PASH)–stained cornea section of a 48-hour-infected LysM-eGFP mouse with infiltrating cells in the stroma and anterior chamber, and few intact fungal hyphae. In contrast, the corneas of neutrophil-depleted mice exhibited reduced cellular infiltrates and prominent fungal hyphae in both the corneal stroma and the anterior chamber.
Figure 1. Neutrophil depletion enhances fungal growth during corneal infection.

(A) Transgenic C57BL/6 mice with neutrophil-specific eGFP expression downstream of the lysozyme promotor (LysM) were depleted of neutrophils (Neuts) with neutrophil-specific NIMPR-14 antibody (i.p.) and infected with 40,000 Af-dsRed conidia. Eyes were imaged at 24 and 48 hours after infection for neutrophil infiltration (eGFP), fungal growth (dsRed), and corneal opacity (BF). In addition, PASH stains were performed on 5-μm sections of corneas at 48 hours after infection. (B) MetaMorph software was used to quantify neutrophil infiltration (eGFP emission) and (C) fungal dsRed expression. (D) At 4 and 48 hours after infection, eyes were homogenized and plated on SDA plates and CFU quantified by direct counting. MetaMorph software was utilized to quantify (E) corneal opacity area and (F) total corneal opacity (described in detail in Supplemental Figure 1). Three independent experiments (n = 5) were performed. *P < 0.05. Original magnification, ×20 (eye images); ×400 (histology).
As a second approach, we utilized two mouse strains with known defects in neutrophil infiltration during infection and adoptively transferred WT naive bone marrow–derived neutrophils (BMNs) into these mice to study the specific role of neutrophils in killing fungi during corneal infection. Cxcr2–/– neutrophils are unable to recognize and respond to ELR+ CXC chemokines, whereas Cd18–/– neutrophils are unable to bind to ICAM-1 on limbal vessel vascular endothelial cells (29, 30).
C57BL/6, Cxcr2+/–, and Cxcr2–/– corneas were infected with A. fumigatus conidia as described above. Figure 2A shows fungal dsRed expression and corneal opacity in C57BL/6 and Cxcr2+/– heterozygous mice, which both increase at 48 hours after infection. In contrast, infected Cxcr2–/– mice had significantly increased fungal dsRed expression (Figure 2B) and lower corneal opacity scores (Figure 2, C and D) at 24 and 48 hours compared with C57BL/6 mice. However, Cxcr2–/– mice given C57BL/6 syngeneic neutrophils i.v. had significantly lower fungal dsRed values (Figure 2B) and higher corneal opacity (Figure 2, C and D) compared with Cxcr2–/– mice not receiving neutrophils, indicating that neutrophils contribute to both fungal killing and corneal opacity. Corneal sections from these mice showed intense cellular infiltration, neutrophil recruitment, and minimal intact fungal hyphae at 48 hours after infection in C57BL/6 mice and Cxcr2+/– mice, whereas Cxcr2–/– corneas exhibited minimal cellular infiltrates, but abundant hyphae in the stroma and anterior chamber (Figure 2A). Following adoptive transfer of C57BL/6 neutrophils into Cxcr2–/– mice, neutrophils were detected in the corneal stroma, and fungal growth in the cornea was lower than in the absence of neutrophils (Figure 2A).
Figure 2. Neutrophil adoptive transfer restricts fungal growth during corneal infection.

(A) C57BL/6, Cxcr2+/–, and Cxcr2–/– mice were infected with 30,000 Af-dsRed conidia. At 24 hours after infection, one group of fungus-infected Cxcr2–/– mice were injected i.v. with 4 × 106 BMNs from C57BL/6 (B6) mice. At 24 hours after infection, corneas were imaged for cellular infiltration, fungal growth, and corneal opacity. At this time point, mice were euthanized, eyes were fixed in formalin, and 5-μm corneal sections were PASH stained. (B) MetaMorph software was used to quantify fungal dsRed expression, (C) corneal opacity area, and (D) total corneal opacity in infected corneas. (E) Similar to Cxcr2–/– mice, C57BL/6 and Cd18–/– mice were infected with Af-dsRed conidia, and at 24 hours after infection one group of infected Cd18–/– mice were given 4 million adoptively transferred BMNs isolated from a LysM-eGFP mouse (eGFP+ Neuts) and eyes were imaged at 48 hours. (F) Fungal dsRed expression, (G) eGFP+ neutrophil infiltration, (H) corneal opacity area, and (I) total corneal opacity were quantified using MetaMorph software. Three independent experiments (n = 5) were performed. *P < 0.05. Original magnification, ×20 (eye images); ×400 (histology).
Similarly, C57BL/6 neutrophils transferred to Cd18–/– mice during infection conferred a protective response with increased cell migration to the cornea and lower fungal load. Figure 2, E and F, shows that fungal dsRed expression at 48 hours was significantly increased in Cd18–/– mice compared with C57BL/6 mice. However, Cd18–/– mice given syngeneic BMNs from transgenic C57BL/6 LysM-eGFP mice exhibited significantly decreased fungal dsRed expression (Figure 2F) along with significantly increased eGFP+ neutrophil infiltration (Figure 2G) and increased corneal opacity (Figure 2, H and I).
Together, these findings demonstrate that both CXCR2 and CD18 regulate neutrophil recruitment to fungus-infected corneas and that neutrophils have an essential role in controlling fungal growth at this site.
Neutrophil NOX activity is required for control of fungal growth during corneal infection.
NOX is an enzyme complex required for reduction of molecular O2 to O2– (31). Superoxide is the limiting reagent in subsequent reactions, leading to the transient synthesis of ROS with greater oxidative and fungal killing potential (21), and individuals with CGD due to impaired NOX function are unable to control microbial infections and often succumb to filamentous fungal infections in the lung (2, 32).
Cybb–/– mice lack the gene encoding the NOX subunit gp91phox and thus do not express a functional NOX complex. These mice are also more susceptible to Aspergillus lung infections (23). To determine the role of NOX in fungal keratitis, we infected Cybb–/– mice with Af-dsRed, and ROS levels in the corneas were measured at 48 hours after infection after intrastromal injection of carboxyfluorescein diacetate (CFDA), which emits green fluorescence upon oxidation. Fungal dsRed expression and corneal opacification were measured by image analysis as described above. Figure 3, A and B, shows CFDA activity in C57BL/6 and Cybb–/– corneas at 48 hours after infection; however, total fluorescence was significantly lower in Cybb–/– corneas (there was no fluorescence in naive corneas injected with CFDA; S.M. Leal Jr., unpublished observation). Conversely, fungal dsRed expression and CFU (Figure 3, C and D) were elevated in Cybb–/– compared with C57BL/6 corneas, indicating impaired fungal clearance in Cybb–/– corneas. Interestingly, Cybb–/– corneas had more severe disease than in C57BL/6 mice, with significantly higher total corneal opacity scores (Figure 3, E and F), which correlated with increased neutrophil infiltration and formation of microabscesses and the presence of intact fungal hyphae (Figure 3G). As there is no defect in the ability of Cybb–/– neutrophils to migrate to the cornea, it is likely that these findings represent “frustrated” neutrophils that are unable to kill hyphae but can still recruit neutrophils to this site. Very similar results were found when mice were infected with other pathogenic Aspergillus and Fusarium species that cause keratitis (Supplemental Figures 2–4), indicating that ROS has a more general role in inhibiting growth of filamentous fungi.
Figure 3. Neutrophil NOX is required for control of A. fumigatus fungal growth during corneal infection.

(A) C57BL/6 mice and Cybb–/– mice were infected with 40,000 A. fumigatus strain Af-dsRed conidia. Eyes were imaged at 48 hours after infection for ROS-mediated CFDA dye oxidation, fungal dsRed expression, and corneal opacity. (B) CFDA dye oxidation, (C) fungal dsRed expression, (D) CFU, (E) corneal opacity area, and (F) total corneal opacity were quantified after infection. (G) 5-μm sections of 48-hour-infected fungal corneas were stained with PASH or neutrophil-specific NIMP antibody. (H) Cd18–/– mice were infected with A. fumigatus strain Af-dsRed conidia. At 2 hours after infection, one set of mice were left untreated, the second set received an i.v. injection of 4 × 106 BMNs isolated from C57BL/6 mice, and a third set received the same number of neutrophils isolated from Cybb–/– mice. (I) At 24 hours after infection, eyes were imaged and fungal dsRed expression quantified using MetaMorph software. Three independent experiments (n = 5) were performed. *P < 0.05. Original magnification, ×20 (eye images); ×400 (histology).
To ascertain directly whether the impaired fungal killing in Cybb–/– mice is due to NOX that is specifically produced by neutrophils, we infected Cd18–/– mice with Af-dsRed as described above and injected BMNs from C57BL/6 or Cybb–/– mice i.v. 2 hours after infection. Corneas were imaged 24 hours after infection. Figure 3, H and I, shows that fungal dsRed expression was significantly reduced following adoptive transfer of C57BL/6 neutrophils, whereas mice given Cybb–/– neutrophils had the same fungal dsRed expression as mice not receiving neutrophils. These data clearly demonstrate that NOX-dependent ROS production by neutrophils is essential for inhibiting fungal growth in the cornea.
iNOS and RNS are not essential for control of fungal growth.
The superoxide produced by NOX can be converted to ROS or alternatively can react with NO produced by the enzymatic cleavage of arginine by iNOS, and can form the highly reactive nitrogen species (RNS) peroxynitrite (ONOO–) (21). If iNOS activity is high and significant amounts of NO are produced, the end products of NOX activity will shift from production of ROS to production of RNS.
To test the hypothesis that RNSs are required for control of fungal growth during fungal keratitis, we injected C57BL/6 mice systemically with 1400W, which is an irreversible pharmacologic inhibitor of iNOS. In addition, the role of iNOS was examined in gene-knockout mice.
Figure 4 shows that there were no significant differences in corneal opacification or fungal CFU between iNOS–/– and C57BL/6 mice, or between untreated and 1400W-treated C57BL/6 mice. These findings indicate that in contrast to NOX and ROS, neither iNOS nor RNS have an essential role in control of fungal growth during corneal infection.
Figure 4. iNOS is not required for control of fungal growth during corneal infection.

(A) C57BL/6, iNOS–/–, and 1400W-treated C57BL/6 mice were infected with A. fumigatus strain Af-BP conidia, and eyes were imaged at 24 and 48 hours after infection. (B) Corneal opacity area, (C) total corneal opacity, and (D) CFU were quantified in infected corneas after infection. Three independent experiments (n = 5) were performed.
Human and murine neutrophil-mediated killing of A. fumigatus, A. flavus, and F. oxysporum hyphae is dependent on NOX, but not iNOS or myeloperoxidase.
As neutrophil NOX is required for control of fungal growth in vivo, but both conidia and hyphae are present during corneal infection, we developed a neutrophil-hypha coincubation assay to determine which neutrophil mediators are required to limit the growth of fungal hyphae. Af-dsRed, A. flavus eGFP, and F. oxysporum FoxL-GFP conidia were incubated in 96-well plates for 6 hours to allow time for germination and hyphal growth. For human neutrophil studies, neutrophils were isolated from the peripheral blood of normal volunteers and added to each well at a 16:1 ratio in the presence of pharmacological inhibitors of NOX, iNOS, or myeloperoxidase (MPO) (NOX: diphenyliodonium [DPI], apocynin [Apo]; iNOS: aminoguanidine HCl [Agd], 2-methyl-2-thiopseudoureasulfate [SMT]; MPO: indomethacin [Indo], 4-aminobenzoic hydrazide [4-AH]). For mouse neutrophil studies, neutrophils were isolated from the peritoneal cavities of C57BL/6, Cybb–/–, and iNOS–/– mice and incubated with fungal hyphae as described above. Fungal growth was observed by fluorescence microscopy and measured by fluorescence spectroscopy.
Figure 5A shows hyphal growth of A. fumigatus and A. flavus, and Supplemental Figure 6A shows hyphal growth of F. oxysporum after 16 hours incubation in RPMI. However, in the presence of normal human neutrophils, fungal growth was clearly reduced, indicating that normal human neutrophils inhibit hyphal growth. However, addition of the NOX inhibitor DPI resulted in hyphal growth levels similar to that with RPMI alone, which is consistent with NOX-dependent killing. Figure 5B shows quantification of the growth of A. fumigatus exposed to human neutrophils for 16 hours in the presence or absence of inhibitors. Neutrophil-mediated killing was inhibited in the presence of the NOX inhibitors DPI and Apo, but not iNOS or MPO inhibitors.
Figure 5. NOX but not iNOS or MPO is required for human and mouse neutrophils to control hyphal growth.

Fungal conidia were cultured in SDB media for 6 hours in 96-well plates. Neutrophils were added to each well containing hyphae, and the cells were coincubated for an additional 16 hours. In certain experiments, the NOX inhibitors (DPI, Apo), iNOS inhibitors (SMT, AgD), or MPO inhibitors (Indo, 4-AH) were added to the wells. (A) Af-dsRed and eGFP-expressing A. flavus (70-GFP) were cultured either alone in PBS or RPMI or coincubated with 2 × 105 human neutrophils in RPMI or the same number of neutrophils in RPMI plus DPI. Fungal fluorescence was imaged at 16 hours after incubation. Original magnification, ×400. (B) Af-dsRed was coincubated with neutrophils as described above for 16 hours, plates were washed and stained with calcofluor white, and fungal chitin content determined via fluorometry. Tx, treatment. (C and D) To examine the role of NOX in mouse neutrophil-mediated killing of fungal hyphae, we grew Af-dsRed conidia as described above for 6 hours and coincubated them with thioglycolate-elicited peritoneal neutrophils from WT C57BL/6, (C) Cybb–/–, and (D) iNOS–/– mice. At 16 hours after infection, chitin content was quantified using fluorometry. (E) Similarly, A. flavus (70-GFP) neutrophil-hypha coincubation assays were performed with human neutrophils and (F) Cybb–/– mice and (G) iNOS–/– mice. Three independent experiments were performed. *P < 0.05.
As a complementary approach, we examined the role of NOX and iNOS in Cybb–/–, iNOS–/–, and C57BL/6 mouse neutrophils. Importantly, ROS was detected after A. fumigatus incubation with C57BL/6, but not Cybb–/–, neutrophils (Supplemental Figure 5), indicating that most of the ROS was dependent on NOX. In contrast, there was no difference in NO production between Cybb–/– and C57BL/6 neutrophils, and ROS production by iNOS–/– neutrophils was not significantly different from that by C57BL/6 neutrophils, while iNOS–/– neutrophils produced minimal NO (Supplemental Figure 5).
As shown in Figure 5C, A. fumigatus growth in the presence of C57BL/6 neutrophils was lower than with RPMI alone. In contrast, fungal growth after incubation with Cybb–/– neutrophils was significantly higher than after incubation with C57BL/6 neutrophils and similar to that after incubation with no neutrophils, and is consistent with impaired ROS activity and fungal killing. However, fungal growth was not significantly different between iNOS–/– and C57BL/6 neutrophils (Figure 5D). Similar results were obtained for A. flavus (Figure 5, E–G) and F. oxysporum (Supplemental Figure 6, B–D). Taken together, these data indicate that inhibition of A. fumigatus, A. flavus, and F. oxysporum hyphal growth by human and mouse neutrophils is dependent on NOX and ROS production but not iNOS or MPO.
ROS production and fungal killing are dependent on CD18 but not dectin-1.
Activation of NOX is regulated by the physical separation of membrane and cytoplasmic components of the complex. However, following cellular activation by pathogen recognition or cytokine receptors, p47phox in the cytoplasm is phosphorylated and translocated along with other cytoplasmic NOX components to the membrane, thereby forming the functional enzyme (33). Dectin-1 and CD18 recognize fungal cell wall β-glucan and can activate NOX (19, 34, 35), and dectin-1–/– and Cd18–/– mice exhibit impaired fungal clearance in cornea infection models (19), suggesting a possible role for these receptors in neutrophil-mediated fungal killing. To examine whether these cell surface receptors mediate NOX activation and ROS production by neutrophils following exposure to fungal hyphae, we loaded naive BMNs with the ROS-sensitive dye CFDA, incubated them with A. fumigatus hyphae for 1 hour, and measured intracellular ROS-mediated CFDA dye oxidation using flow cytometry. Figure 6A shows that 91.5% of C57BL/6 neutrophils and 94.2% of dectin-1–/– neutrophils exhibited high levels of ROS, compared with 31.4% of Cd18–/– neutrophils, indicating that ROS production is dependent on CD18. Figure 6B shows mean fluorescence intensity quantification of these data.
Figure 6. CD18-dependent neutrophil NOX activity is required for killing of A. fumigatus hyphae.

C57BL/6, Cd18–/–, and dectin-1–/– BMNs were isolated, pre-loaded with the ROS-sensitive dye CFDA, and exposed to A. fumigatus hyphae for 1.5 hours. (A) CFDA dye oxidation analyzed by flow cytometry and (B) mean fluorescence intensities were graphed. (C) Thioglycolate-elicited C57BL/6, Cd18–/–, and dectin-1–/– peritoneal neutrophils were purified and exposed to A. fumigatus hyphae for 16 hours, and fungal chitin was quantified using calcofluor white. Three independent experiments were performed. *P < 0.05.
We also examined the role of neutrophil-expressed CD18 and dectin-1 in mediating killing of fungal hyphae by coincubating A. fumigatus hyphae with purified thioglycolate-elicited peritoneal neutrophils from C57BL/6, Cd18–/–, or dectin-1–/– mice. Figure 6C shows significant hyphal growth at 16 hours after infection when hyphae were incubated in RPMI alone but not in PBS. These parameters were used as positive and negative controls. Hyphae coincubated with either C57BL/6 or dectin-1–/– neutrophils showed limited growth, whereas hyphae coincubated with Cd18–/– neutrophils showed significant hyphal growth similar to that observed with RPMI alone (Figure 6C). Taken together, these findings identify a major role for CD18, but not dectin-1, in neutrophil NOX activation, ROS production, and killing of A. fumigatus hyphae.
A. fumigatus antioxidant resistance to human neutrophils is dependent on superoxide dismutase and Yap1, but not catalases, gliotoxin, or LaeA-regulated secondary metabolites.
As neutrophil NOX is essential for control of fungal growth in the cornea, and NOX is required for both human and mouse neutrophil-mediated control of hyphal growth, we hypothesize that anti-oxidative mediators produced by Aspergillus and Fusarium will scavenge ROS or inhibit NOX and thereby impair neutrophil killing.
The transcription factor Yap1 is activated in A. fumigatus exposed to oxidative conditions and regulates production of intracellular antioxidants and enzymes that convert ROS into less reactive products (36). For example, A. fumigatus superoxide dismutases (SOD1/2/3) convert superoxide to the less-reactive hydrogen peroxide (37), whereas A. fumigatus catalases (CatA/1/2) convert hydrogen peroxide to H2O (38). In addition to ROS-catabolizing enzymes, filamentous fungi including A. fumigatus produce secondary metabolites such as gliotoxin and fumagillin, which inhibit NOX and are controlled by the master transcriptional regulator protein LaeA (39–41). We therefore examined the susceptibility of A. fumigatus strains with mutations in the Yap1 transcription factor (Δyap1), superoxide dismutases (Δsod1/2/3), catalases (ΔcatA, Δcat1/2), or NOX-inhibiting secondary metabolites (ΔgliP, ΔgliZ, ΔlaeA) to human neutrophils.
To determine whether Yap1 is required for hyphal survival in the presence of human neutrophils, we incubated Δyap1 A. fumigatus hyphae with 1 × 105 human neutrophils, which are not sufficient to kill wild-type Aspergillus (2 × 105 neutrophils were used above), and measured fungal biomass by calcofluor white binding as before. Figure 7, A–E, shows that all WT and mutant strains grew similarly after 16 hours in RPMI medium, suggesting that there are no intrinsic growth defects in the mutant strains. Figure 7A shows that the relative fluorescence unit (RFU) level of WT Dal hyphae grown in the presence of 1 × 105 human neutrophils was not significantly different from that of those grown in RPMI alone, indicating that WT Dal hyphae were not killed by 1 × 105 human neutrophils. In contrast, the RFU level of Δyap1 mutants (derived from WT Dal) incubated with the same number of neutrophils was significantly lower than that of WT Dal, which is consistent with increased susceptibility of Δyap1 mutants to killing by neutrophils. Inhibition of neutrophil NOX using the inhibitor DPI resulted in significant growth of the Δyap1 mutant strain in the presence of neutrophils, indicating that Yap1 is required for optimal antioxidant defense against NOX-mediated oxidative stress. Similarly, the RFU level of the Δsod1/2/3 mutant was significantly lower than that of the WT Ku80 strain, and DPI treatment enabled Δsod1/2/3 mutant growth in the presence of neutrophils (Figure 7B). However, there were no significant differences in RFU/fungal growth between ΔcatA or Δcat1/Δcat2 double mutants and WT G10 (Figure 7C), between ΔgliP and WT B-5233 A. fumigatus strains (Figure 7D), or between ΔgliZ or ΔlaeA mutants and WT Af293 (Figure 7E).
Figure 7. Yap1, SOD1/2/3, but not catalases or secondary metabolites mediate hypha growth upon exposure to purified human neutrophils.

(A) The Δyap1 and WT Dal1 A. fumigatus strains (12,500/well) were cultured for 6 hours to obtain hyphae and subsequently coincubated with a sublethal MOI of human neutrophils (1 × 105) for 16 hours, at which time fungal growth was quantified by calcofluor white staining and fluorometry. In addition, the DPI inhibitor was used to inhibit neutrophil NOX. (B) Similarly, the fungal growth of the Δsod1/2/3, WT Ku80, (C) ΔcatA, Δcat1/2, WT G10, (D) ΔgliP, gliPR, WT B-5233, and (E) ΔgliZ, gliZR, ΔlaeA, laeA-R, WT Af293 A. fumigatus strains were quantified 16 hours after exposure to human neutrophils. Three independent experiments were performed. *P < 0.05.
Taken together, these data show that the anti-oxidative pathways regulated by the transcription factor Yap1 and SOD1/2/3 are essential for optimal fungal growth in the presence of human neutrophils, whereas CatA, Cat1, Cat2, gliotoxin, and LaeA-regulated secondary metabolites have either a redundant or no role in blocking neutrophil-mediated killing.
Yap1 and SOD1/2/3 are required for fungal survival in vivo.
Since the A. fumigatus transcription factor Yap1 and SOD1/2/3 regulate the survival of hyphae in the presence of human neutrophils, we next examined the role of these mediators in corneal infection. WT or mutant conidia were injected into the corneal stroma of C57BL/6 mice, and corneal disease and fungal survival were examined as described above. We found that there was a significantly lower CFU level in corneas infected with Δyap1 mutants compared with those infected with WT Dal (Figure 8A). These mutants also induced significantly less corneal opacity than WT Dal (Figure 8, B–D). Similarly, the Δsod1/2/3 mutant CFU level was significantly lower than that in WT Ku80 (Figure 8E), although there was no detectable difference in corneal opacity (Figure 8, F–H).
Figure 8. Yap1, SOD1/2/3, but not catalases or secondary metabolites mediate fungal growth during corneal infection.

(A) C57BL/6 mice were infected with 40,000 A. fumigatus conidia isolated from either Δyap1 or the WT Dal1 strain, and fungal CFU were quantified at 4 and 48 hours after infection. (B) Eyes were imaged at 24 and 48 hours after infection. (C) Corneal opacity area and (D) total corneal opacity were quantified. (E) C57BL/6 mice were infected as described above with either the Δsod1/2/3 or the WT Ku80 strain, and fungal CFU were quantified at 4 and 48 hours after infection. (F) Eyes were imaged at 24 and 48 hours after infection. (G) Corneal opacity area and (H) total corneal opacity were quantified. (I) C57BL/6 mice were infected as described with ΔcatA, Δcat1/2, or the WT G10 strain, and CFU were quantified in infected corneas after infection. (J) C57BL /6 mice were infected as described with ΔgliZ, gliZR, ΔlaeA, laeA-R, WT Af293 or ΔgliP, gliPR, WT B-5233, and CFU were quantified in infected corneas after infection. Three independent experiments (n = 5) were performed. *P < 0.05.
In contrast to Δyap1 and Δsod1/2/3 mutants, and consistent with the human neutrophil studies, mice infected with ΔcatA or Δcat1/2 showed no significant differences in CFU compared with WTG10 (Figure 8I), and there were no significant differences in CFU between corneas infected with ΔgliZ, ΔgliP, or ΔlaeA compared with either WT or reconstituted strains (Figure 8J and Supplemental Figure 7). Taken together, these data reveal a critical role for Yap1-regulated antioxidant pathways and SOD1/2/3 in fungal keratitis, but either a redundant or no role for catalases, gliotoxin, or LaeA-regulated secondary metabolites.
Fungal thioredoxin mediates resistance to human neutrophils, and oxidative stress and is required for fungal survival during corneal infection.
Exposure of A. fumigatus to oxidative stress activates the transcription factor Yap1, which upregulates genes involved in anti-oxidative processes (36, 42). During oxidative stress, the Yap1-dependent proteins allergen aspf3 and peroxiredoxin Prx1 are highly upregulated. These are putative thioredoxin peroxidases (peroxiredoxins) in the thioredoxin antioxidant pathway (36), and their peroxidatic cysteine residues reduce H2O2 to H2O and are then oxidized and nonfunctional (43, 44). Subsequently, thioredoxin protein reduces peroxiredoxins to their functional state, allowing further detoxification of H2O2 (44). Thioredoxin is then further reduced by the enzyme thioredoxin reductase using H+ equivalents from NADPH+ (44).
Our bioinformatic analysis identified 5 putative thioredoxins encoded in the A. fumigatus genome (Afu5g11320/aspf29, Afu6g10300/aspf28, Afu3g14970, Afu8g01090, and Afu4g09090), two of which are known human allergens, suggesting high expression during infection (45). To inhibit total thioredoxin function in A. fumigatus, we utilized the anticancer drug PX-12 (2-[(1-methylpropyl)dithio]-1H-imidazole), which binds to the active site of thioredoxin and inhibits its ability to mediate redox reactions with target proteins and ultimately quench ROS (46–48). Figure 9A shows that A. fumigatus hyphae survived incubation with sublethal doses of human neutrophils; however, upon inhibition of total thioredoxin protein with 10 μM or 100 μM PX-12, fungal growth was decreased, indicating that thioredoxin is required for hyphae survival. To determine whether PX-12 sensitizes hyphae to ROS, we grew conidia for 6 hours in SDB and incubated them with hydrogen peroxide and PX-12 (1 μM). Figure 9B shows that 5 mM and 10 mM H2O2 killed hyphae in the presence or absence of PX-12, whereas 1 mM H2O2 alone had no effect. However, in the presence of 1 μM PX-12, this dose of H2O2 had antifungal activity. These findings demonstrate that Aspergillus thioredoxin mediates resistance to neutrophil and H2O2 oxidative stress.
Figure 9. Thioredoxin is required for hyphal survival during neutrophil exposure, oxidative stress, and corneal infection.

(A) To ascertain the role of the 5 putative thioredoxin proteins encoded in the A. fumigatus genome, hyphae were coincubated with a sublethal MOI of human neutrophils in RPMI or neutrophils plus varying doses of the thioredoxin inhibitor PX-12. (B) To examine the effect of thioredoxin inhibition on fungal growth during oxidative stress, Af-dsRed was coincubated with PX-12 and lethal and sublethal doses of H2O2. (C) To test the role of thioredoxin in mediating fungal survival during corneal infection, C57BL/6 mice were infected with Af-dsRed. At 0 and 6 hours after infection, 3 mM PX-12 or vehicle was applied topically to the infected mouse corneas. At 24 hours after infection, corneas were imaged, and (D) fungal dsRed expression and (E) CFU were quantified after infection. Three independent experiments (n = 5) were performed. *P < 0.05.
Pharmacokinetic studies during a recent PX-12 phase I clinical trial revealed that PX-12 given at 400 mg/m2/d for 72 hours is safe to administer to patients (49). However, to determine whether PX-12 exhibits cytotoxicity to immune cells and cornea-specific cell types, we incubated several human myeloid and cornea cell lines for 16 hours with a dose-response curve of PX-12, at which point the release of lactate dehydrogenase (LDH) into the surrounding medium was assayed. In these assays, LDH release in the presence of PX-12 was not significantly different from that with medium alone, indicating there was no cytotoxic effect of PX-12 (S.M. Leal Jr., unpublished observations).
To determine whether thioredoxin also impairs neutrophil-mediated killing in vivo, we infected C57BL/6 mice with Af-dsRed and gave them 8-μl eyedrops of 3-mM PX-12 or vehicle at 0 and 6 hours after infection. Mice were sacrificed at 24 hours after infection and imaged. Figure 9C shows no difference in corneal opacity between vehicle and PX-12 treatment; however, a significant reduction in fungal dsRed and CFU (Figure 9, D and E) was observed in PX-12–treated mice. Taken together with in vitro findings with human neutrophils and H2O2, these data clearly demonstrate that A. fumigatus thioredoxin proteins function as antioxidants during infection, impair neutrophil-mediated fungal killing, and are required for fungal growth during infection of the cornea.
Discussion
In this study, we identified the molecular interactions between neutrophils and fungal hyphae that result in fungal death. Conclusions from these studies are illustrated in the context of neutrophil oxidase production and antioxidants produced by hyphae (Figure 10). Unlike most leukocytes, neutrophils exhibit pronounced extracellular microbicidal activity and are likely required to kill all genera and species of pathogenic filamentous fungi (17). In addition, the anti-oxidative defense mechanisms shown to be important in this study are highly conserved (50); therefore, results from the current study are very likely relevant to fungal infections of other tissues in addition to other fungal pathogens.
Figure 10. Oxidative stress responses at the neutrophil-hypha interface.

Cat, catalase; GR, glutaredoxin; GSH, glutathione; GSSG, dimeric glutathione; GTR, glutathione reductase; HOCl, hypochlorous acid; ox, oxidized; red, reduced; TR, thioredoxin reductase.
Both dectin-1 and CR3 (CD11b/CD18) recognize fungal β-glucan (51, 52); however, we identified an essential role for CD18, but not dectin-1, in ROS production and killing of A. fumigatus hyphae in vitro. These findings are consistent with observations that CR3 is the major receptor on human neutrophils for β-glucan (35, 53) and, more recently, that CR3 induces ROS production in response to A. fumigatus hyphae (54). The role of CD18 in neutrophil-mediated hyphal killing in vivo could not be determined in the current model due to the requirement for CD18 for neutrophil extravasation (55). However, we have shown that dectin-1–/– mice exhibit defects in fungal killing in vivo (34, 56). In addition, we and others have shown that dectin-1 is required for neutrophil NOX activation in response to conidia and macrophage-mediated phagocytosis of conidia (57, 58). It is therefore likely that during infection, dectin-1 on macrophages and neutrophils mediates killing of conidia, whereas CD18 on neutrophils mediates killing of hyphae.
The requisite roles for CD18 and dectin-1 in fungal killing can potentially be explained by enhanced CR3 surface expression on neutrophils compared with macrophages (59). Resting neutrophils and macrophages express CR3 on their surface; however, activation of neutrophils, unlike macrophages, results in progressive degranulation, CD11b translocation to the cell surface, and CD11b/CD18 heterodimer (CR3) formation (60). Alternative explanations include differences in the ability of CD18 and dectin-1 to induce granule exocytosis and translocate gp91/p22phox to the cell surface (60) or differential CD18 and dectin-1 recognition of distinct β-glucan conformations variably expressed in hyphae and conidia (61). Both CR3 and dectin-1 signal through spleen tyrosine kinase (Syk), which mediates downstream activation of serine-threonine kinases (MAPKs: ERK, p38, JNK) and can potentially mediate p47phox phosphorylation and NOX activation (33, 62). Studies in our laboratory are currently addressing these hypotheses.
In addition to the role of CD18, we reported that Tlr4–/–, but not MD-2–/– or Tlr2–/–, mice exhibit increased Aspergillus and Fusarium growth during fungal keratitis (20, 56), indicating an MD-2–independent role for TLR4. However, Tlr4–/– neutrophils killed A. fumigatus hyphae in vitro as efficiently as control neutrophils (S.M. Leal Jr., unpublished observations), indicating no role for TLR4 in hyphal killing. As TLR4 binds A. fumigatus galactomannan and mediates o-linked mannose-dependent recognition of Candida albicans (63, 64), it is possible that these sugar residues are not expressed on living hyphae in sufficient number or the proper conformation to activate neutrophil TLR4. It is also possible that, since TLR4 is also activated by endogenous molecules released from lysed cells (65), host-mediated inflammatory products such as heat shock proteins induce TLR4 activation and TLR4-dependent fungal killing in vivo. Similar to the role of TLR4 in fungal killing in vivo (56), MD-2 is not required for TLR4 activation by dead cells (6, 7, 66).
CD11b/CD18 activation results in translocation of cytoplasmic NOX components to the plasma membrane and formation of a functional NOX enzyme (33). As illustrated in Figure 10, NOX subsequently produces short-lived superoxide (O2•) within nanometer proximity to the fungal cell wall (33). O2• can oxidize cell wall components directly or be converted to the more stable H2O2, and both can enter the fungal cytoplasm through porins and anion channels on the fungal plasma membrane (21, 67). H2O2 can also be converted extracellularly by MPO into hypohalous acids, which are likely too short lived to enter the fungal cytoplasm or react with iNOS-derived NO forming ONOO– (21). Our finding that the fungal cytoplasmic antioxidant thioredoxin and superoxide dismutases, but not catalases, are essential for hyphal survival suggests that during infection, neutrophil-derived O2• enters the fungal cytoplasm and along with other ROS derivatives (H2O2, etc.) mediates oxidation of essential cytoplasmic proteins and lipids, leading to death of fungal hyphae.
Using NOX-deficient Cybb–/– mice and adoptive transfer of Cybb–/– neutrophils, we demonstrate that neutrophil-specific expression of NOX and O2• production are essential for controlling the growth of A. fumigatus in vivo. In addition, we demonstrated using specific inhibitors and gene-knockout neutrophils that NOX is required for human and murine neutrophils to kill A. fumigatus, A. flavus, and F. oxysporum hyphae. However, in contrast to NOX, we found no role for either iNOS or MPO in killing Aspergillus or Fusarium hyphae. Similar findings were obtained in experimental A. fumigatus lung infections in which NOX but not MPO or iNOS were found to be required to control fungal growth (21, 23, 25, 68). Our findings are also in agreement with the enhanced susceptibility of CGD patients, but not MPO-deficient patients, to filamentous fungal infections (2, 32). However, our findings differ from a study showing no role for NOX in killing A. nidulans in vitro (27). Given that iNOS is required for optimal intracellular killing of Candida and Cryptococcus yeasts (69, 70), and that MPO is required for optimal responses to Candida (68), it is possible that these enzymes are more effective against intracellular yeast rather than extracellular hyphae.
Figure 10 also illustrates fungal antioxidant pathways. Oxidation by neutrophils induces nuclear translocation of Yap1, as well as upregulation of the H2O2-catabolizing thioredoxin-dependent peroxiredoxin aspf29 and prx and secreted Cat1/2 (36). The current study identified the Yap1 transcription factor as essential for hyphal survival during corneal infection and following exposure to neutrophils. These findings are in agreement with the results of a report on C. albicans in which the Yap1 homolog Cap1p was required for anti-oxidative defenses (71). However, our results differ from A. fumigatus lung infection studies that showed no role for Yap1 during infection of immunocompromised mice or after exposure of neutrophils to swollen conidia (36, 42). These differences are likely explained by differences in the immune status of the infected mice, although there may also be a morphotype-specific role for Yap1.
In the current study, we did not detect a role for Yap1-regulated and -secreted Cat1/2 or for cytoplasmic CatA in hyphal survival. These findings are consistent with studies on experimental Aspergillus lung infection and are likely due to the lack of CatA expression in hyphae as opposed to conidia and the unavailability of secreted Cat1/2 for cytoplasmic antioxidant defense by hyphae (38). Further, Yap1 induces high levels of intracellular peroxiredoxins following oxidative stress (42), and similar to catalases, peroxiredoxins reduce H2O2 to H2O and are then oxidized (44). Subsequently, peroxiredoxins are cyclically reduced to their functional state by thioredoxin protein, which is itself reduced by thioredoxin reductase (44). The importance of thioredoxin protein in fungal antioxidant defense is evidenced by (a) elevated A. fumigatus thioredoxin reductase expression found after exposure to normal versus CGD neutrophils (72); and (b) the essential role of thioredoxin in anti-oxidative responses in A. nidulans (73). In the current study, bioinformatic analysis revealed 5 putative thioredoxins in the A. fumigatus genome, including the human allergens aspf29 and aspf28, which are highly expressed during human infection (45). We constructed a single-knockout strain of aspf29 and did not observe a difference in fungal survival in vivo, likely due to redundancy by the other 4 putative thioredoxins encoded in the A. fumigatus genome (S.M. Leal Jr., unpublished observations). We therefore utilized PX-12 to inhibit all 5 putative thioredoxins and showed that thioredoxin is required for hyphal survival in the presence of neutrophils in vitro and that PX-12 enhances H2O2-mediated killing (Figure 9 and ref. 48). Further, we found that topical application of PX-12 was sufficient to restrict fungal growth in vivo, thereby indicating that this or similar compounds that target the thioredoxin pathway can block fungal thioredoxins and inhibit the predominant antioxidant defense utilized by these pathogens during infection.
Figure 10 also illustrates that A. fumigatus produces 3 superoxide dismutases that catalyze the conversion of superoxide to H2O2 (37, 38). SOD1 and SOD3 are both cytoplasmic, whereas SOD2 is restricted to the mitochondrial membrane (37). In this study, we identified a role for the superoxide dismutases in mediating hyphal survival during oxidation by neutrophils and during infection. Our findings are consistent with increased cytoplasmic SOD1 and SOD3 but not mitochondrial SOD2 expression by A. fumigatus hyphae in the presence of normal versus CGD neutrophils (72) and the increased susceptibility of A. fumigatus Δsod1/2/3 mutants to killing by alveolar macrophages (37). However, Δsod1/2/3 mutants showed no difference in survival in an immunocompromised lung infection model (37). Given that SOD3 is the most highly expressed SOD in hyphae under oxidative conditions, it is likely that SOD3 is functionally dominant in A. fumigatus hyphae. Future studies will address this hypothesis.
Lastly, although secondary metabolites upregulated by the LaeA transcription factor are reported to have immunosuppressive effects, including gliotoxin, fumagillin, fumagatin, and helvolic acid (39–41, 74), we did not detect a role for these toxins in hyphal survival following exposure to neutrophils or during infection. Given that secondary metabolite production by A. fumigatus in culture peaks at 48 hours after infection (75), it is possible that secondary metabolites may mediate fungal survival in patients, but not mice, due to the more chronic nature of human disease — typically weeks to months — compared with our acute murine model of fungal keratitis.
In conclusion, we have identified superoxide production by neutrophil NOX as the optimal effector for hyphal killing and shown that superoxide dismutase and thioredoxin are the key antioxidant agents used by hyphae during infection. One approach to exploiting these findings is to block the antioxidant activity of the fungus, thereby increasing hyphal sensitivity to oxidative stress and decreasing the threshold required for neutrophils to kill hyphae during infection. As noted earlier, PX-12 blocks hyphal resistance to ROS, and as PX-12 has successfully completed two phase I clinical safety trials for cancer therapy (46, 47), it is possible that targeting fungal thioredoxin by PX-12 or related compounds could be utilized to treat a broad range of fungal infections.
Methods
Source of mice.
C57BL/6 mice (6–12 weeks old) and Cybb–/– mice were purchased from The Jackson Laboratory. Cd18–/– mice were provided by Claire Doerschuk (University of North Carolina, Chapel Hill, North Carolina, USA), and Cxcr2–/– mice were provided by Richard Ransohoff (Cleveland Clinic, Cleveland, Ohio, USA). Dectin-1–/– mice were provided by Yoichiro Iwakura (Tokyo University, Tokyo, Japan). All mice used in this study were on a C57BL/6 background.
Fungal strains, media, and growth conditions.
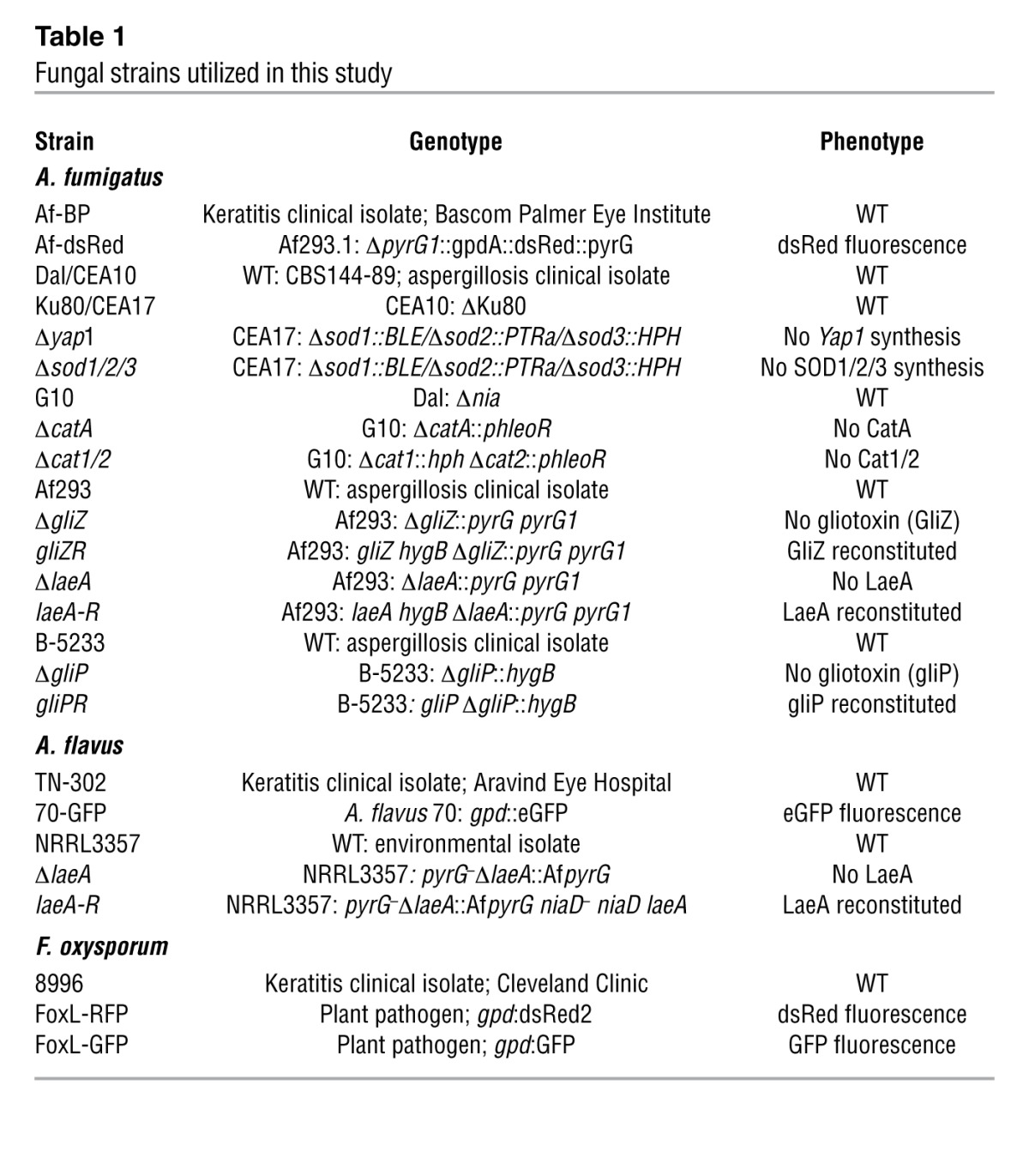
Table 1 lists the genotype and phenotype of all strains utilized in this study. A. fumigatus and A. flavus strains used in this study were cultured on Vogel’s minimal medium (VMM) with or without 2% agar unless otherwise stated. F. oxysporum and F. solani strains were cultured on Sabouraud dextrose medium. To visualize fungi in the transparent murine cornea, we utilized fluorescent strains of Aspergillus or Fusarium species expressing either RFP or GFP. Af-dsRed constitutively expresses enhanced monomeric dsRed protein (gpdA promotor driven; pyrG1 selection marker) (56). The strain A. flavus 70-GFP constitutively expresses GFP (gpdA promotor driven; niaD selection marker; provided by Rajah Rajasekaran, USDA, New Orleans, Louisiana, USA) (76). The strains FoxL-RFP and FoxL-GFP are F. oxysporum lycopersici strains constitutively expressing either RFP or GFP, which were provided by Seogchan Kang (Pennsylvania State University, University Park, Pennsylvania, USA). The A. fumigatus strain Af-BP was isolated from a keratitis patient at Bascom Palmer Eye Institute and provided by Darlene Miller (University of Miami, Miami, Florida, USA) (56). The A. flavus strain TN-302 was isolated from a keratitis patient at Aravind Eye Hospital, Tamil Nadu, India, and provided by Lalitha Prajna. The F. oxysporum strain 8996 was isolated from a keratitis patient at the Cole Eye Institute, Cleveland Clinic (20). For our studies on gliotoxin, June Kwon-Chung (NIAID, Bethesda, Maryland, USA) provided A. fumigatus strains B-5233 (WT), ΔgliP, and gliPR (77). Additionally, Nancy Keller (University of Wisconsin, Madison, Wisconsin, USA) provided the A. fumigatus strains Af293 (WT), ΔgliZ, gliZR, ΔlaeA, laeA-R, as well as the Aspergillus flavus strains A. flavus NRRL3357 (WT), ΔlaeA, and laeA-R (78, 79). For ROS scavenging studies, we used A. fumigatus strains G10 (WT), ΔcatA, and Δcat1/2, and Ku80 (WT), Δsod1/2/3, and Dal (WT), Δyap1 (36–38).
Table 1.
Fungal strains utilized in this study
Mouse model of Aspergillus and Fusarium keratitis.
Aspergillus strains were cultured for 2–3 days on VMM in 25-cm2 tissue culture flasks. Fresh conidia were disrupted with a bacterial L-loop, harvested in 5 ml PBS, and filtered through sterile, PBS-soaked cotton gauze in a 10-ml syringe to obtain pure conidial suspensions. Conidia were quantified using a hemocytometer and adjusted in PBS to a final stock solution: A. fumigatus, 20,000 conidia/μl; A. flavus, 20,000 conidia/μl; F. oxysporum, 25,000 conidia/μl. Subsequently, mice were anesthetized with 1.25% 2,2,2-tribromoethanol. The corneal epithelium of anesthetized mice was abraded using a 30-gauge needle, through which a 2-μl injection containing conidia was released into the corneal stroma using a 33-gauge Hamilton syringe (total: A. fumigatus, 40,000 conidia; A. flavus, 40,000; F. oxysporum, 50,000). Mice were examined daily under a stereomicroscope for corneal opacification, ulceration, perforation, and fungal fluorescent protein expression. At set time points, animals were euthanized by CO2 asphyxiation, and eyes were either placed in 10% formalin and embedded in paraffin and sectioned at 5-μm intervals or excised and placed in 1 ml sterile saline and homogenized for quantitative culture. For depletion of neutrophils in mice, 400 μg of anti-mouse neutrophil antibody (NIMP-R14) was injected at day 1 prior to infection. To detect ROS production in the mouse cornea during fungal infection, 2 μl of 12.5-ng/ml CFDA (Invitrogen) was injected into the stroma of anesthetized mice and eyes imaged under standard GFP filters after 10 minutes incubation at 25°C. Subsequently, mice were euthanized, and eyes processed accordingly. To inhibit RNS production during corneal infection, 500 μg of the irreversible iNOS inhibitor 1400W was injected i.p. into C57BL/6 mice at 6, 24, and 48 hours after infection. To inhibit thioredoxin in vivo, PX-12 (Sigma-Aldrich) was dissolved at 3 mM in a proprietary eyedrop formulation provided by Alcon and applied topically at 0 and 6 hours after infection. All animals were bred under specific pathogen–free conditions and maintained according to institutional guidelines.
Neutrophil-specific adoptive transfer mouse model used to study fungal killing.
In vivo Cxcr2–/– and Cd18–/– mice were anesthetized as described above, and conidia from a fluorescent fungal strain were injected into the corneal stroma strain (30,000 Af-dsRed conidia). At 2 hours after infection, 4 million naive BMNs isolated from the femurs and tibias of donor mice were injected i.v. into recipient mice. At 24 hours after infection, infected mice were euthanized, and fungal growth in the cornea was imaged using a fluorescence stereomicroscope. The level of fluorescence emitted from infecting fungi was quantified using image analysis software (MetaMorph, Molecular Devices; described below) and used as a measure of fungal growth during infection.
Imaging and quantification of light reflected or emitted from infected mouse corneas.
Mice were euthanized by CO2 asphyxiation and positioned in a 3-point stereotactic mouse restrainer. Corneal opacity (brightfield [BF]), fungal proliferation (RFP/GFP), cellular infiltration (GFP), and CFDA dye oxidation (GFP spectra) were visualized in the intact cornea using a high-resolution stereo fluorescence MZFLIII microscope (Leica Microsystems) and Spot RT Slider KE camera (Diagnostics Instruments). All images were obtained using the same Spot Advanced Software under the same magnification (×20), exposure (BF, 0.4 seconds; RFP, 10 seconds; eGFP, 2 seconds), gain (BF, 1; RFP/eGFP, 4/16), and gamma (BF/RFP/eGFP, 1.85) parameters. MetaMorph imaging software was used to quantify the percent area of opacity and the integrated corneal opacity as described in the legend to Supplemental Figure 1.
Quantification of Aspergillus CFU.
For assessment of fungal viability, whole eyes were homogenized under sterile conditions in 1 ml PBS using the Mixer Mill MM300 (Retsch) at 33 Hz for 4 minutes. Subsequently, serial log dilutions were performed and plated onto bacteriologic-grade Sabouraud dextrose agar (SDA) plates (BD). Following incubation for 24 hours at 37°C (Aspergillus) or 30°C (Fusarium), the number of CFU was determined by direct counting.
Identification of fungal growth patterns and neutrophil recruitment into the cornea.
Eyes were enucleated and fixed in 10% formalin in PBS (Fisher) for 24 hours. Five-micrometer sections from the center of the cornea (as determined by noncontiguous iris morphology) were cut and stained with PASH for identification of fungi and inflammatory cell recruitment. To detect infiltrating neutrophils, sections were immunostained using monoclonal rat anti-mouse neutrophil IgG (NIMP-R14, Abcam) and Alexa Fluor 488–tagged rabbit anti-rat IgG (Invitrogen). All histology slides were imaged and shown at an original magnification of ×400.
Isolation of human neutrophils from peripheral blood.
Human neutrophils were isolated from normal, healthy donors using Ficoll-Paque Plus (GE) density centrifugation. Peripheral blood (20 ml) was obtained, and rbc were separated from whole blood via incubation at 1 g for 20 minutes with 3% dextran in PBS (Sigma-Aldrich). The top clear layer containing leukocytes was transferred to a fresh 50-ml conical tube and 10 ml Ficoll-Paque Plus was underlain. The cell suspension was centrifuged at 500 g for 20 minutes at 4°C to separate mononuclear cells from neutrophils and the remaining rbc. The overlying plasma and PBMC layers were aspirated, and the neutrophil/rbc pellet was resuspended in RBC Lysis Buffer (eBioscience) (8.3 g NH4Cl, 1 g KHCO3, 0.09 g EDTA/1 l ddH2O), incubated at 37°C for 10 minutes to lyse remaining rbc, and spun at 300 g for 5 minutes at 4°C. The lysis procedure was repeated as needed to obtain sufficient rbc lysis in cell preparations. Subsequently, cells were washed twice in PBS and ultimately resuspended in RPMI plus l-glutamine without phenol red (Hyclone). The neutrophil cell suspension was counted using a hemocytometer, and samples were collected by Cytospin and stained by Wright-Giemsa (Fisher). Using this approach, neutrophils were routinely found to be greater than 97% of the final cell preparation.
Isolation of peritoneal and bone marrow–derived murine neutrophils.
To isolate peritoneal neutrophils, mice were injected with 1 ml 4% thioglycolate 16 and 3 hours prior to peritoneal lavage with cold 1× PBS. Cells were transferred to a fresh 50-ml conical tube in a total volume of 30 ml, and 10 ml Ficoll-Paque Plus was underlain. Cells were then centrifuged at 1,200 g for 20 minutes at 25°C. The upper monocytic cell layer was aspirated, and the underlying neutrophil layer was washed 3 times with 50 ml PBS and resuspended in RPMI plus l-glutamine without phenol red (Hyclone). The resulting cell suspension routinely yielded greater than 95% neutrophils. To obtain BMNs, mice were euthanized by CO2 asphyxiation, and femurs and tibias were removed, cleaned, and centrifuged at 5,000 g for 45 seconds at 4°C. Contaminating rbc were lysed in 5 ml RBC Lysis Buffer, and remaining bone marrow cells were pipetted onto a discontinuous Percoll gradient (GE) of 52%, 69%, and 78%. Cells were centrifuged for 30 minutes at 1,500 g at 25°C. Following centrifugation, the neutrophils suspended in Percoll at the 69%/78% interface and below were harvested, washed twice in PBS in 50-ml conical tubes, and resuspended in RPMI medium plus l-glutamine without phenol red. Neutrophil purity of greater than 98% was obtained routinely using this protocol.
In vitro neutrophil/hypha growth inhibition assay.
An in vitro assay was developed to study the ability of mouse and human neutrophils to inhibit the growth of fungal hyphae from A. fumigatus, A. flavus, F. oxysporum, and F. solani species. Purified conidia (12,500/well for Aspergillus species and 100,000/well for Fusarium species) from fluorescent or non-fluorescent fungal strains were cultured in 200 μl SDA medium in black-walled 96-well plates with an optically clear bottom (CoStar 3720) until early germ tubes were observed (A. fumigatus, 6 hours; A. flavus, 4 hours; F. oxysporum, 6 hours; and F. solani, 6 hours). Wells were washed twice with sterile ddH2O and incubated 16 hours with either RPMI medium without phenol red (positive control), PBS (negative control), or RPMI with 1 × 105 murine BMNs or 2 × 105 human peripheral blood neutrophils from healthy donors. For assays in which the desired phenotype was survival of the WT fungus, 1 × 105 human neutrophils/well were used. At 16 hours after exposure, fungal growth of fluorescent strains was visualized directly using standard GFP and RFP filters in an upright microscope (Zeiss). In order to visualize and quantify fungal chitin content by non-fluorescent fungal strains, 50 μl calcofluor white stain (binds chitin; Fluka 18909) was added to each well for 5 minutes in the dark. Subsequently, plates were washed 3 times with ddH2O and imaged as stated above using standard DAPI filters or quantified via fluorometry (360/440 nm; Synergy HT; Biotek). To inhibit neutrophil NOX, we used DPI (200 μM) and Apo (30 mM). To inhibit iNOS, we used the pharmacologic inhibitors Agd (1 mM) and SMT (100 μM). To inhibit MPO we used Indo (200 μM) and 4-AH (100 μM). All fungal growth images were taken and shown at an original magnification of ×400.
Quantification of extracellular ROS and NO.
ROS was assayed using a dye that fluoresces upon oxidation (CFDA, Invitrogen), and NO was assayed using the Griess reagent (Invitrogen). For both assays, hyphae were incubated with neutrophils in RPMI as described above for 2 hours at 37°C and 5% CO2. After 2 hours, 50 μl supernatant was transferred to wells containing either 50 μl CFDA (25 ng/ml) or 250 μl Griess reagent. For CFDA, plates were incubated in the dark for 10 minutes and read using a fluorescence spectrophotometer at excitation/emission of 485/520 nm γ (CFDA/DAF-FM). For Griess reactivity, plates were incubated in the dark for 30 minutes at 37°C, and absorbance was read at 548 nm.
Quantification of neutrophil intracellular ROS by flow cytometry.
BMNs were isolated as described above and incubated with 10 μM CFDA at 37°C for 10 minutes. CFDA-containing neutrophils (2 × 105) were added to 96-well plates containing hyphae, and plates were spun at 300 g for 1 minute to enhance cell contact. Neutrophils were incubated at 37°C for 1 hour, collected, and analyzed by flow cytometry (excitation/emission of 485/520 nm γ). CFDA-loaded BMNs incubated for 1 hour in empty wells were used to control for background fluorescence.
H2O2 fungal killing assay.
Fungi were grown for 4 hours at 37°C in 200 μl SDB in 96-well plates as described above, and H2O2 was added at final concentrations of 1 mM–10 mM in the presence of PX-12 (0–100 μM) for an additional 2 hours. After 16 hours, fungal growth was examined by phase-contrast microscopy, stained with calcofluor white, and quantified by fluorometry as described above.
Statistics.
Statistical analysis was performed for each experiment using 1-way ANOVA with a Tukey post hoc analysis (Prism, GraphPad Software). A P value less than 0.05 was considered significant.
Study approval.
All animals were treated in accordance with the guidelines provided in the Association for Research in Vision and Ophthalmology (ARVO) statement for the Use of Animals in Ophthalmic and Vision Research; and protocols were approved by the Case Western Reserve University IACUC. The protocol for the use of human peripheral blood from normal healthy volunteers was approved by the Institutional Review Board of University Hospitals of Cleveland. Informed consent was obtained from each volunteer.
Supplementary Material
Acknowledgments
This work was supported by NIH grants F31 EY019841 (to S.M. Leal Jr.), R01 EY018612 (to E. Pearlman), P30 EY011373 (to E. Pearlman), and R21 AI074846-02 (to M. Momany). These studies were also supported by a Research to Prevent Blindness Medical Student Fellowship (to S.M. Leal Jr.), and unrestricted grants from the Research to Prevent Blindness Foundation and the Ohio Lions Eye Research Foundation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We would like to thank Brandon Leibell and the Case Western Reserve University visual sciences core facility managers for excellent technical assistance.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2012;122(7):2482–2498. doi:10.1172/JCI63239.
References
- 1.Milner JD, Sandler NG, Douek DC. Th17 cells, Job’s syndrome and HIV: opportunities for bacterial and fungal infections. Curr Opin HIV AIDS. 2010;5(2):179–183. doi: 10.1097/COH.0b013e328335ed3e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Antachopoulos C. Invasive fungal infections in congenital immunodeficiencies. Clin Microbiol Infect. 2010;16(9):1335–1342. doi: 10.1111/j.1469-0691.2010.03289.x. [DOI] [PubMed] [Google Scholar]
- 3.Thomas PA. Fungal infections of the cornea. Eye. 2003;17(8):852–862. doi: 10.1038/sj.eye.6700557. [DOI] [PubMed] [Google Scholar]
- 4.Gower EW, et al. Trends in fungal keratitis in the United States, 2001 to 2007. Ophthalmology. 2010;117(12):2263–2267. doi: 10.1016/j.ophtha.2010.03.048. [DOI] [PubMed] [Google Scholar]
- 5.Liesegang TJ, Forster RK. Spectrum of microbial keratitis in South Florida. Am J Ophthalmol. 1980;90(1):38–47. doi: 10.1016/s0002-9394(14)75075-5. [DOI] [PubMed] [Google Scholar]
- 6.Rosa RH, Jr, Miller D, Alfonso EC. The changing spectrum of fungal keratitis in south Florida. Ophthalmology. 1994;101(6):1005–1013. doi: 10.1016/s0161-6420(94)31225-5. [DOI] [PubMed] [Google Scholar]
- 7.Xie L, Zhong W, Shi W, Sun S. Spectrum of fungal keratitis in north China. Ophthalmology. 2006;113(11):1943–1948. doi: 10.1016/j.ophtha.2006.05.035. [DOI] [PubMed] [Google Scholar]
- 8.Bharathi MJ, Ramakrishnan R, Meenakshi R, Padmavathy S, Shivakumar C, Srinivasan M. Microbial keratitis in South India: influence of risk factors, climate, and geographical variation. Ophthalmic Epidemiol. 2007;14(2):61–69. doi: 10.1080/09286580601001347. [DOI] [PubMed] [Google Scholar]
- 9.Bhartiya P, Daniell M, Constantinou M, Islam FM, Taylor hour Fungal keratitis in Melbourne. Clin Experiment Ophthalmol. 2007;35(2):124–130. doi: 10.1111/j.1442-9071.2006.01405.x. [DOI] [PubMed] [Google Scholar]
- 10.Dunlop AA, et al. Suppurative corneal ulceration in Bangladesh. A study of 142 cases examining the microbiological diagnosis, clinical and epidemiological features of bacterial and fungal keratitis. Aust N Z J Ophthalmol. 1994;22(2):105–110. doi: 10.1111/j.1442-9071.1994.tb00775.x. [DOI] [PubMed] [Google Scholar]
- 11.Saha R, Das S. Mycological profile of infectious keratitis from Delhi. Indian J Med Res. 2006;123(2):159–164. [PubMed] [Google Scholar]
- 12.Perez-Balbuena AL, Vanzzini-Rosano V, Valadez-Virgen Jde J, Campos-Moller X. Fusarium keratitis in Mexico. Cornea. 2009;28(6):626–630. doi: 10.1097/ICO.0b013e31819bc2ea. [DOI] [PubMed] [Google Scholar]
- 13.Chang DC, et al. Multistate outbreak of Fusarium keratitis associated with use of a contact lens solution. JAMA. 2006;296(8):953–963. doi: 10.1001/jama.296.8.953. [DOI] [PubMed] [Google Scholar]
- 14.Srinivasan M. Fungal keratitis. Curr Opin Ophthalmol. 2004;15(4):321–327. doi: 10.1097/00055735-200408000-00008. [DOI] [PubMed] [Google Scholar]
- 15.Prajna NV, et al. Comparison of natamycin and voriconazole for the treatment of fungal keratitis. Arch Ophthalmol. 2010;128(6):672–678. doi: 10.1001/archophthalmol.2010.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pham CT. Neutrophil serine proteases: specific regulators of inflammation. Nat Rev Immunol. 2006;6(7):541–550. doi: 10.1038/nri1841. [DOI] [PubMed] [Google Scholar]
- 17.Nauseef WM. How human neutrophils kill and degrade microbes: an integrated view. Immunol Rev. 2007;219:88–102. doi: 10.1111/j.1600-065X.2007.00550.x. [DOI] [PubMed] [Google Scholar]
- 18.Karthikeyan RS, et al. Expression of innate and adaptive immune mediators in human corneal tissue infected with Aspergillus or fusarium. J Infect Dis. 2011;204(6):942–950. doi: 10.1093/infdis/jir426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leal SM, Jr, Cowden S, Hsia YC, Ghannoum MA, Momany M, Pearlman E. Distinct roles for Dectin-1 and TLR4 in the pathogenesis of Aspergillus fumigatus keratitis. PLoS Pathog. 2011;6:e1000976. doi: 10.1371/journal.ppat.1000976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tarabishy AB, et al. MyD88 regulation of Fusarium keratitis is dependent on TLR4 and IL-1R1 but not TLR2. J Immunol. 2008;181(1):593–600. doi: 10.4049/jimmunol.181.1.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Winterbourn CC. Reconciling the chemistry and biology of reactive oxygen species. Nat Chem Biol. 2008;4(5):278–286. doi: 10.1038/nchembio.85. [DOI] [PubMed] [Google Scholar]
- 22.Segal BH, Romani LR. Invasive aspergillosis in chronic granulomatous disease. Med Mycol. 2009;47(suppl 1):S282–290. doi: 10.1080/13693780902736620. [DOI] [PubMed] [Google Scholar]
- 23.Morgenstern DE, Gifford MA, Li LL, Doerschuk CM, Dinauer MC. Absence of respiratory burst in X-linked chronic granulomatous disease mice leads to abnormalities in both host defense and inflammatory response to Aspergillus fumigatus. J Exp Med. 1997;185(2):207–218. doi: 10.1084/jem.185.2.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pollock JD, et al. Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat Genet. 1995;9(2):202–209. doi: 10.1038/ng0295-202. [DOI] [PubMed] [Google Scholar]
- 25.Philippe B, et al. Killing of Aspergillus fumigatus by alveolar macrophages is mediated by reactive oxidant intermediates. Infect Immun. 2003;71(6):3034–3042. doi: 10.1128/IAI.71.6.3034-3042.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rex JH, Bennett JE, Gallin JI, Malech HL, Melnick DA. Normal and deficient neutrophils can cooperate to damage Aspergillus fumigatus hyphae. J Infect Dis. 1990;162(2):523–528. doi: 10.1093/infdis/162.2.523. [DOI] [PubMed] [Google Scholar]
- 27.Henriet SS, et al. Human leukocytes kill Aspergillus nidulans by reactive oxygen species-independent mechanisms. Infect Immun. 2011;79(2):767–773. doi: 10.1128/IAI.00921-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Faust N, Varas F, Kelly LM, Heck S, Graf T. Insertion of enhanced green fluorescent protein into the lysozyme gene creates mice with green fluorescent granulocytes and macrophages. Blood. 2000;96(2):719–726. [PubMed] [Google Scholar]
- 29.Viola A, Luster AD. Chemokines and their receptors: drug targets in immunity and inflammation. Annu Rev Pharmacol Toxicol. 2008;48:171–197. doi: 10.1146/annurev.pharmtox.48.121806.154841. [DOI] [PubMed] [Google Scholar]
- 30.Wilson RW, et al. Gene targeting yields a CD18-mutant mouse for study of inflammation. J Immunol. 1993;151(3):1571–1578. [PubMed] [Google Scholar]
- 31.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4(3):181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 32.Andrews T, Sullivan KE. Infections in patients with inherited defects in phagocytic function. Clin Microbiol Rev. 2003;16(4):597–621. doi: 10.1128/CMR.16.4.597-621.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.El-Benna J, Dang PM, Gougerot-Pocidalo MA. Priming of the neutrophil NADPH oxidase activation: role of p47phox phosphorylation and NOX2 mobilization to the plasma membrane. Semin Immunopathol. 2008;30(3):279–289. doi: 10.1007/s00281-008-0118-3. [DOI] [PubMed] [Google Scholar]
- 34.Hohl TM, et al. Aspergillus fumigatus triggers inflammatory responses by stage-specific beta-glucan display. PLoS Pathog. 2005;1(3):e30. doi: 10.1371/journal.ppat.0010030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Bruggen R, et al. Complement receptor 3, not Dectin-1, is the major receptor on human neutrophils for beta-glucan-bearing particles. Mol Immunol. 2009;47(2–3):575–581. doi: 10.1016/j.molimm.2009.09.018. [DOI] [PubMed] [Google Scholar]
- 36.Lessing F, et al. The Aspergillus fumigatus transcriptional regulator AfYap1 represents the major regulator for defense against reactive oxygen intermediates but is dispensable for pathogenicity in an intranasal mouse infection model. Eukaryot Cell. 2007;6(12):2290–2302. doi: 10.1128/EC.00267-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lambou K, Lamarre C, Beau R, Dufour N, Latge JP. Functional analysis of the superoxide dismutase family in Aspergillus fumigatus. Mol Microbiol. 2010;75(4):910–923. doi: 10.1111/j.1365-2958.2009.07024.x. [DOI] [PubMed] [Google Scholar]
- 38.Paris S, et al. Catalases of Aspergillus fumigatus. Infect Immun. 2003;71(6):3551–3562. doi: 10.1128/IAI.71.6.3551-3562.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fallon JP, Reeves EP, Kavanagh K. Inhibition of neutrophil function following exposure to the Aspergillus fumigatus toxin fumagillin. J Med Microbiol. 2010;59(pt 6):625–633. doi: 10.1099/jmm.0.018192-0. [DOI] [PubMed] [Google Scholar]
- 40.Tsunawaki S, Yoshida LS, Nishida S, Kobayashi T, Shimoyama T. Fungal metabolite gliotoxin inhibits assembly of the human respiratory burst NADPH oxidase. Infect Immun. 2004;72(6):3373–3382. doi: 10.1128/IAI.72.6.3373-3382.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perrin RM, et al. Transcriptional regulation of chemical diversity in Aspergillus fumigatus by LaeA. PLoS Pathog. 2007;3(4):e50. doi: 10.1371/journal.ppat.0030050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qiao J, et al. Afyap1, encoding a bZip transcriptional factor of Aspergillus fumigatus, contributes to oxidative stress response but is not essential to the virulence of this pathogen in mice immunosuppressed by cyclophosphamide and triamcinolone. Med Mycol. 2008;46(8):773–782. doi: 10.1080/13693780802054215. [DOI] [PubMed] [Google Scholar]
- 43.Wood ZA, Schroder E, Robin Harris J, Poole LB. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem Sci. 2003;28(1):32–40. doi: 10.1016/S0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- 44.Koharyova M, Kolarova M. Oxidative stress and thioredoxin system. Gen Physiol Biophys. 2008;27(2):71–84. [PubMed] [Google Scholar]
- 45.Glaser AG, Menz G, Kirsch AI, Zeller S, Crameri R, Rhyner C. Auto- and cross-reactivity to thioredoxin allergens in allergic bronchopulmonary aspergillosis. Allergy. 2008;63(12):1617–1623. doi: 10.1111/j.1398-9995.2008.01777.x. [DOI] [PubMed] [Google Scholar]
- 46.Ramanathan RK, et al. A phase I trial of PX-12, a small-molecule inhibitor of thioredoxin-1, administered as a 72-hour infusion every 21 days in patients with advanced cancers refractory to standard therapy. Invest New Drugs. doi: 10.1007/s10637-011-9739-9. [published online ahead of print August 24, 2011]. doi: 10.1007/s10637-011-9739-9 . [DOI] [PubMed] [Google Scholar]
- 47.Ramanathan RK, et al. A Phase I pharmacokinetic and pharmacodynamic study of PX-12, a novel inhibitor of thioredoxin-1, in patients with advanced solid tumors. Clin Cancer Res. 2007;13(7):2109–2114. doi: 10.1158/1078-0432.CCR-06-2250. [DOI] [PubMed] [Google Scholar]
- 48.Welsh SJ, Williams RR, Birmingham A, Newman DJ, Kirkpatrick DL, Powis G. The thioredoxin redox inhibitors 1-methylpropyl 2-imidazolyl disulfide and pleurotin inhibit hypoxia-induced factor 1alpha and vascular endothelial growth factor formation. Mol Cancer Ther. 2003;2(3):235–243. [PubMed] [Google Scholar]
- 49.Ramanathan RK, et al. A randomized phase II study of PX-12, an inhibitor of thioredoxin in patients with advanced cancer of the pancreas following progression after a gemcitabine-containing combination. Cancer Chemother Pharmacol. 2011;67(3):503–509. doi: 10.1007/s00280-010-1343-8. [DOI] [PubMed] [Google Scholar]
- 50.Chauhan N, Latge JP, Calderone R. Signalling and oxidant adaptation in Candida albicans and Aspergillus fumigatus. Nat Rev Microbiol. 2006;4(6):435–444. doi: 10.1038/nrmicro1426. [DOI] [PubMed] [Google Scholar]
- 51.Brown GD, Herre J, Williams DL, Willment JA, Marshall AS, Gordon S. Dectin-1 mediates the biological effects of beta-glucans. J Exp Med. 2003;197(9):1119–1124. doi: 10.1084/jem.20021890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vetvicka V, Thornton BP, Ross GD. Soluble beta-glucan polysaccharide binding to the lectin site of neutrophil or natural killer cell complement receptor type 3 (CD11b/CD18) generates a primed state of the receptor capable of mediating cytotoxicity of iC3b-opsonized target cells. J Clin Invest. 1996;98(1):50–61. doi: 10.1172/JCI118777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lavigne LM, Albina JE, Reichner JS. Beta-glucan is a fungal determinant for adhesion-dependent human neutrophil functions. J Immunol. 2006;177(12):8667–8675. doi: 10.4049/jimmunol.177.12.8667. [DOI] [PubMed] [Google Scholar]
- 54.Boyle KB, et al. Class IA phosphoinositide 3-kinase beta and delta regulate neutrophil oxidase activation in response to Aspergillus fumigatus hyphae. J Immunol. 2011;186(5):2978–2989. doi: 10.4049/jimmunol.1002268. [DOI] [PubMed] [Google Scholar]
- 55.Abram CL, Lowell CA. The ins and outs of leukocyte integrin signaling. Annu Rev Immunol. 2009;27:339–362. doi: 10.1146/annurev.immunol.021908.132554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Leal SM, Jr, Cowden S, Hsia YC, Ghannoum MA, Momany M, Pearlman E. Distinct roles for Dectin-1 and TLR4 in the pathogenesis of Aspergillus fumigatus keratitis. PLoS Pathog. 2010;6:e1000976. doi: 10.1371/journal.ppat.1000976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Werner JL, et al. Requisite role for the dectin-1 beta-glucan receptor in pulmonary defense against Aspergillus fumigatus. J Immunol. 2009;182(8):4938–4946. doi: 10.4049/jimmunol.0804250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Goodridge HS, et al. Activation of the innate immune receptor Dectin-1 upon formation of a ‘phagocytic synapse’. Nature. 2011;472(7344):471–475. doi: 10.1038/nature10071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vedder NB, Harlan JM. Increased surface expression of CD11b/CD18 (Mac-1) is not required for stimulated neutrophil adherence to cultured endothelium. J Clin Invest. 1988;81(3):676–682. doi: 10.1172/JCI113372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. Neutrophil function: from mechanisms to disease. Annu Rev Immunol. 2012;Β30:459–489. doi: 10.1146/annurev-immunol-020711-074942. [DOI] [PubMed] [Google Scholar]
- 61.Goodridge HS, Wolf AJ, Underhill DM. Beta-glucan recognition by the innate immune system. Immunol Rev. 2009;230(1):38–50. doi: 10.1111/j.1600-065X.2009.00793.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mocsai A, Ruland J, Tybulewicz VL. The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat Rev Immunol. 2010;10(6):387–402. doi: 10.1038/nri2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chai LY, et al. Aspergillus fumigatus cell wall components differentially modulate host TLR2 and TLR4 responses. Microbes Infect. 2011;13(2):151–159. doi: 10.1016/j.micinf.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 64.Netea MG, et al. Immune sensing of Candida albicans requires cooperative recognition of mannans and glucans by lectin and Toll-like receptors. J Clin Invest. 2006;116(6):1642–1650. doi: 10.1172/JCI27114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10(12):826–837. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chun KH, Seong SY. CD14 but not MD2 transmit signals from DAMP. Int Immunopharmacol. 2010;10(1):98–106. doi: 10.1016/j.intimp.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 67.Dickinson BC, Chang CJ. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat Chem Biol. 2011;7(8):504–511. doi: 10.1038/nchembio.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Aratani Y, et al. Relative contributions of myeloperoxidase and NADPH-oxidase to the early host defense against pulmonary infections with Candida albicans and Aspergillus fumigatus. Med Mycol. 2002;40(6):557–563. doi: 10.1080/mmy.40.6.557.563. [DOI] [PubMed] [Google Scholar]
- 69.Elahi S, Pang G, Ashman RB, Clancy R. Nitric oxide-enhanced resistance to oral candidiasis. Immunology. 2001;104(4):447–454. doi: 10.1046/j.1365-2567.2001.01331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.de Jesus-Berrios M, Liu L, Nussbaum JC, Cox GM, Stamler JS, Heitman J. Enzymes that counteract nitrosative stress promote fungal virulence. Curr Biol. 2003;13(22):1963–1968. doi: 10.1016/j.cub.2003.10.029. [DOI] [PubMed] [Google Scholar]
- 71.Bahn YS, Sundstrom P. CAP1, an adenylate cyclase-associated protein gene, regulates bud-hypha transitions, filamentous growth, and cyclic AMP levels and is required for virulence of Candida albicans. J Bacteriol. 2001;183(10):3211–3223. doi: 10.1128/JB.183.10.3211-3223.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sugui JA, et al. Genes differentially expressed in conidia and hyphae of Aspergillus fumigatus upon exposure to human neutrophils. PLoS One. 2008;3(7):e2655. doi: 10.1371/journal.pone.0002655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Thon M, Al-Abdallah Q, Hortschansky P, Brakhage AA. The thioredoxin system of the filamentous fungus Aspergillus nidulans: impact on development and oxidative stress response. J Biol Chem. 2007;282(37):27259–27269. doi: 10.1074/jbc.M704298200. [DOI] [PubMed] [Google Scholar]
- 74.Spikes S, et al. Gliotoxin production in Aspergillus fumigatus contributes to host-specific differences in virulence. J Infect Dis. 2008;197(3):479–486. doi: 10.1086/525044. [DOI] [PubMed] [Google Scholar]
- 75.Belkacemi L, Barton RC, Hopwood V, Evans EG. Determination of optimum growth conditions for gliotoxin production by Aspergillus fumigatus and development of a novel method for gliotoxin detection. Med Mycol. 1999;37(4):227–233. [PubMed] [Google Scholar]
- 76.Chapman RW, Phillips JE, Hipkin RW, Curran AK, Lundell D, Fine JS. CXCR2 antagonists for the treatment of pulmonary disease. Pharmacol Ther. 2009;121(1):55–68. doi: 10.1016/j.pharmthera.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 77.Sugui JA, et al. Gliotoxin is a virulence factor of Aspergillus fumigatus: gliP deletion attenuates virulence in mice immunosuppressed with hydrocortisone. Eukaryot Cell. 2007;6(9):1562–1569. doi: 10.1128/EC.00141-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bok JW, et al. GliZ, a transcriptional regulator of gliotoxin biosynthesis, contributes to Aspergillus fumigatus virulence. Infect Immun. 2006;74(12):6761–6768. doi: 10.1128/IAI.00780-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kale SP, Milde L, Trapp MK, Frisvad JC, Keller NP, Bok JW. Requirement of LaeA for secondary metabolism and sclerotial production in Aspergillus flavus. Fungal Genet Biol. 2008;45(10):1422–1429. doi: 10.1016/j.fgb.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.