

Abstract
The association of alcohol consumption and breast cancer is more pronounced in cases that are positive for estrogen receptor (ER+) than in cases that are negative (ER−). Its mechanism remains to be determined. Deregulation of RNA polymerase III (Pol III) transcription enhances cellular tRNAs and 5S rRNA production, increasing translational capacity to promote cell transformation and tumor formation. Here, we report that alcohol increases Pol III gene transcription in both normal and cancer breast cell lines. The induction in ER+ breast cancer cells (MCF-7) is significantly higher than in ER− normal breast cells (MCF-10A, MCF-10F and MCF-12A) and is correlated with ER expression. E2 causes <2-fold increase in Pol III gene transcription. The addition of ethanol to this system now produces a 10–15-fold increase. Ethanol increases ERα expression, resulting in an increase in Brf1 protein and mRNA levels. In addition, ethanol markedly stimulates phosphorylation of JNK1, but not JNK2. Inhibition of JNK1 decreases ERE-Luc reporter activity and represses expression of ERα, Brf1 and Pol III genes. Reduction of ERα by its small interfering RNA represses Brf1 and Pol III gene transcription. Ethanol with E2 produces larger and more numerous colonies. Repression of ERα or Brf1 inhibits alcohol-induced cell transformation. Together, these results support the idea that alcohol increases ERα expression through JNK1 to elevate Brf1 expression and Pol III gene transcription to bring about greater phenotypic changes. These studies demonstrate that ERα mediates Pol III gene transcription through Brf1, suggesting that ERα may play a critical role in alcohol-induced deregulation of Pol III genes in ER+ breast cancer development.
Introduction
Alcohol is the dietary factor, which is most consistently associated with breast cancer risk (1–4). This association involves the estrogen receptor (ER), which is overexpressed (ER+) in approximately 80% of breast cancer cases (5,6). Alcohol is known to promote mammary tumorigenesis (7–9). Cancer cells have a consistent cytological feature of nucleolar hypertrophy. rRNAs are synthesized by RNA polymerase (Pol) I and III within this nucleolar compartment. Pathologists have been using enlarged nucleoli as a strong diagnostic indicator of cell transformation and neoplasia. This indicates that transformation in situ is tightly linked to the deregulation of RNA Pol I and III gene transcription because the size of the nucleolus reflects the levels of rRNA synthesis (10). Although alcohol-associated breast cancer is widely studied, the molecular mechanism remains to be addressed.
RNA Pol III transcribes a variety of untranslated RNAs, including tRNAs, 5S rRNAs, 7SL RNA, 7SK RNA and U6 RNA (11–13), whereas tRNAs and 5S rRNAs control the translational and growth capacity of cells (10,14). Oncogenic proteins, such as Ras, c-Jun and c-Myc, stimulate RNA Pol III gene transcription (15–17), whereas tumor suppressors, such as pRb, p53, PTEN and Maf1, repress transcription of this class of genes (10,17,18). Studies have indicated that RNA Pol III transcription products are elevated in both transformed and tumor cells suggesting that they play a crucial role in tumorigenesis (10,18,19). Consistent with this idea, enhanced Pol III transcription is required for oncogenic transformation (20). The ability of these oncogenic and tumor suppressor proteins to alter Pol III transcription results from their capacity to regulate the TFIIIB complex. The TFIIIB complex consists of TATA box-binding protein (TBP) and its associated factors, Brf1 and Bdp1. TFIIIB, together with TFIIIC and RNA Pol III, are required to transcribe tRNA genes, whereas TFIIIB, together with TFIIIA, TFIIIC and RNA Pol III, are required to transcribe 5S rRNA genes.
High translational capacity is necessary for rapid growth and proliferation of tumor cells; Pol III gene transcripts have been found to be increased in ovarian tumor and breast cancer (19,21). Furthermore, expression of the Pol III gene, BC200, was elevated in breast squamous cell carcinoma tissues (22). Our recent studies using both cell culture model and animal models have revealed that alcohol induces transcription of tRNALeu and 5S rRNA (23). This induction in mice fed with ethanol is associated with liver tumor development (23). This implies that alcohol-induced deregulation of Pol III genes may play a critical role in tumor development. However, very little is known about the role of ERα in Pol III gene transcription. To explore the role of ERα in this relationship, we treated normal and breast cancer cell lines with ethanol. Our results indicate that ethanol-induced tRNA and 5S rRNA transcription in a breast cell lines is correlated with ER expression. Repression of ERα decreases alcohol-induced Brf1 expression, Pol III gene transcription and cell transformation. Further analysis reveals that ethanol increases ERα expression through the JNK1 pathway. Inhibition of JNK1 by a chemical inhibitor (SP600125) or JNK1 small interfering RNA (siRNA) reduces ERα and Brf1 expression and Pol III gene transcription. These studies support the idea that ERα may mediate the regulation of ethanol-induced Brf1 expression and Pol III gene transcription. Our results demonstrate, for the first time, that alcohol induces deregulation of Pol III gene transcription via ERα. These novel findings will be of great interest both to the basic and clinical research communities and provide a potential approach of treatment for alcohol-associated breast cancer patients.
Materials and methods
Cell lines, reagents and antibodies
ER− human breast non-tumorigenic epithelial cell lines (MCF-10A, MCF-10F and MCF-10-2A), ER+ human breast cancer cell lines (MCF-7 and T-47D) and ER− human breast cancer cell lines (MDA-MB231 and SKBR-3) were from ATCC. Cell culture medium Dulbecco’s modified Eagle’s medium (DMEM)/F12, Lipofectin reagent, Lipofectamine 2000, TRIzol reagent and OPTI-MEM were from Invitrogen. Antibodies against TBP and β-actin were obtained from Santa Cruz. Brf1 antibody was from Bethyl laboratories. JNK and phosphor-JNK antibodies were from Cell Signaling. JNK inhibitor, SP600125, was from A.G. Scientific. The sequences of Brf1 primers and siRNA and ERα siRNAs were listed in Supplementary Tables 1–3. E2 (17β-estradiol) was from Sigma–Aldrich. Plasmid of ERα expression and ERE-Luc reporter construct were kindly provided by Dr Baruch Frenkel (University of Southern California).
RNA isolation and real-time qPCR
Total RNA was isolated from MCF-10A and MCF-7 treated with ethanol using single step extraction method TRIzol reagent (Invitrogen). Total RNA samples were quantified and reverse transcribed in a 20 µl reaction containing 1× reverse transcription buffer. After first-strand cDNA synthesis, the cDNAs were diluted in DNase-free water and real-time qPCR (RT-qPCR) was performed with specific primers (Supplementary Table 1, available at Carcinogenesis Online) and PCR reagent kits (Bio-Rad Biotech) in the ABI prism 7700 Sequence Detection System. Precursor of tRNALeu and 5S rRNA transcripts and mRNAs of Brf1, TBP and ERα were measured by RT-qPCR as described previously (15,23).
Transfection and ERE-Luc reporter assays
For transient transfection assays, cells were transfected with plasmids and/or siRNAs as described previously (16). Serum-free medium was added to each dish with Lipofectin–DNA or Lipofectamine 2000–siRNA complexes, and cells were further incubated for 4h. The medium was changed with 10% fetal bovine serum/DMEM/F12 [phenol red-free (24)] and cells were incubated for 48h before harvesting. Protein concentrations of the resultant lysates were measured by the Bradford method. For ERE-Luc reporter assays, cells were transfected with 0.2 µg of the ERE-Luc constructs or plus JNK1 expression construct, or JNK1 and ERα siRNA for 24h. Cells were starved in DMEM/F12 for 3h and treated with 25mM ethanol for 60min. Cell pellets were resuspended in Promega reporter lysis buffer. The lysates were analyzed for luciferase activity using a luminometer and the Promega Luciferase Assay System as described (Promega). Resultant luciferase activities were normalized to the amount of protein in each lysate as described (23). The fold change in luciferase activity was calculated by determining the level of luciferase activity in the absence of alcohol. This value will be set at 1 for each independent experiment. Values are means ± SE of at least three independent experiments.
Cell-anchorage-independent growth
MCF-10A cells were transfected with mismatch RNA (mmRNA), Brf1 or ERα siRNAs (Supplementary Table 1, available at Carcinogenesis Online) as described (23). The transfected MCF-10A cells or parent MCF-10A cells (1×104 cells/well in 6-well plates) were suspended in 0.35% (w/v) agar in 10% fetal bovine serum/DMEM/F12 with or without 5nM E2, 25mM ethanol or both E2 and ethanol over a bottom layer of media with 0.5% (w/v) agar. Cells were fed fresh complete media with E2 or/and ethanol twice weekly. Colonies were counted 2–3 weeks or longer after plating as described previously (25).
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis and immunoblot analysis
Human breast cells were incubated with 25mM ethanol for 60min after starvation for 3h. Cells were collected with lysis buffer and sonicated. The suspensions were centrifuged to save the supernatants. Protein concentrations were determined by the Bradford method using Fluostar Omega spectrometer (Cell Biology Core Laboratory of University of Southern California Research Center for Liver Diseases, P30DK DK48522). Lysates (50 µg of protein) were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis. Proteins were transferred from the sodium dodecyl sulfate–polyacrylamide gel electrophoresis gel to Hybond-P membrane and immunoblot analysis was performed with specific antibodies. Membranes were probed with either antibodies against TBP, Brf1, JNKs, phosphor-JNK, ERα and β-actin as described (15). Bound primary antibody was visualized using horseradish peroxidase-conjugated secondary antibody (Vector Laboratories) and enhancing chemiluminscence reagents (Amersham).
Results
Alcohol-induced RNA Pol III gene transcription is ER dependent
Our recent study has demonstrated that alcohol-induced RNA Pol III-dependent transcription in vitro and in vivo by using cell culture model and animal model (23). To investigate the mechanism of alcohol-associated breast cancer, human breast cells were treated with ethanol and the amounts of precursor tRNALeu and 5S rRNA transcript were measured by RT-qPCR. Ethanol treatment of MCF-10A cells resulted in a concentration-dependent increase in pre-tRNALeu (Figure 1A) and 5S rRNA (Figure 1B) transcription, where the maximum response for ethanol-mediated induction was observed at the ethanol concentration of 25mM for 60min. Thus, this condition was used for the entire study unless stated otherwise. We determined in different ER− normal breast cell lines (MCF-10F, MCF-12A, MCF-10A) that the induction of pre-tRNALeu and 5S rRNA by ethanol is 2–3-fold (Figure 1C and 1D). MCF-7 treated with ethanol displayed a dramatic increase in pre-tRNALeu (Figure 1E) and 5S rRNA (Figure 1F) transcription. The induction of Pol III genes by alcohol in ER+ MCF-7 cells was 5–8-fold higher than in ER− normal breast cells. Furthermore, we also determined other ER− breast cancer cell lines (MDA-MB231, SK-BR-3) and ER+ breast cancer cell line (T-47D) (Figure 1G and 1H). These results reveal that induction of Pol III genes in ER+ breast cancer cells was higher than either ER− breast cancer cells or ER− normal breast cells. These results suggest that alcohol-induced Pol III gene transcription is correlated with ER expression.
Fig. 1.

Alcohol induces RNA Pol III-dependent transcription. (A and B) Ethanol enhances transcription in MCF-10A cells. MCF-10A cells were starved in DMEM/F12 for 3h. Cells were treated with or without different amounts of ethanol. (C and D) Ethanol induces transcription in ER− non-tumorigenic cells. MCF-10F, MCF-12A and MCF-10A cells were treated with or without 25mM ethanol; *P < 0.05, **P < 0.01 control compared with ethanol. (E and F) Ethanol stimulates transcription in MCF-7 cells. MCF-7 and MCF-10A cells were treated with or without 25mM ethanol. **P < 0.01 MCF-7 compared with MCF-10A. (G and H) Ethanol increases transcription in different breast cancer cell lines. ER+ breast cell line T-47D and ER− breast cell lines (MDA-MB231 and SKBR-3) were treated with ethanol. **P < 0.01 ER+ cancer cells compared with ER− cancer cells. RNA was isolated from these cells and RT-qPCR was performed to measure the amounts of pre-tRNALeu (A, C, E, G) and 5S rRNA (B, D, F, H). The fold change was calculated by normalizing to the amount of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA. The bars represent mean ± SE of at least three independent determinations.
Given that alcohol intake increased the transcriptional activity of ERα and activated E2 signaling pathway (26,27), we have examined the ERα effect on alcohol-stimulated induction of Pol III genes. The result indicates that alcohol enhanced the activity of ERE-Luc promoter and E2 alone increased 3-fold of this activity (Figure 2A, left), whereas ethanol plus E2 produced a 4.5-fold increase in its activity of MCF-7 cells (Figure 2A, left). In addition, we performed RT-qPCR and immunoblot analysis to measure the amount of ERα mRNA and protein. Ethanol increased ERα expression, either mRNA or protein (Figure 2A, middle and right). We next established whether ERα expression affects Pol III gene transcription. As indicated in Figure 2B, E2 alone elevated (~2-fold) tRNALeu and 5S rRNA transcription, but E2 plus ethanol produced an 11–14-fold of increase in the Pol III genes (Figure 2B,). In contrast, repressing ERα expression by its siRNA decreased the cellular levels of ERα mRNA and protein (Figure 2C, middle and right) and also reduced tRNALeu and 5S rRNA transcription (Figure 2D). These studies support the conclusion that ERα mediates Pol III gene transcription.
Fig. 2.

Enhanced ERα expression is required for alcohol-mediated induction of Pol III genes. (A) Ethanol increased ERE-dependent promoter activity and ERα expression. MCF-7 cells were transiently transfected with an ERE-Luc reporter plasmid for 24h, starved in DMEM/F12 for 3h and treated with ethanol or ethanol plus 5nM E2. Resultant protein lysates were subjected to determine luciferase activity (left). MCF-7 cells were treated with ethanol. The total RNA and cell lysates from these cells were extracted to determine mRNA and protein of ERα by RT-qPCT and immunoblot analysis. The antibodies were indicated (right). (B) Ethanol-enhanced RNA Pol III-dependent transcription is ER dependent. MCF-7 cells were treated with ethanol or ethanol + E2 as indicated at A. RT-qPCR was performed on RNA derived from these cells to measure pre-tRNALeu (left), 5S rRNA (right) and GAPDH transcripts. *P < 0.05 ethanol compared with ethanol + E2. (C) ERα expression is repressed by its siRNA. MCF-7 cells were transfected with ERE-Luc reporter plus 100nM mmRNA or ERα siRNA (left), or MCF-7 cells transfected with ERα siRNA or mmRNA. Luciferase activity (left), ERα mRNA (middle) and proteins (right) were determined from these cell lysates or total RNAs. (D) Reduction of ERα represses Pol III gene transcription. MCF-7 cells were transfected with ERα siRNA or mmRNA to measure pre-tRNALeu (left), 5S rRNA (right) and GAPDH transcripts. The fold change was calculated by normalizing to the amount of GAPDH. The bars represent mean ± SE of at least three independent determinations. *P < 0.05 mmRNA compared with ERα siRNA.
Alteration of ERα affects TFIIIB subunit Brf1 expression
To determine the mechanism by which ERα mediates Pol III gene transcription, we examined potential changes in the Pol III transcription machinery. Previous studies have demonstrated that change in TFIIIB subunits is associated with deregulation of Pol III genes, cell transformation and tumor formation (20,23). Our earlier study indicated that alteration of cellular levels of TBP affected Bdp1 expression, but did not affect Brf1 expression (28). Given that alteration of ERα affected Pol III gene transcription (Figure 2), we further investigated whether ERα was able to alter expression of TBP and Brf1. The results indicate that alcohol increased the cellular levels of Brf1 and TBP mRNAs and proteins (Figure 3A and 3B). E2 caused <1.5-fold of increase in the mRNA levels of Brf1 and TBP, whereas ethanol with E2 strongly increased 5-fold of Brf1 transcription, but slightly increased TBP (Figure 3C). In contrast, decreasing ERα expression repressed Brf1, but did not significantly affect TBP transcription (Figure 3D). We also established expression of TFIIIC63, but the level of TFIIIC63 was not affected by E2 and ethanol (Supplementary Figure 1, available at Carcinogenesis Online). To confirm the specific role of ERα on Pol III genes in a manner of Brf1 dependent, we have determined whether E2 and ethanol affects type III gene, U6, which is regulated by Brf2 (29). The result reveals that either E2 or E2 working with ethanol does not significantly alter U6 transcription (Supplementary Figure 2, available at Carcinogenesis Online). Ethanol + E2 increases in the level of Brf1 mRNA compared with ethanol alone, while produces 2-fold more increase in Pol III gene transcription (Figures 2 and 3). This implies that ERα selectively mediates Brf1 transcription.
Fig. 3.

Alcohol-mediated induction of Brf1, but not TBP, expression requires ERα. (A and B) Ethanol induces an increase in Brf1 and TBP expression. MCF-7 cells were treated with or without ethanol. Immunoblot analysis and RT-qPCR was performed using protein lysates and RNAs derived from these cells and antibodies were used to probe the proteins as designated. Left are cellular levels of Brf1 protein and mRNA. Right are the levels of TBP. (C) E2 increases alcohol-induced Brf1 expression. MCF-7 cells were treated with ethanol or ethanol plus E2. RT-qPCR was performed to determine cellular levels of Brf1 (left) and TBP (right) mRNA. (D) Repression of ERα expression decreases alcohol-stimulated Brf1 expression. MCF-7 cells were transfected with ERα siRNA or mmRNA for 48h and treated with ethanol. RT-qPCR was performed to measure Brf1 (left) and TBP (right) mRNA. The values represent mean ± SE from three independent experiments. *P < 0.05 as indicated.
JNK1 mediates ERα activity to modulate expression of Brf1 and Pol III genes
Given that alcohol has been shown to induce JNK activation (23,30) and that the JNKs play an important role in regulating Pol III gene transcription (28), we examined the role of JNKs in alcohol-mediated ERα expression. Ethanol induced a strong activation of JNK1, but a weaker activation of JNK2 in the MCF-7 cells (Figure 4A). Next, we assessed whether alcohol-activated JNK1 mediated ERα transcription. The results reveal that overexpression of JNK1 by its expression construct increased ERE-dependent Luc promoter activity (Figure 4B, left), whereas JNK inhibitor SP600125 and a specific JNK1 siRNA reduced ethanol-increased ERE-dependent Luc activity (Figure 4B, middle and right). The above results indicate that ERα modulates Brf1 expression, but not TBP (Figure 3). Therefore, we further determined whether JNK1 mediated Brf1 expression. Increasing JNK1 by its expression construct elevated the level of Brf1 mRNA (Figure 4C, left), whereas inhibition of JNK1 by SP600125 abrogated the ethanol-mediated increase in Brf1 transcription (Figure 4C, right). Decreasing JNK1 by its siRNA reduced the levels of either ERα or Brf1 protein and mRNA (Figure 4D). Furthermore, repression of ERα by its siRNA decreased alcohol-stimulated induction of ERα mRNA and protein and also reduced the cellular amounts of Brf1 mRNA and protein (Figure 4D). We next assessed whether the alcohol-mediated JNK1 activation was required for induction of Pol III gene transcription. Inhibition of JNK1 by SP600125 or JNK1 siRNA abrogated the induction of tRNALeu and 5S rRNA genes by ethanol (Figure 5A and 5B). However, ERKs inhibitor (U0126) or p38 kinase inhibitor (SB 200190) did not significantly reduce the induction of the Pol III genes by ethanol (data not shown). Conversely, JNK1 expression increased in the presence of ethanol produced a robust increase in Pol III gene transcription (Figure 5C). Together, these results support the idea that alcohol-mediated ERα and Brf1 expression requires the activation of JNK1. It suggests that alcohol-mediated Brf1 expression and Pol III gene transcription in MCF-7 cells go through JNK1 and ERα pathway.
Fig. 4.

Alcohol-mediated activation of JNK1 is required for induction of ERα and Brf1. (A) Ethanol induces JNK1 activation. MCF-7 cells were treated with or without ethanol. Immunoblot analysis was performed using protein lysates derived from these cells and antibodies against phosphorylated JNK1 and 2, and JNK1 and JNK2 as designated. A representative blot from three independent determinations is shown. (B) JNK1 mediates alcohol-induced ERα transcription. MCF-7 cells were transfected with ERE-Luc reporter plus JNK1 expression (left) construct or JNK1 siRNA (right) for 48h; MCF-7 cells were pretreated with 5 µM SP600125 (JNK inhibitor) for 1h (middle). Cells were then treated with or without ethanol. The cell lysates were extracted from these cells and luciferase activity assay was performed to measure the ERE-dependent promoter activity. (C) Ethanol-activated JNK1 mediates Brf1 expression. MCF-7 cells were transfected with JNK1 expression plasmid for 48h (left) or MCF-7 cells pretreated with 5 µM SP600125 for 1h (right). And then these cells were treated with or without ethanol for another 1h. Brf1 mRNA was determined by using the RNA from these cells. (D) Inhibition of JNK1 represses ethanol-induced ERα expression. Immunoblot analysis was performed using the lysates derived from MCF-7 cells transfected with either JNK1 siRNA or ERα siRNA (left panel) or RT-qPCR was carried out to measure the levels of ERα and Brf1 mRNA (middle and right panels) as indicated in (D). A representative blot from three independent determinations is shown (left). The values represent mean ± SE from three independent experiments. *P < 0.05 as indicated.
Fig. 5.

Alcohol-activated JNK1 mediates transcription of Pol III genes. (A) JNK inhibitor SP600125 inhibits alcohol-induced Pol III gene transcription. MCF-7 cells were pretreated with 5 µM SP600125 and then treated with or without ethanol. RT-qPCR was performed to determine pre-tRNALeu (left) and 5S rRNA (right). (B) Repression of JNK1 decreases transcription. MCF-7 cells were transfected with either mmRNA or JNK1-specific siRNA for 48h and then treated with ethanol. RNAs were derived from these cells and RT-qPCR was performed to measure the amounts of pre-tRNALeu (left), 5S rRNA (right) and GAPDH transcripts. (C) Overexpression of JNK1 enhances transcription. MCF-7 cells were transfected with either JNK1 expression construct or vector for 48h and treated with ethanol. RNA was isolated to determine the amounts of pre-tRNALeu (left), 5S rRNA (right) and GAPDH transcripts. The fold change was calculated by normalizing to the amount of GAPDH mRNA. The values represent mean ± SE of three independent determinations. *P < 0.05 as indicated.
Alcohol enhanced the occupancy of ERα in Brf1 promoter and reduction of ERα decreased Pol III gene transcription and repressed cell transformation
Next, we determined whether ERα mediates Brf1 transcription, which is a subunit of TFIIIB and specifically regulates Pol III gene transcription. We performed a ChIP assay to determine if ERα occupied the Brf1 promoter. The results reveal that ethanol increased the occupancy of Brf1 promoters by ERα in MCF-7 cells (Figure 6A). It suggests that ERα is able to directly regulate Brf1 to modulate Pol III gene transcription. Our results further reveal that ethanol increased the occupancy of tRNALeu and 5S rRNA promoters by Brf1 (Figure 6B). In addition, the results reveal that ethanol also enhances ERα to Pol III gene promoters (Supplementary Figure 3, available at Carcinogenesis Online). This indicates that ERα can directly and indirectly modulates Pol III gene transcription. It suggests that ERα and Brf1 may be targets in alcohol-induced response.
Fig. 6.

Ethanol induces occupancy of ERα to Brf1 promoters to affect cell transformation. (A) Ethanol-mediated binding of ERα to the Brf1 promoter. Schematic of the Brf1 promoter and primers used for ChIP assays are designated relative to the ERα site (top). MCF-7 cells were treated with or without ethanol and ChIP assays were performed using ERα and histone H3 antibodies and qPCR to quantify the amplified DNA. The relative occupancy of the proteins was calculated based on the control (no ethanol treatment). All values shown are the means ± SE of at least three independent chromatin preparations. (B) Ethanol enhances Brf1 recruitment to tRNALeu and 5S rRNA genes. Cells were treated with or without ethanol and ChIP assays were performed to measure the occupancy of Brf1 and H3 on the tRNALeu and 5S rRNA gene promoter. (C) Repression of Brf1 decreases Pol III gene transcription. MCF-7 cells were transfected with either mmRNA or Brf1 siRNA for 48h and then treated with ethanol. RNA was isolated and RT-qPCR was performed to measure the amounts of pre-tRNALeu (left), 5S rRNA (right) and GAPDH transcripts. The fold change was calculated by normalizing to the amount of GAPDH mRNA. The values represent + SE of three independent determinations. (D) Down regulating ERα and Brf1 expression decreases ethanol-induced anchorage-independent growth. Parent MCF-10A cells and MCF-10A cells expressing ERα or Brf1 siRNAs were poured in triplicate into 6-well plates with 0.35% agar containing 25mM ethanol, 5nM E2 or ethanol plus E2. Cells were incubated at 37°C in 5% CO2 for 2–3 weeks or longer and were fed with fresh complete media with or without ethanol, E2 or ethanol plus E2 twice weekly. Colonies were counted at 1–2 weeks after plating. Values are the means ± SE (n ≥ 3). *P < 0.05 as indicated.
Previous studies demonstrated that increasing Brf1 expression resulted in enhancement of Pol III gene transcription and was sufficient for cell transformation (20,23). Reduction of Brf1 expression decreases anchorage-independent colonies to form and repressed tumor formation in mouse (20,25). To further assess potential alterations in Pol III gene transcription and Brf1 expression by alcohol-induced ERα activity, we performed soft agar assay and determined cell-anchorage-independent growth. In the present study, we demonstrated that ERα modulated Brf1 expression, which in turn regulated tRNA and 5S rRNA transcription (Figures 2–4 and 6C). Thus, we investigated whether ERα affected ethanol-induced cell transformation. Previous studies demonstrated that E2 treatment increases cellular levels of estrogen receptor of MCF-10A cells (31) and the cells were responsive to E2-induced colony formation in soft agar (31,32). Here, the results indicate that ethanol alone had no significant effect on transformation of MCF-10A cells, whereas E2 alone was able to induce cell transformation. However, ethanol in combination with E2 strongly induced MCF-10A cell-anchorage-independent growth (Figure 6D). Given that inhibiting ERα expression by its siRNAs decreased cellular levels of Brf1 protein and mRNA (Figure 3), as well as Pol III gene transcription (Figure 2), reduction of Brf1 expression by its siRNA significantly decreased ethanol-induced Pol III gene transcription (Figure 6C) and colony formation of MCF-10A cells, compared with mmRNA (Figure 6D). These results demonstrate that alcohol increases cellular ERα, which mediates Brf1 expression to enhance tRNA and 5S rRNA transcription, thereby affecting alcohol-promoted transformation of MCF-10A cells.
Discussion
This study presents a mechanistic analysis characterizing how ERα mediates the endogenous Pol III gene, tRNA and 5S rRNA, transcription. In this study, results demonstrate that alcohol-induced deregulation of Pol III genes in breast cell lines is correlated with ERα expression. Alcohol increases ERα expression through the JNK1 pathway. Our studies identified the mechanism, by which alcohol increased occupancy of ERα in the Brf1 promoter, mediating Brf1 expression, which in turn upregulates Pol III gene transcription. Repression of ERα decreases Brf1 expression and Pol III gene transcription. Reduction of ERα and Brf1 was sufficient to repress alcohol-induced anchorage-independent cell growth. These findings support the notion that ERα increases Brf1 expression, but not TBP, to regulate alcohol-induced Pol III gene transcription, resulting in a change in phenotype.
Alcohol consumption has consistently been associated with an increased risk for breast cancer in both premenopausal and postmenopausal women (3,4). Studies by Wang et al. (33) have demonstrated that alcohol increased MCP-1 and CRR2 expression, which promoted mammary tumor growth in alcohol-fed mice. Epidemiologic studies indicated that alcohol consumption was associated with ER+ breast cancer cases more than to ER− cases (5,6,34). A recent study indicates that alcohol increased ERα expression to promote breast tumor formation in mice (27). However, the exact mechanism, by which alcohol promotes development of ER+ breast cancer, is still unknown. A previous study demonstrated that alcohol down regulated the expression of BRCA1, a potent inhibitor of ERα, thereby contributing to breast cancer (34). Alcohol intake was also shown to increase the transcriptional activity of ERα (26). Studies have indicated that Ral-dependent recruitment of ERα to the AP-1 binding site stimulated JNK1 enzymatic activity (35). A study indicates that ethanol activated JNKs in the engineered HB2 cells overexpressing ErbB2 (36). Here, we established that alcohol activated JNK1, but not JNK2 in ER+ MCF-7 cells. This implies that alcohol-activated JNK pathway is associated with either ErbB2 or ERα expression. Given AP-1, which is modulated by JNK mediates ERα activity, it suggests that alcohol-activated JNK1 is more specific in ER+ breast cancer cells. This result is consistent with our recent study, where alcohol induced JNK1 activation in HepG2-ADH cells (23). Since JNK1 positively mediates Pol III gene transcription (28), it suggests that alcohol-induced activation of JNK1 in both breast and liver cells may be a common signaling pathway to mediate Pol III gene transcription.
Our studies have demonstrated that epidermal growth factor increased TFIIIB subunit, such as TBP, Brf1 and Bdp1, expression and enhanced Pol III gene transcription in JB6 cells (15,16). Regulation of Bdp1, but not Brf1, occurred through JNK1-mediated alterations in TBP expression (28), suggesting that Brf1 and Bdp1 may be regulated independently. Our recent study demonstrates that alcohol-induced Pol III gene transcription in vivo and in vitro, where this induction promoted tumor development in liver of NS5A transgenic mouse (23). This indicates that deregulation of Pol III genes by alcohol promotes liver tumor development. However, little is known concerning the mechanism by which ERα mediates alcohol-induced deregulation of Pol III gene transcription. Studies have indicated that oncogenic proteins or tumor suppressors interacted with TFIIIB to enhance or repress Pol III gene transcription (10,15–18). TBP interacts with the N-terminal activation domain of ERα, where it can induce and/or stabilized an ordered structure in the N-terminal regions of ERα (37). Ethanol-stimulated ERα increases Brf1 expression, but not TBP (Figure 3). This indicates that ERα does not affect TBP expression, whereas the interaction between ERα and TBP may increase Pol III gene transcription. In contrast, change in cellular level of ERα by ethanol caused an alteration of Brf1 expression. ERα directly occupied the Brf1 promoter to modulate its expression. This finding is consistent with a previous study using human breast cancer biopsies, in which Brf1 expression in ER+ breast cancer cases is higher than in ER− cases (38). This suggests that Brf1 may be a target modulated by ERα. The ERα-mediated alteration of Brf1 may play an important role in cell transformation and alcohol-associated tumor formation.
Previous studies have demonstrated that alcohol intake increased the transcriptional activity of ERα (26), as well as level of AP-1 expression (39). We established that alcohol treatment increased c-Jun, a subunit of AP-1, expression and enhanced occupancy of TBP, Brf1 and tRNALeu promoters by c-Jun to elevate Pol III gene transcription in HepG2-ADH cells (23). In the present study, the results indicate that 25mM ethanol in MCF-7 cells was able to produce higher induction (11–15-fold) of Pol III gene transcription than 50mM ethanol in HepG2-ADH cells (4–5-fold). This indicates that breast cancer cells are more sensitive to ethanol than liver cells. Given that the interaction between ERα and c-Jun resulted in elevation of transcription of AP-1-dependent genes (40,41), this interaction may produce higher induction of Brf1 expression and Pol III gene transcription in MCF-7 cells. Epidemiological studies revealed that alcohol consumption was associated with over a dozen human cancers (42,43). However, it is not clear how alcohol promotes human cancer development in different organs. Our studies using liver and breast cells indicate a possible common mechanism by which alcohol induces deregulation of Pol III genes to promote tumor development (23). Since ERα is significantly expressed in ER+ breast cancer cells and breast tissues, whereas ER− breast cancer cells do not express detectable levels of ERα, ERα-mediated deregulation of Pol III genes explains why alcohol consumption is associated more with ER+ breast cancer cases than with ER− cases.
In summary, the present study provides evidence that alcohol-induced JNK1 activation enhances ERα expression, increasing ERα occupancy in the Brf1 promoter to enhance Brf1 expression, resulting in increasing Pol III gene transcription and the rate of cell transformation (Figure 7). This is the first report that ERα mediates RNA Pol III-dependent transcription induced by alcohol. The novel findings suggest the possibility that inhibition of Brf1 expression may be a potential approach to repress alcohol-promoted cell transformation and breast cancer development.
Fig. 7.
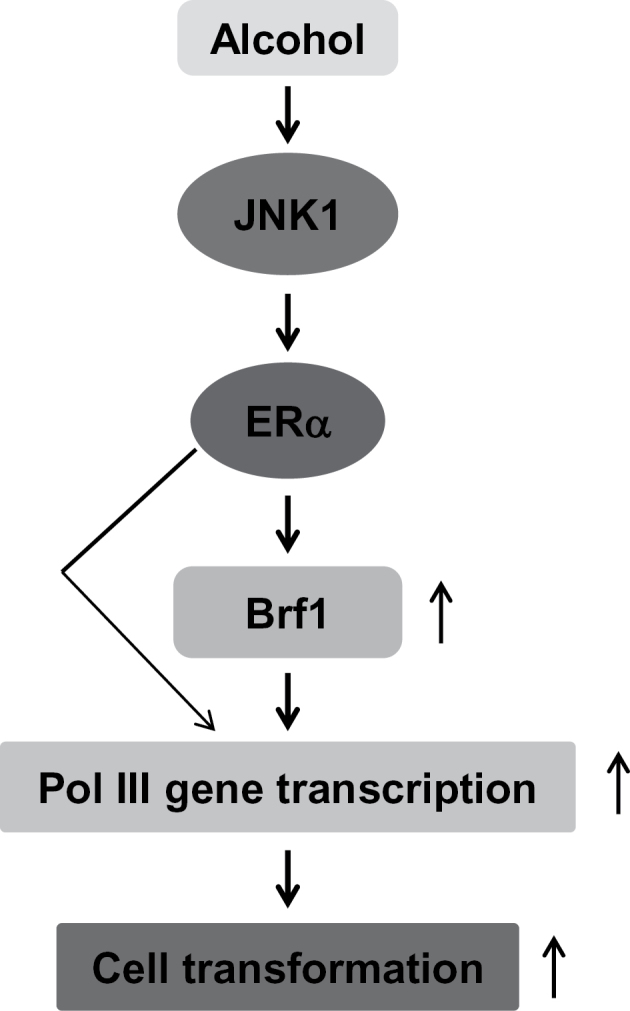
Schematic illustration of ERα mediating Brf1 expression and Pol III gene transcription. Ethanol enhances ERα activity through the JNK1 pathway. ERα increases Brf1 expression, which in turn regulates Pol III gene transcription to promote alcohol-induced cell transformation.
Supplementary material
Supplementary Figures 1–3 and Tables 1–3 can be found at http://carcin.oxfordjournals.org/
Funding
National Institute of Health/National Institute on Alcohol Abuse and Alcoholism (AA017288 and AA021114 to Shuping Zhong).
Supplementary Material
Acknowledgements
We want to thank Drs M.R.Stallcup and D.L.Johnson (University of Southern California) for scientific discussions. We would like to thank Dr B.Frenkel (USC), who provide ERα expression and ERE-dependent reporter constructs.
Conflict of Interest Statement: None declared.
Glossary
Abbreviations:
- DMEM
Dulbecco’s modified Eagle’s medium
- ER
estrogen receptor
- ERE-Luc
estrogen receptor response element-luciferase
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- mmRNA
mismatch RNA
- Pol
polymerase
- RT-qPCR
real-time qPCR
- siRNA
small interfering RNA.
References
- 1. Hamajima N., et al. (2002). Alcohol, tobacco and breast cancer—collaborative reanalysis of individual data from 53 epidemiological studies, including 58,515 women with breast cancer and 95,067 women without the disease. Br. J. Cancer 87 1234–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. MacMahon B. (2006). Epidemiology and the causes of breast cancer Int. J. Cancer 118 2373–2378 Review [DOI] [PubMed] [Google Scholar]
- 3. Petri A., et al. (2004). Alcohol intake, type of beverage, and risk of breast cancer in pre- and postmenopausal women. Alcohol. Clin. Exp. Res. 28 1084–1090 [DOI] [PubMed] [Google Scholar]
- 4. Singletary K.W., et al. (2001). Alcohol and breast cancer: review of epidemiologic and experimental evidence and potential mechanisms. JAMA 286 2143–2151 [DOI] [PubMed] [Google Scholar]
- 5. Suzuki R., et al. (2008). Alcohol intake and risk of breast cancer defined by estrogen and progesterone receptor status—a meta-analysis of epidemiological studies. Int. J. Cancer 122 1832–1841 [DOI] [PubMed] [Google Scholar]
- 6. Deandrea S., et al. (2008). Alcohol and breast cancer risk defined by estrogen and progesterone receptor status: a case-control study. Cancer Epidemiol. Biomarkers Prev. 17 2025–2028 [DOI] [PubMed] [Google Scholar]
- 7. Singletary K.W., et al. (1991). Ethanol consumption and DMBA-induced mammary carcinogenesis in rats. Nutr. Cancer 16 13–23 [DOI] [PubMed] [Google Scholar]
- 8. Singletary K.M., et al. (1995). Enhancement by chronic ethanol intake of N-methyl-N-nitrosourea-induced rat mammary tumorigenesis. Carcinogenesis 16 959–964 [DOI] [PubMed] [Google Scholar]
- 9. Watabiki Y., et al. (2000). Long-term ethanol consumption in ICR mice causes mammary tumor in females and liver fibrosis in males Alcohol Clin. Exp. Res. 24 117S–122S [PubMed] [Google Scholar]
- 10. White R.J. (2001). RNA polymerase III transcription and cancer. Oncogene 23 3208–3216 [DOI] [PubMed] [Google Scholar]
- 11. Ullu E., et al. (1984). Alu sequences are processed 7SL RNA genes. Nature 312 171–172 [DOI] [PubMed] [Google Scholar]
- 12. Dieci G., et al. (2007). The expanding RNA polymerase III transcriptome. Trends Genet. 23 614–622 [DOI] [PubMed] [Google Scholar]
- 13. Raha D., et al. (2010). Close association of RNA polymerase II and many transcription factors with Pol III genes. Proc. Natl. Acad. Sci. U.S.A. 107 3639–3644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Goodfellow S.J., et al. (2006). Regulation of RNA polymerase III transcription during hypertrophic growth. EMBO J. 25 1522–1533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhong S., et al. (2004). Epidermal growth factor enhances cellular TBP levels and induces RNA polymerase I- and III-dependent gene activity. Mol. Cell. Biol. 24 5119–5129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhong S., et al. (2007). TBP is differentially regulated by JNK1 and JNK2 through Elk-1, controlling c-Jun expression and cell proliferation. Mol. Cell. Biol. 27 54–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Johnson D.L., et al. (2008). Cell biology, RNA metabolism and oncogenesis. Science 320 461–462 [DOI] [PubMed] [Google Scholar]
- 18. Woiwode A., et al. (2008). PTEN represses RNA polymerase III-dependent transcription by targeting the TFIIIB complex. Mol. Cell. Biol. 28 4204–4214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Winter A., et al. (2007). RNA polymerase III transcription factor TFIIIC2 is overexpressed in ovarian tumors. Proc. Natl. Acad. Sci. U.S.A. 97 12619–12624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Johnson S.A., et al. (2008). Enhanced RNA polymerase III-dependent transcription is required for oncogenic transformation. J. Biol. Chem. 283 19184–19191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kamath R.V., et al. (2005). Perinucleolar compartment prevalence has an independent prognostic value for breast cancer. Cancer Res. 65 246–253 [PubMed] [Google Scholar]
- 22. Chen W., et al. (1997). Expression of neural BC200 RNA in human tumours. J. Pathol. 83 345–351 [DOI] [PubMed] [Google Scholar]
- 23. Zhong S., et al. (2011). Alcohol induces RNA polymerase III-dependent transcription through c-jun by coregulating TBP and Brf1 expression J. Biol. Chem. 286 2393–2401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Berthois Y., et al. (1986). Phenol red in tissue culture media is a weak estrogen: implications concerning the study of estrogen-responsive cells in culture. Proc. Natl. Acad. Sci. U.S.A. 83 2496–2500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang Q., et al. (2011). Phosphorylation of histone H3 serine 28 modulates RNA polymerase III-dependent transcription. Oncogene 30 3943–3952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fab S., et al. (2000). Alcohol stimulates estrogen receptor signaling in human breast cancer cell lines Cancer Res. 60 5635–5639 [PubMed] [Google Scholar]
- 27. Wong A.W., et al. (2012). Alcohol promotes mammary tumor development via the estrogen pathway in estrogen receptor alpha-negative HER2/neu mice. Alcohol. Clin. Exp. Res. 36 577–587 [DOI] [PubMed] [Google Scholar]
- 28. Zhong S., et al. (2009). The JNKs differentially regulate RNA polymerase III transcription by coordinately modulating the expression of all TFIIIB subunits. Proc. Natl. Acad. Sci. U.S.A. 106 12682–12687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schramm L., et al. (2002). Recruitment of RNA polymerase III to its target promoters. Genes Dev. 16 2593–2620 [DOI] [PubMed] [Google Scholar]
- 30. Luedemann C.E., et al. (2005). Ethanol modulation of TNF-alpha biosynthesis and signaling in endothelial cells: synergistic augmentation of TNF-alpha mediated endothelial cell dysfunctions by chronic ethanol. Alcohol. Clin. Exp. Res. 29 930–938 [DOI] [PubMed] [Google Scholar]
- 31. Liu S., et al. (2004). Transformation of MCF-10A human breast epithelial cells by zeranol and estradiol-17beta. Breast J. 10 514–521 [DOI] [PubMed] [Google Scholar]
- 32. Park S.A., et al. (2009). 4-Hydroxyestradiol induces anchorage-independent growth of human mammary epithelial cells via activation of IkappaB kinase: potential role of reactive oxygen species. Cancer Res. 69 2416–2424 [DOI] [PubMed] [Google Scholar]
- 33. Wang S., et al. (2012). Ethanol promotes mammary tumor growth and angiogenesis: the involvement of chemoattractant factor MCP-1 Breast Cancer Res. Treat., 133(3):1037––1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dumitrescu R.G., et al. (2005). The etiology of alcohol-induced breast cancer. Alcohol 35 213–225 [DOI] [PubMed] [Google Scholar]
- 35. Fogarty E.A., et al. (2012). Activation of estrogen receptor α by raloxifene through an activating protein-1-dependent tethering mechanism in human cervical epithelial cancer cells: a role for c-Jun N-terminal kinase. Mol. Cell. Endocrinol. 348 331–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ma C., et al. (2003). Overexpression of ErbB2 enhances ethanol-stimulated intracellular signaling and invasion of human mammary epithelial and breast cancer cells in vitro. Oncogene 22 5251–5290 [DOI] [PubMed] [Google Scholar]
- 37. Warmmark A., et al. (2001). The N-terminal regions of estrogen receptor alpha and beta are unstructured in vitro and show different TBP binding properties J. Biol. Chem. 276 45939–45944 [DOI] [PubMed] [Google Scholar]
- 38. Julka P.K., et al. (2008). A phase II study of sequential neoadjuvant gemcitabine plus doxorubicin followed by gemcitabine plus cisplatin in patients with operable breast cancer: prediction of response using molecular profiling. Br. J. Cancer 98 1327–1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chiu R., et al. (1988). The c-fos protein interacts with c-Jun AP-1 to stimulate transcription of AP-1 responsive genes. Cell 54 541–552 [DOI] [PubMed] [Google Scholar]
- 40. Teyssier C., et al. (2001). Characterization of the physical interaction between estrogen receptor alpha and JUN proteins. J. Biol. Chem. 276 36361–36369 [DOI] [PubMed] [Google Scholar]
- 41. Cheung E., et al. (2005). Altered pharmacology and distinct coactivator usage for estrogen receptor-dependent transcription through activating protein-1. Proc. Natl. Acad. Sci. U.S.A. 102 559–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Purohit V., et al. (2005). Mechanisms of alcohol-associated cancers: introduction and summary of the symposium. Alcohol 35 155–160 [DOI] [PubMed] [Google Scholar]
- 43. Bagnardi V., et al. (2001). A meta-analysis of alcohol drinking and cancer risk. Br. J. Cancer 85 1700–1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.