

Abstract
We investigated the spatiotemporal expression of vascular endothelial growth factor receptor–3 (VEGFR-3) in the spinal cord of Lewis rats with experimental autoimmune encephalomyelitis (EAE), an animal model for multiple sclerosis. VEGFR-3 mRNA and protein were constitutively expressed in gray matter neurons and in a few white matter astrocytes. Induction of VEGFR-3 occurred predominantly in perivascular infiltrated macrophages in the spinal cord white matter during the inductive phase of EAE. VEGFR-3 expression was also induced in activated microglial cells in the gray and white matter, mainly in the peak phase. In addition, reactive astrocytes in the white matter, but not in the gray matter, expressed VEGFR-3 as disease severity increased. These data suggest that VEGFR-3 is involved in the recruitment of monocytic macrophages and in glial reactions during EAE.
Keywords: VEGFR-3, macrophage, microglia, reactive astrocyte, experimental autoimmune encephalomyelitis
Experimental autoimmune encephalomyelitis (EAE) is a well-established animal model of multiple sclerosis (MS) in which the immune system attacks myelin protein of the central nervous system (CNS) and leads to inflammatory demyelination and oligodendrocyte loss (Frohman et al. 2006; Cassan and Liblau 2007; Compston and Coles 2008; Centonze et al. 2010; Moore 2010). Although the precise trigger for MS remains elusive, resident microglia and blood-derived monocytes or macrophages seem to play a central role in the pathogenesis of MS and EAE (for review, see Gandhi et al. 2010; Almolda et al. 2011; Chastain et al. 2011). Elimination of macrophages or microglia suppresses clinical and histopathological manifestations in rodent models of MS (Brosnan et al. 1981; Huitinga et al. 1990; Bauer et al. 1995; Polfliet et al. 2002; Heppner et al. 2005), and infiltrating blood-borne monocytes trigger EAE progression to the severe form of the disease (Ajami et al. 2011). In addition, astrocytes, CNS-resident glial cells, play an active role in the pathogenesis of EAE and MS (Voskuhl et al. 2009; Toft-Hansen et al. 2011), and their activation is a prominent feature in EAE and MS (Aquino et al. 1988; Liedtke et al. 1998).
Recent studies have shown that vascular endothelial growth factor (VEGF) participates in the pathogenesis of MS and EAE by inducing focal blood-brain barrier breakdown, angiogenesis, and inflammatory infiltration into lesions (Proescholdt et al. 2002; Kirk and Karlik 2003; Su et al. 2006; Tham et al. 2006; Sasaki et al. 2010; Seabrook et al. 2010). Most of these studies have focused on VEGF-A and its two tyrosine kinase receptors: VEGFR-1 and VEGFR-2. We recently reported that VEGFR-3 (also known as fms-like tyrosine kinase-4; Flt-4), the receptor for VEGF-C and VEGF-D, was induced in reactive astrocytes and possibly in infiltrating blood-borne macrophages after focal ischemia (Shin et al. 2010) in a rat model of stroke. These data suggest that in addition to its role as a trophic factor for neural progenitors in the developing and adult brain (Le Bras et al. 2006; Calvo et al. 2011), VEGF-C/VEGFR-3 may be involved in the glial reaction and possibly in the recruitment of monocytic macrophages during ischemic insults, contributing to inflammation in the ischemic brain. In support of this hypothesis, VEGFR-3 is thought to be expressed in bone marrow–derived cells, including macrophages, monocytes, and dendritic cells (Skobe et al. 2001; Hamrah et al. 2003, 2004; Stepanova et al. 2007). Furthermore, VEGF-C affects macrophage migration via VEGFR-3 (Skobe et al. 2001; Stepanova et al. 2007), and VEGFR-3 modulates adaptive immunity by mediating chemotaxis of antigen-presenting (dendritic) cells (Chen et al. 2007). Therefore, we speculate that VEGF-C is involved in the glial reaction and the recruitment of monocytic macrophages via VEGFR-3 during EAE. However, little is known about the pathophysiological role of VEGF-C in EAE animal models.
Therefore, we investigated the spatiotemporal regulation of VEGF-C and VEGFR-3 mRNA in the spinal cords of Lewis rats with acute EAE using quantitative real-time reverse transcriptase–polymerase chain reaction (qRT–PCR) analysis. In addition, we examined the cellular localization of VEGFR-3 mRNA and protein during the course of EAE, using in situ hybridization and immunohistochemical analysis. Double- and triple-labeling techniques were used to identify the phenotypes of cells expressing VEGFR-3. Our results provide a better understanding of the role of VEGF-C during the acquired autoimmune inflammation associated with EAE.
Materials and Methods
Animal Preparation and Assessment of Clinical Signs of EAE
All surgical interventions and presurgical and postsurgical animal care were provided in accordance with the Laboratory Animal Welfare Act, the Guide for the Care and Use of Laboratory Animals, and the Guidelines and Policies for Rodent Survival Surgery provided by the IACUC (Institutional Animal Care and Use Committee) in the School of Medicine, The Catholic University of Korea.
EAE was induced in adult female Lewis rats (180–200 g). Animals were immunized by a single subcutaneous injection into the dorsal base of the tail with 200 µl of an emulsion containing equal parts of myelin basic protein (MBP, 1 mg/ml; Sigma, St. Louis, MO) and complete Freund’s adjuvant (CFA) supplemented with Mycobacterium tuberculosis H37Ra (5 mg/ml; Difco, Detroit, MI). Animals injected with the same emulsion without MBP were used as CFA controls. All MBP-injected rats were evaluated daily for the presence of clinical symptoms using the following clinical scoring scale: 0, no clinical sign; E1, incomplete tail paralysis; E2, complete tail paralysis; E3, unsteady gait or incomplete paraplegia; E4, complete paraplegia; and E5, hind limb and forelimb paralysis (Gao et al. 2010). EAE rats were sacrificed at different phases of the EAE course: control, onset (EAE score, E1 to E2, 7–10 days postimmunization [PI]), peak (EAE score, E3 to E4, 13–15 days PI), and recovery (EAE score, 0, 30 days PI). For histological studies, seven animals were included in each group. To examine the distribution of VEGFR-3 expression over the chronic interval of 10 weeks after immunization, three rats were allowed to live for 10 weeks PI.
Animals were deeply anesthetized with 16.9% urethane (10 ml/kg) and killed by transcardial perfusion with a fixative containing 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer (PB, pH 7.4). The lumbar segment of the spinal cord was removed and postfixed for 2 hr in the same fixative. After treatment with 30% sucrose, spinal cords were embedded in OCT compound and stored at −70C. For qRT-PCR and immunoblot analysis, six rats from each of the three groups (control, peak, and recovery) were deeply anesthetized and perfused with 0.1 M PB (pH 7.4). White matter from the lumbar spinal cord was excised under a microscope and immediately frozen in liquid nitrogen. Samples were stored at −70C until further processing. Spinal cord sections were routinely stained with hematoxylin-eosin and luxol fast blue for histological studies.
Quantitative Real-Time RT-PCR
Control rats and EAE rats at peak and recovery stages were sacrificed as described above. Total RNA was extracted from the white matter of the lumbar spinal cord with RNAlater (Ambion, Inc., Austin, TX), and the purity and yield of the RNA were determined spectrophotometrically. One microgram of total RNA from each sample was transcribed into first-strand cDNA using reverse-transcriptase M-MLV (Takara Korea Biomedical, Inc., Seoul, Korea) in a total volume of 20 µl according to the manufacturer’s instructions. Equal amounts (1 µl) of cDNA were real-time amplified in a Rotor-Gene RG-6000 (Corbett Research, Mortlake, Australia) using the SYBR Premix EX Taq kit (RR420A, Takara Korea Biomedical, Inc.). The primers were designed at the Internet site provided by Integrated DNA Technologies, Inc. (Coralville, IA; http://eu.idtdna.com/Scitools/Applications/RealTimePCR/). The sequences of the primers used were as follows: VEGFR-3, accession number NM_053652, forward CGAGACTGGAAGG AGGTGA and reverse TGTACATGGCTGACACATTGG; VEGF-C, accession number NM_053653, forward CGCT GTGTCCCATCATATTG and reverse CCATGGTCCC ACAGAGTCTT; and GAPDH, accession number NM_ 017008, forward GGATGGAATTGTGAGGGAGA and reverse GTGGACCTCATGGCCTACAT. The primers were verified to generate a single PCR product using gel electrophoresis after conventional PCR. The PCR conditions were as follows: incubation for 10 min at 95C, followed by 45 cycles of denaturation at 95C for 10 sec, annealing at 60C for 15 sec, and extension at 72C for 20 sec. A reaction mixture lacking cDNA was used as the negative control. The data were analyzed using Rotor-Gene 6000 Series software (version 1.7.75; Corbett Research). The expression level of VEGF-C or VEGFR-3 was calculated using the comparative threshold cycle method (2−ΔΔCt) with GAPDH as the control gene.
In Situ Hybridization Histochemistry and Double/Triple Labeling
Specific sequences for VEGFR-3 were prepared using RT-PCR, and antisense and sense riboprobes were labeled with digoxigenin (DIG) using in vitro transcription, as described previously (Shin et al. 2008). Coronal cryostat sections (25 µm thick) of the lumbar spinal cords were cut and processed free floating for in situ hybridization. Each well contained spinal cord sections representing the different phases of the EAE course, to ensure consistent in situ hybridization conditions. The sections were hybridized with antisense or sense probes diluted in hybridization solution (150 ng/ml) at 52C for 18 hr. Hybridization was visualized using an alkaline phosphatase-conjugated sheep anti-DIG antibody (Roche, Grenzach-Wyhlen, Germany; dilution 1:2000) with 4-nitroblue tetrazolium chloride (0.35 mg/ml) and 5-bromo-4-chloro-3-indolyl phosphate (0.18 mg/ml) as substrates. Tissue sections were visualized using a microscope and photographed using a digital camera (Jenoptik, Berlin, Germany). Images were converted to TIFF format and contrast levels adjusted using Adobe Photoshop v. 7.0 (Adobe Systems, San Jose, CA). The same intensity of light in the microscope and the same parameters in the digital camera were used when images were acquired, with minor adjustments to ensure uniform contrast for all figures.
After hybridization, as described above, some sections were incubated with biotin-conjugated mouse monoclonal anti-DIG antibody (Jackson ImmunoResearch, West Grove, PA; dilution 1:200) overnight at 4C. For double or triple immunofluorescence, sections were incubated at 4C overnight with the following antibodies: mouse monoclonal antibodies to ED1 (CD68) (Serotec, Oxford, UK; dilution 1:100), glial fibrillary acidic protein (GFAP; Millipore Corporation, Temecula, CA; dilution 1:500), and rabbit polyclonal antibodies to VEGFR-3 (Abnova, Taipei, Taiwan; # PAB11653; dilution 1:70), ionized calcium- binding adaptor molecule 1 (Iba1; Wako Pure Chemical Industries Ltd., Tokyo, Japan; dilution 1:500), laminin (Sigma; dilution 1:100), and neuron-specific enolase (NSE; Millipore Corporation; dilution 1:500). Antibody staining was visualized with Cy3-conjugated streptavidin (Jackson ImmunoResearch; dilution 1:1500), Alexa Fluor 488 goat anti-mouse (Molecular Probes, Eugene, OR; dilution 1:300), Alexa Fluor 488 goat anti-rabbit (Molecular Probes; dilution 1:300), Alexa Fluor 647 goat anti-mouse (Molecular Probes; dilution 1:300), and Alexa Fluor 647 goat anti- rabbit (Molecular Probes; dilution 1:300). The specificity of immunoreactivity was confirmed by the absence of an immunohistochemical reaction in sections from which primary or secondary antibodies were omitted. Counterstaining of cell nuclei was carried out by incubating the sections with DAPI (4′,6-diamidino-2′-phenyindole; Roche; dilution 1:1000) for 10 min. Slides were viewed with a confocal microscope (LSM 700; Carl Zeiss Co., Ltd., Jena, Germany) equipped with four lasers (Diode 405, Argon 488, HeNe 543, HeNe 633). Images were converted to TIFF format and contrast levels adjusted using Adobe Photoshop version 7.0.
Immunoblot Analysis
Control rats and EAE rats at peak and recovery stages were sacrificed as described above. White matter from the lumbar spinal cord was homogenized in ice-cold RIPA buffer (50 mM Tris buffer, pH 8.0; 150 mM NaCl; 1% NP-40; 0.5% deoxycholate; and 0.1% SDS). Equal amounts (20 µg) of total protein from the spinal cord were separated on 7% or 10% SDS–polyacrylamide gels and blotted onto nitrocellulose membranes. Immunostaining of the blots was performed using rabbit polyclonal antibody to VEGFR-3 (Abnova; dilution 1:1000). Antibody staining was visualized with peroxidase-conjugated donkey anti rabbit antibody (Jackson ImmunoResearch; dilution 1:2000). Bands were developed with an enhanced chemiluminescence system (ECL kit; Amersham, Arlington Heights, IL).
Immunohistochemistry and Double Labeling
Free-floating sections (25 µm thick) were processed for immunohistochemistry. After blocking with 10% normal goat serum for 1 hr, sections were incubated at 4C overnight with primary antibodies as follows: rabbit polyclonal antibodies to VEGFR-3 (Abnova; dilution 1:70), amyloid precursor protein (APP; Sigma; dilution 1:1000), and degraded myelin basic protein (dMBP; Chemicon International, Inc., Temecula, CA; dilution 1:2000). Primary antibody binding was visualized using peroxidase-labeled goat anti-rabbit IgG (Millipore Corporation; dilution 1:200) and 0.05% 3,3′-diaminobenzidine tetrahydrochloride (DAB) and 0.01% H2O2 as the substrate. Five cryostat sections per each animal were used for each antibody. Reproducible changes in both the staining intensity and the distribution of these immunoreactivities were observed during the course of EAE. The specificity of these antibodies was confirmed by the absence of an immunohistochemical reaction in sections from which the primary or secondary antibodies were omitted or in which the primary antibody was substituted with nonspecific rabbit IgG.
For double-immunofluorescence histochemistry, sections were incubated at 4C overnight with a mix of rabbit polyclonal anti-VEGFR-3 antibody and one of the following monoclonal antibodies: mouse anti-rat endothelial cell antigen-1 (RECA-1; Serotec; dilution 1:200), mouse anti-rat CD45 (Serotec; dilution 1:100), or rat anti-MECA-32 (BD Pharmingen, La Jolla, CA; dilution 1:20). Antibody staining was visualized with Cy3-conjugated anti-rabbit antibody (Jackson ImmunoResearch; dilution 1:1500) and Alexa Fluor 488 goat anti-mouse antibody (Molecular Probes; dilution 1:300). In controls, the primary antibody was omitted from the incubation solution. Counterstaining of cell nuclei was carried out by incubating the sections with DAPI for 10 min. Control sections were prepared as described above.
To detect blood-brain barrier (BBB) leakage of MBP-injected rats, we used the method of Morita and Miyata (2012) with minor modifications. CFA control and EAE rats at onset and peak stages were deeply anesthetized and perfused with the mixture of 1% Evans Blue (Sigma) and 4% PFA in 0.1 M PB for 20 min, followed by perfusion with 4% PFA in 0.1 M PB for 40 min. Double labeling with VEGFR-3 and Evans Blue staining was achieved by processing the sections for VEGFR-3 immunohistochemistry as described above but using Alexa Fluor 488 goat anti-rabbit (Molecular Probes; dilution 1:300) as a secondary antibody. The sections were then cover-slipped with mounting medium and observation was performed using a confocal microscope (LSM 700). Counterstaining of cell nuclei was carried out with DAPI for 10 min. Control sections were prepared as described above.
Results
As previously reported (Gao et al. 2010), most MBP-immunized Lewis rats showed initial neurological signs of EAE on days 7 to 10 after immunization, followed by a peak of clinical symptoms on days 13 to 15. Animals were fully recovered by days 18 to 19 after immunization. Control rats showed no evidence of cellular infiltration and a paucity of myelin, indicative of demyelination in the spinal cord (Suppl. Fig. S1A, B, E, G) as determined by routine pathology with luxol fast blue and immunostaining for APP, a marker of axonal damage (Gentleman et al. 1993), or dMBP, a marker for damaged myelin (Matsuo et al. 1997; Moxon-Emre and Schlichter 2010). However, EAE rats showed considerable cellular infiltrates in the meninges, in the perivascular space, and throughout the white matter in which punctate APP- and dMBP-immunoreactive profiles, indicating damaged axons and myelin, had accumulated inside myelinated bundles (Suppl. Fig. S1C,D,F,H). These general pathological features are consistent overall with data reported previously (Lassmann and Wisniewski 1979; Itoyama and Webster 1982).
Quantitative Real-Time RT-PCR for VEGF-C and VEGFR-3 during the Course of EAE
Real-time RT-PCR revealed that mRNA expression of the VEGF-C gene was significantly increased in the lumbar spinal cord white matter of EAE rats compared with those of CFA controls. The levels increased to maximum values during the peak stage of EAE and then declined, although the enhanced expression appeared maintained until the recovery stage (Fig. 1A). The expression of VEGFR-3 mRNA in the white matter tissue obtained from control and EAE rats showed a temporal pattern similar to that of VEGF-C (Fig. 1B).
Figure 1.
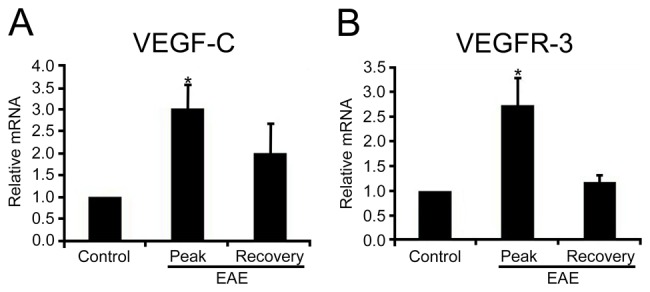
Quantitative real-time reverse transcriptase–polymerase chain reaction for (A) vascular endothelial growth factor–C (VEGF-C) and (B) vascular endothelial growth factor receptor–3 (VEGFR-3) in the lumbar spinal cord white matter of complete Freund’s adjuvant (CFA) controls and experimental autoimmune encephalomyelitis (EAE)–affected rats. The expression level of VEGF-C and VEGFR-3 was calculated using the comparative threshold cycle method (2−ΔΔCt) with GAPDH as the control gene. The expression level of VEGF-C and VEGFR-3 in the CFA control was set to 1. Both VEGF-C and VEGFR-3 expression increased and reached a peak during the peak stage of EAE and then declined at the recovery stage. *p<0.05 compared with CFA controls.
Spatiotemporal Relationships of VEGFR-3 mRNA and Protein during the Course of EAE
The distribution and cellular localization of VEGFR-3 mRNA during the course of EAE were examined in the lumbar spinal cord of Lewis rats using in situ hybridization histochemistry. In normal (Fig. 2A, E) and CFA controls (Suppl. Fig. S2A), VEGFR-3 mRNA was found in abundance in gray matter neurons and at a weaker intensity in some scattered cells in the white matter. Specificity was verified by the lack of signals when hybridization was carried out in the presence of an excess of a sense-stranded probe, even at a concentration three times that of the antisense probe (data not shown). During the course of EAE, VEGFR-3 mRNA hybridization was upregulated in gray and white matter by the onset stage and became more evident at the peak stage. In the onset stage, prominent enrichment was seen in patchy accumulations of cells preferentially in the white matter (Fig. 2B). As shown at higher magnifications, these cells in the white matter seemed to be glial cells (Fig. 2F). In the peak stage, intense hybridization signals were found in small cells with glial morphology throughout the gray and white matter (Fig. 2C, G). During the recovery phase, the labeling intensity of VEGFR-3 mRNA, although still more evident than in control spinal cords, was considerably reduced from that at the peak (Fig. 2D, H). Over the chronic interval of 10 weeks after immunization, VEGFR-3 expression declined toward control levels (Suppl. Fig. S2C, D).
Figure 2.

Spatial and temporal expression of vascular endothelial growth factor receptor–3 (VEGFR-3) mRNA (A–H) and protein (I–N) in the lumbar spinal cords of control and experimental autoimmune encephalomyelitis (EAE)–affected rats. (A, E) In control sections, hybridization signals for VEGFR-3 were observed in gray matter neurons and some scattered cells in the white matter. Upregulation of VEGFR-3 mRNA expression was induced in the spinal cords of EAE-affected rats from the onset stage (B, F) and significantly increased at the peak stage (C, G). (D, H) During the recovery stage, expression of VEGFR-3 mRNA was decreased but still more evident than that in the controls. (E–H) Higher magnification views of the boxed areas in A–D, respectively. Changes in VEGFR-3 immunoreactivity in the spinal cord of controls (I, M) and EAE rats at onset (J), peak (K, N), and recovery stages (L) closely matched those of VEGFR-3 mRNA expression. (M, N) Higher magnification views of the boxed areas in I and K, respectively. (O) Representative results of immunoblot analysis of VEGFR-3 expression showing that VEGFR-3, detected at 125 kDa and 170 kDa, in rat spinal cord seemed to increase at the peak stage of EAE and then declined, but remained at a high level, at the recovery stage compared with normal controls. Scale bars = 500 µm for A–D, I–L; 200 µm for E–H, M, N.
The spatiotemporal distribution pattern of VEGFR-3 immunoreactivity closely matched that of VEGFR-3 mRNA. Immunoreactivity for VEGFR-3 in the control spinal cord could be seen in gray matter neurons and in some glia-like cells in the white matter (Fig. 2I, M and Suppl. Fig. S2B). At the onset stage (Fig. 2J), VEGFR-3 immunoreactivity increased in the white matter and, at the peak stage (Fig. 2K, N), immunoreactivity had preferentially increased throughout the gray and white matter. VEGFR-3 immunoreactivity then declined during the recovery stage of EAE but was still intense compared with controls (Fig. 2L). The distribution pattern and morphology of these immunoreactive cells suggested that they are glial cells.
These results of the immunohistochemical analysis were further corroborated by immunoblot analysis of VEGFR-3 expression (Fig. 2O). Immunoblotting revealed two major bands, about 125 kDa and 170 kDa, in protein extracts from the lumbar spinal cord white matter of control and EAE rats, which is in agreement with observations in previous studies (Lee et al. 1996; Petrova et al. 2008). The intensity of the VEGFR-3 protein seemed to increase during the peak stage of EAE and then declined but remained at a high level during the recovery stage as compared with normal controls.
To define the precise localization and relationship of VEGFR-3 gene transcripts and protein in the spinal cord with EAE, double labeling using in situ hybridization and immunohistochemistry was performed. In the controls, cells positive for VEGFR-3 mRNA and protein were mainly gray matter neurons and some glia-like cells in the white matter (data not shown). In the onset stage of EAE, rounded cells that appeared to be brain macrophages adjacent to vascular profiles expressed both VEGFR-3 mRNA and protein (Fig. 3A–D). In addition, the distribution of VEGFR-3 mRNA overlapped well with its protein expression in gray and white matter of the spinal cord during the peak stage of EAE (Fig. 3I–L). Double labeling using VEGFR-3 and RECA-1, a marker for blood vessels, revealed that VEGFR-3 expression was closely associated with the vascular wall in EAE-affected rats (Fig. 3E–H,M–P). Thus, VEGFR-3 mRNA and protein generally co-localized on the same populations of neurons and glia-like cells located in the gray and white matter in the spinal cord of control and EAE-affected rats, and VEGFR-3 expression was often associated with blood vessels.
Figure 3.

Spatiotemporal relationship of vascular endothelial growth factor receptor–3 (VEGFR-3) mRNA and protein in the lumbar spinal cords of experimental autoimmune encephalomyelitis (EAE)–affected rats at onset (A–H) and peak stages (I–P). Note that VEGFR-3 mRNA and protein are generally co-localized in the same population of neurons and glia-like cells located in the gray and white matter in the spinal cords of EAE rats. Also note that rounded cells resembling brain macrophages adjacent to rat endothelial cell antigen–1 (RECA-1)–positive blood vessels expressed both VEGFR-3 mRNA and protein in EAE rats. (B–D, F–H, J–L, N–P) Higher magnification views of the boxed areas in A, E, I, and M, respectively. Scale bars = 200 µm for A, I; 100 µm for E, M; 50 µm for B–D, F–H, J–L, N–P.
The Phenotype of VEGFR-3–Expressing Cells during the Course of EAE
To clarify the phenotype of cells expressing VEGFR-3 in control and EAE-affected rats, double or triple labeling using cell type–specific markers was performed. As described above, VEGFR-3 mRNA was observed in the gray and white matter of control spinal cords, where GFAP-positive astrocytes showed thin astroglial processes and Iba1-positive microglia exhibited a ramified morphology (Fig. 4A–C). As shown at higher magnifications of control spinal cords (Fig. 4D–I), constitutive expression of VEGFR-3 mRNA was observed in a subset of astrocytes in the white matter, especially near the surface of the spinal cord, but not in gray matter astrocytes. By contrast, Iba1-positive microglia located in both gray and white matter were devoid of specific VEGFR-3 expression (Fig. 4D–I). Instead, intense VEGFR-3 expression was observed in gray matter neurons showing the neuronal cell marker NSE (Fig. 4J–L).
Figure 4.

Identification of phenotypes of vascular endothelial growth factor receptor–3 (VEGFR-3)–expressing cells in the lumbar spinal cords of normal controls. (A–C) Low-magnification views of control spinal cord showing triple labeling for VEGFR-3, glial fibrillary acidic protein (GFAP), and ionized calcium-binding adaptor molecule 1 (Iba1). (D–F) Higher magnification views of the white matter of control spinal cord. Note that a subset of astrocytes in the white matter (arrows), especially near the surface of the spinal cord, showed VEGFR-3 expression, whereas Iba1-positive microglia with ramified morphology (arrowheads) were devoid of specific VEGFR-3 expression. (G–L) Higher magnification views of the gray matter of control spinal cord. (G–I) Note that neither astrocytes with thin astroglial processes (arrows) nor microglia with ramified morphology (arrowheads) showed VEGFR-3 expression. (J–L) Also note that intense labeling for VEGFR-3 was observed in gray matter neurons showing the neuronal cell marker NSE (neuron-specific enolase). Scale bars = 200 µm for A–C; 50 µm for D–L.
We also defined the phenotype of cells expressing VEGFR-3 in the onset stage of EAE-affected rats. Triple labeling with VEGFR-3, GFAP, and Iba1 indicated that virtually all of the perivascular cells expressing VEGFR-3 in the white matter were indeed Iba1-immunoreactive microglia/macrophages, whereas some astrocytes in close proximity with these perivascular clusters showed VEGFR-3 expression (Fig. 5A–H). We further characterized these microglia/macrophages expressing VEGFR-3 by triple labeling with VEGFR-3, Iba1, and ED1. VEGFR-3 expression was localized in Iba1+/ED1+ cells associated with vessels; these cells had a large, round shape and fewer shorter processes, resembling amoeboid-like brain macrophages, whereas most Iba1+/ED1− cells were devoid of specific labeling for VEGFR-3 (Fig. 5I–P). In addition, triple labeling with VEGFR-3, ED1, and laminin confirmed that ED1-positive macrophages expressing VEGFR-3 were often associated with laminin-positive vascular profiles (Fig. 6A–F). In addition, double labeling using VEGFR-3 and the pan-hematopoietic marker CD45, which is expressed on all nucleated hematopoietic cells or leukocytes (Penninger et al. 2001), revealed that nearly all of CD45-positive cells coexpressed VEGFR-3 (Fig. 6G–J). VEGFR-3/CD45 double-labeled cells were associated with blood vessels or distributed intraparenchymally and appeared to be macrophage-like cells, in agreement with the previous study that blood-derived macrophages expressed CD45 (Guillemin and Brew 2004).
Figure 5.

Identification of phenotypes of vascular endothelial growth factor receptor–3 (VEGFR-3)–expressing cells in the lumbar spinal cords at the onset stage of experimental autoimmune encephalomyelitis (EAE)–affected rats. (A–H) Triple labeling with VEGFR-3, glial fibrillary acidic protein (GFAP), and ionized calcium-binding adaptor molecule 1 (Iba1) showing that virtually all of the perivascular cells expressing VEGFR-3 in the white matter were indeed Iba1-immunoreactive microglia/macrophages. Note that some astrocytes (arrows in E–H) showed VEGFR-3 expression. (E–H) Higher magnification views of the boxed areas in A–D, respectively. (I–P) Triple labeling with VEGFR-3, Iba1, and ED1 showing that VEGFR-3 expression was localized in Iba1/ED1 double-labeled cells. Note that most Iba1+/ED1− cells were devoid of specific labeling for VEGFR-3. (M–P) Higher magnification views of the boxed areas in I–L, respectively. Scale bars = 200 µm for A–D; 50 µm for E–L; 20 µm for M–P.
Figure 6.

Identification of phenotypes of vascular endothelial growth factor receptor–3 (VEGFR-3)–expressing cells in the lumbar spinal cords at the onset stage of experimental autoimmune encephalomyelitis (EAE)–affected rats. (A–F) Triple labeling with VEGFR-3, ED1, and laminin showing that ED1-positve macrophages expressing VEGFR-3 (asterisks in E and F) were often associated with laminin-positive vascular profiles. (E, F) Higher magnification views of the boxed areas in A–C, respectively. (G–J) Double labeling using VEGFR-3 and CD45 showing that nearly all of CD45-positive cells coexpressed VEGFR-3. (H–J) Higher magnification views of the boxed area in G. Scale bars = 100 µm for A–D, G; 50 µm for H–J; 20 µm for E, F.
Finally, we identified the phenotype of cells expressing VEGFR-3 in the spinal cord with the peak stage of EAE rats, where increased immunoreactivity for astrocytes and microglia/macrophages was observed compared with those of control rats (compare Fig. 7A–C with Fig. 4A–C). As shown at higher magnifications of the white matter of these EAE rats (Fig. 7D–F), most of the reactive astrocytes and microglia/macrophages expressed hybridization signals for VEGFR-3. In the gray matter (Fig. 7G–I), however, cells expressing VEGFR-3 strongly were neurons or activated stellate microglial cells with thick and short processes, whereas reactive astrocytes with thick glial processes were devoid of specific labeling for VEGFR-3.
Figure 7.

Identification of phenotypes of vascular endothelial growth factor receptor–3 (VEGFR-3)–expressing cells in the lumbar spinal cords at the peak stage of experimental autoimmune encephalomyelitis (EAE)–affected rats. (A–C) Low-magnification views of EAE-affected spinal cords showing triple labeling for VEGFR-3, glial fibrillary acidic protein (GFAP), and ionized calcium-binding adaptor molecule 1 (Iba1). (D–E) Higher magnification views of the white matter. (F) Higher magnification view of the boxed areas in D and E. Three-dimensional confocal analysis demonstrated that hybridization signals for VEGFR-3 were observed in reactive astrocytes (arrow) and microglia/macrophages (arrowhead). (G–I) Higher magnification views of the boxed areas in the gray matter in A–C, respectively. Note that intense labeling for VEGFR-3 was observed in neurons (asterisks in G) and activated stellate microglial cells with thick and short processes (arrowheads) but not in reactive astrocytes (arrows). Scale bars = 200 µm for A–C; 50 µm for D, E, G–I; 20 µm for F.
To determine whether vessel-associated VEGFR-3 expression could be related to BBB damage that occurred in EAE rats, animals were perfused intracardially with Evans Blue added in a fixative solution. The observation by confocal microscopy of Evans Blue fluorescence revealed that although CFA control rats showed no noticeable extravascular fluorescence (data not shown), EAE rats showed considerable red Evans Blue fluorescence around the vessels in the white matter (Suppl. Fig. S3A–C), indicating that the BBB is impaired and extravascular leakage of fluorescence is possible (Saria and Lundberg 1983; del Valle et al. 2008; Strbian et al. 2008; Morita and Miyata 2012). Double labeling with VEGFR-3 and Evans Blue staining revealed that VEGFR-3 expression appeared associated with vascular profiles fluorescing with Evans Blue in the white matter of EAE rats (Suppl. Fig. S3D–F). To further confirm whether VEGFR-3 expression was associated with blood vessels in which the BBB was disrupted, double labeling using VEGFR-3 and MECA-32, a marker of immature/leaky cerebral vessels (Milner et al. 2008; Li et al. 2012), was performed. As shown in Supplementary Fig. S3G–O, VEGFR-3 expression was localized or in close vicinity to the blood vessels showing MECA-32 immunoreactivity in EAE-affected rats.
Discussion
In the present study, for the first time to our knowledge, we demonstrated the significant increase in the expression of VEGF-C and its tyrosine kinase receptor, VEGFR-3, in the white matter of EAE rats. In addition, we observed a detailed characterization of the time course and cellular localization of VEGFR-3 mRNA and protein expression in the spinal cord during the course of EAE. Constitutive expression of VEGFR-3 mRNA in the spinal cord was observed in gray matter neurons and in a subset of white matter astrocytes, but neither resting ramified microglia nor gray matter astrocytes showed specific labeling for VEGFR-3. In animals afflicted with EAE, upregulation of VEGFR-3 expression was readily detected in the spinal cord, becoming increasingly prominent during disease progression. The spatiotemporal expression of VEGFR-3 mRNA and protein exhibited overlapping patterns in control and EAE-affected spinal cords, except for sustained immunoreactivity during the recovery stage of EAE. Consistent with these findings, immunoblot data showed that upregulation of VEGFR-3 protein in the microdissected white matter was more pronounced during the peak stage of EAE compared with normal controls, and this pattern remained in the recovery stage.
In EAE-affected rats, upregulation of VEGFR-3 expression was induced in association with microglia/macrophages, which showed different characteristics during the course of disease progression. During the inductive stage of EAE, VEGFR-3 was induced in almost all Iba1/ED1 double-labeled cells, which were predominantly localized in the perivascular infiltrated area in the white matter and had the morphology of large, round, ameboid-like macrophages, as observed previously (Bauer et al. 1995). Thus, these characteristics of VEGFR-3–expressing brain macrophages suggest that, although infiltrating blood-borne monocytes or macrophages and reactive microglia are not distinguishable by morphological criteria due to a lack of discriminating specific markers (Guillemin and Brew 2004; Graeber and Streit 2010; Kofler and Wiley 2010), these cells might be infiltrating macrophages. In support of this, VEGFR-3 is expressed in bone marrow–derived cells, including macrophages, monocytes, and dendritic cells (Skobe et al. 2001; Hamrah et al. 2003, 2004; Stepanova et al. 2007), and in infiltrating monocytic macrophages within the ischemic brain (Shin et al. 2008, 2010). In agreement with these previous reports, our observations demonstrated that a population of round VEGFR-3–expressing cells appeared in the vicinity of blood vessels with the BBB impairment, as shown by red Evans Blue fluorescence or MECA-32 immunoreactivity during the inductive and peak stages of EAE. This suggests that VEGFR-3 expression could be induced in line with the BBB leakage in EAE rats. In addition, double labeling using VEGFR-3 and CD45 revealed that nearly all of round and amoeboid CD45-positive cells around blood vessels coexpressed VEGFR-3. Taken together, these results suggest that these VEGFR-3–expressing cells may correspond to blood-borne monocytes and macrophages.
In addition to these blood-borne macrophages, we also found that, mainly during peak EAE disease, VEGFR-3 expression occurred in Iba1-positive stellate-shaped cells with thick and short processes. Considering that the progressive activation of microglial cells is characterized by morphological transformation from ramified resting cells to bush-like cells during early activation, and ultimately to phagocytic cells (Ladeby et al. 2005), it seems that the induction of VEGFR-3 expression occurred in activated resident microglia. Thus, it is probable that, during the inductive phase of EAE, VEGFR-3 expression was upregulated predominantly in infiltrating macrophages; a further increase in expression was also observed in activated microglia with increasing disease severity.
The rapid recruitment of blood-borne macrophages and activation of resident microglia and perivascular macrophages are among the most consistent changes observed in MS and EAE (Brosnan et al. 1981; Matsumoto et al. 1992; Bauer et al. 1995; Raivich and Banati 2004; Centonze et al. 2010; Slavin et al. 2010). These cells can present antigens; secrete a wide range of molecules such as cytokines, chemokines, and reactive oxygen radicals; and may be involved in the initiation of the immune response (Benveniste 1997; Raivich and Banati 2004; Almolda et al. 2009, 2011). In addition, a recent study by Ajami et al. (2011) suggested that although T cells and endogenous microglia are responsible for disease initiation, infiltrating monocytes trigger EAE progression to the severe form of the disease. In this regard, several lines of evidence raise the possibility that VEGF-C may be involved in the pathogenesis of EAE. VEGF-C affects macrophage migration via VEGFR-3 (Skobe et al. 2001; Stepanova et al. 2007), and VEGFR-3 modulates adaptive immunity by mediating chemotaxis of antigen-presenting cells (Chen et al. 2007). In addition, specific functional roles of VEGF in the pathogenesis of EAE have been proposed, although most reports have concentrated on VEGF-A. VEGF-A exacerbates inflammatory demyelinating lesions by inducing focal blood-brain barrier breakdown and monocyte recruitment in EAE rats (Proescholdt et al. 2002; Zhu et al. 2008; Sasaki et al. 2010). Collectively, these results, including our new findings, suggest that, in addition to VEGF-A, the VEGF-C/VEGFR-3 system may mediate the recruitment of monocytic macrophages to the inflamed spinal cord, contributing to inflammation during EAE, and that this system may be involved in the activation and migration of intrinsic microglia during disease progression. Thus, further studies using macrophage-specific VEGFR-3 null or overexpressing mice could help to further elucidate the role of VEGFR-3 in microglia/macrophages in EAE.
Induction of VEGFR-3 mRNA and protein was also noted on reactive astrocytes in the white matter during the inductive stage of EAE, and this expression progressively increased during the peak stage. This is in accordance with our previous report that VEGFR-3 induction in reactive astrocytes is a generalized phenomenon following ischemic injury (Shin et al. 2008, 2010). There are several studies demonstrating the role of astrocytes in forming scar-like barriers that serve to restrict leukocytes to perivascular clusters and limit the infiltration of leukocytes into adjacent CNS parenchyma (Liedtke et al. 1998; Voskuhl et al. 2009; Toft-Hansen et al. 2011). In this sense, our observation that reactive astrocytes surrounding perivascular clusters of brain macrophages showed VEGFR-3 expression suggests that VEGFR-3 may be involved in the astroglial reactions associated with critical barriers to leukocytes during EAE. However, gray matter astrocytes of EAE-affected spinal cords were devoid of specific labeling for VEGFR-3, despite their increased GFAP immunoreactivity. Although inflammatory lesions preferentially occur in the spinal cord white matter in EAE, the activation of astrocytes also occurs in the gray matter of EAE animals in the absence of any detectable motoneuron loss and distinct inflammatory infiltrates (Aquino et al. 1988; Matsumoto et al. 1992; Wu et al. 2008). It is unclear why VEGFR-3 is upregulated in reactive astrocytes in the white matter of EAE-affected spinal cords and not in gray matter astrocytes. Therefore, further investigation is needed to determine whether the apparent absence of VEGFR-3 expression in gray matter astrocytes is related to the absence of lymphocyte infiltrates in the gray matter (Bø et al. 2003) and also whether upregulation of VEGFR-3 in white matter astrocytes reflects a function in the astroglial reaction during EAE.
Our data show that the expression of VEGFR-3 progressively increased at transcriptional and protein levels in EAE-affected spinal cords, in close relationship with disease progression. In summary, our study revealed the following: 1) induction of VEGFR-3 occurred predominantly in perivascular infiltrated macrophages in the spinal cord white matter during the inductive phase of EAE; 2) VEGFR-3 expression was also induced in activated microglial cells in the gray and white matter, mainly in the peak phase; and 3) reactive astrocytes in the white matter, but not in the gray matter, expressed VEGFR-3. These data suggest that VEGFR-3 may be involved in the recruitment of monocytic macrophages and in glial reactions in the course of EAE.
Supplementary Material
Footnotes
Declaration of Conflicting Interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was partially supported by the Korea Healthcare technology R&D Project, Ministry for Health, Welfare & Family Affairs, Republic of Korea (A092258), and Mid-career Researcher Program through NRF grant funded by the MEST (2011–0028319).
Supplementary material for this article is available on the Journal of Histochemistry & Cytochemistry Web site at http://jhc.sagepub.com/supplemental.
References
- Ajami B, Bennett JL, Krieger C, McNagny KM, Rossi FM. 2011. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat Neurosci. 14:1142–1149 [DOI] [PubMed] [Google Scholar]
- Almolda B, Costa M, Montoya M, González B, Castellano BJ. 2009. CD4 microglial expression correlates with spontaneous clinical improvement in the acute Lewis rat EAE model. J Neuroimmunol. 209:65–80 [DOI] [PubMed] [Google Scholar]
- Almolda B, Gonzalez B, Castellano B. 2011. Antigen presentation in EAE: role of microglia, macrophages and dendritic cells. Front Biosci. 16:1157–1171 [DOI] [PubMed] [Google Scholar]
- Aquino DA, Chiu FC, Brosnan CF, Norton WT. 1988. Glial fibrillary acidic protein increases in the spinal cord of Lewis rats with acute experimental autoimmune encephalomyelitis. J Neurochem. 51:1085–1096 [DOI] [PubMed] [Google Scholar]
- Bauer J, Huitinga I, Zhao W, Lassmann H, Hickey WF, Dijkstra CD. 1995. The role of macrophages, perivascular cells, and microglial cells in the pathogenesis of experimental autoimmune encephalomyelitis. Glia. 15:437–446 [DOI] [PubMed] [Google Scholar]
- Benveniste EN. 1997. Role of macrophages/microglia in multiple sclerosis and experimental allergic encephalomyelitis. J Mol Med. 75:165–173 [DOI] [PubMed] [Google Scholar]
- Bø L, Vedeler CA, Nyland H, Trapp BD, Mørk SJ. 2003. Intracortical multiple sclerosis lesions are not associated with increased lymphocyte infiltration. Mult Scler. 9:323–331 [DOI] [PubMed] [Google Scholar]
- Brosnan CF, Bornstein MB, Bloom BR. 1981. The effects of macrophage depletion on the clinical and pathologic expression of experimental allergic encephalomyelitis. J Immunol. 126:614–620 [PubMed] [Google Scholar]
- Calvo CF, Fontaine RH, Soueid J, Tammela T, Makinen T, Alfaro-Cervello C, Bonnaud F, Miguez A, Benhaim L, Xu Y, et al. 2011. Vascular endothelial growth factor receptor 3 directly regulates murine neurogenesis. Genes Dev. 25:831–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassan C, Liblau RS. 2007. Immune tolerance and control of CNS autoimmunity: from animal models to MS patients. J Neurochem. 100:883–892 [DOI] [PubMed] [Google Scholar]
- Centonze D, Muzio L, Rossi S, Furlan R, Bernardi G, Martino G. 2010. The link between inflammation, synaptic transmission and neurodegeneration in multiple sclerosis. Cell Death Differ. 17:1083–1091 [DOI] [PubMed] [Google Scholar]
- Chastain EM, Duncan DS, Rodgers JM, Miller SD. 2011. The role of antigen presenting cells in multiple sclerosis. Biochim Biophys Acta. 1812:265–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Hamrah P, Cursiefen C, Zhang Q, Pytowski B, Streilein JW, Dana MR. 2007. Vascular endothelial growth factor receptor–3 mediates induction of corneal alloimmunity. Ocul Immunol Inflamm. 15:275–278 [DOI] [PubMed] [Google Scholar]
- Compston A, Coles A. 2008. Multiple sclerosis. Lancet. 372:1502–1517 [DOI] [PubMed] [Google Scholar]
- del Valle J, Camins A, Pallàs M, Vilaplana J, Pelegrí C. 2008. A new method for determining blood-brain barrier integrity based on intracardiac perfusion of an Evans Blue–Hoechst cocktail. J Neurosci Methods. 174:42–49 [DOI] [PubMed] [Google Scholar]
- Frohman EM, Racke MK, Raine CS. 2006. Multiple sclerosis—the plaque and its pathogenesis. N Engl J Med. 354:942–955 [DOI] [PubMed] [Google Scholar]
- Gandhi R, Laroni A, Weiner HL. 2010. Role of the innate immune system in the pathogenesis of multiple sclerosis. J Neuroimmunol. 221:7–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H, Gao Y, Li X, Shen A, Yan M. 2010. Spatiotemporal patterns of dexamethasone-induced Ras protein 1 expression in the central nervous system of rats with experimental autoimmune encephalomyelitis. J Mol Neurosci. 41:198–209 [DOI] [PubMed] [Google Scholar]
- Gentleman SM, Nash MJ, Sweeting CJ, Graham DI, Roberts GW. 1993. Beta-amyloid precursor protein (beta APP) as a marker for axonal injury after head injury. Neurosci Lett. 160:139–144 [DOI] [PubMed] [Google Scholar]
- Graeber MB, Streit WJ. 2010. Microglia: biology and pathology. Acta Neuropathol. 119:89–105 [DOI] [PubMed] [Google Scholar]
- Guillemin GJ, Brew BJ. 2004. Microglia, macrophages, perivascular macrophages, and pericytes: a review of function and identification. J Leukoc Biol. 75:388–397 [DOI] [PubMed] [Google Scholar]
- Hamrah P, Chen L, Cursiefen C, Zhang Q, Joyce NC, Dana MR. 2004. Expression of vascular endothelial growth factor receptor–3 (VEGFR-3) on monocytic bone marrow–derived cells in the conjunctiva. Exp Eye Res. 79:553–561 [DOI] [PubMed] [Google Scholar]
- Hamrah P, Chen L, Zhang Q, Dana MR. 2003. Novel expression of vascular endothelial growth factor receptor (VEGFR)–3 and VEGF-C on corneal dendritic cells. Am J Pathol. 163:57–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heppner FL, Greter M, Marino D, Falsig J, Raivich G, Hövelmeyer N, Waisman A, Rülicke T, Prinz M, Priller J, et al. 2005. Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat Med. 11:146–152 [DOI] [PubMed] [Google Scholar]
- Huitinga I, van Rooijen N, de Groot CJ, Uitdehaag BM, Dijkstra CD. 1990. Suppression of experimental allergic encephalomyelitis in Lewis rats after elimination of macrophages. J Exp Med. 172:1025–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoyama Y, Webster HD. 1982. Immunocytochemical study of myelin-associated glycoprotein (MAG) and basic protein (BP) in acute experimental allergic encephalomyelitis (EAE). J Neuroimmunol. 3:351–364 [DOI] [PubMed] [Google Scholar]
- Kirk SL, Karlik SJ. 2003. VEGF and vascular changes in chronic neuroinflammation. J Autoimmun. 21:353–363 [DOI] [PubMed] [Google Scholar]
- Kofler J, Wiley CA. 2010. Microglia: key innate immune cells of the brain. Toxicol Pathol. 39:103–114 [DOI] [PubMed] [Google Scholar]
- Ladeby R, Wirenfeldt M, Garcia-Ovejero D, Fenger C, Dissing-Olesen L, Dalmau I, Finsen B. 2005. Microglial cell population dynamics in the injured adult central nervous system. Brain Res Brain Res Rev. 48:196–206 [DOI] [PubMed] [Google Scholar]
- Lassmann H, Wisniewski HM. 1979. Chronic relapsing experimental allergic encephalomyelitis: morphological sequence of myelin degradation. Brain Res. 169:357–368 [DOI] [PubMed] [Google Scholar]
- Le Bras B, Barallobre MJ, Homman-Ludiye J, Ny A, Wyns S, Tammela T, Haiko P, Karkkainen MJ, Yuan L, Muriel MP, et al. 2006. VEGF-C is a trophic factor for neural progenitors in the vertebrate embryonic brain. Nat Neurosci. 9:340–348 [DOI] [PubMed] [Google Scholar]
- Lee J, Gray A, Yuan J, Luoh SM, Avraham H, Wood WI. 1996. Vascular endothelial growth factor-related protein: a ligand and specific activator of the tyrosine kinase receptor Flt4. Proc Natl Acad Sci U S A. 93:1988–1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Liu F, Welser-Alves JV, McCullough LD, Milner R. 2012. Upregulation of fibronectin and the α5β1 and αvβ3 integrins on blood vessels within the cerebral ischemic penumbra. Exp Neurol. 233:283–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedtke W, Edelmann W, Chiu FC, Kucherlapati R, Raine CS. 1998. Experimental autoimmune encephalomyelitis in mice lacking glial fibrillary acidic protein is characterized by a more severe clinical course and an infiltrative central nervous system lesion. Am J Pathol. 152:251–259 [PMC free article] [PubMed] [Google Scholar]
- Matsumoto Y, Ohmori K, Fujiwara M. 1992. Microglial and astroglial reactions to inflammatory lesions of experimental autoimmune encephalomyelitis in the rat central nervous system. J Neuroimmunol. 37:23–33 [DOI] [PubMed] [Google Scholar]
- Matsuo A, Lee GC, Terai K, Takami K, Hickey WF, McGeer EG, McGeer PL. 1997. Unmasking of an unusual myelin basic protein epitope during the process of myelin degeneration in humans: a potential mechanism for the generation of autoantigens. Am J Pathol. 150:1253–1266 [PMC free article] [PubMed] [Google Scholar]
- Milner R, Hung S, Erokwu B, Dore-Duffy P, LaManna JC, del Zoppo GJ. 2008. Increased expression of fibronectin and the alpha 5 beta 1 integrin in angiogenic cerebral blood vessels of mice subject to hypobaric hypoxia. Mol Cell Neurosci. 38:43–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore GR. 2010. Current concepts in the neuropathology and pathogenesis of multiple sclerosis. Can J Neurol Sci. 37(Suppl 2):S5–15 [DOI] [PubMed] [Google Scholar]
- Morita S, Miyata S. 2012. Different vascular permeability between the sensory and secretory circumventricular organs of adult mouse brain. Cell Tissue Res. 349:589–603 [DOI] [PubMed] [Google Scholar]
- Moxon-Emre I, Schlichter LC. 2010. Evolution of inflammation and white matter injury in a model of transient focal ischemia. J Neuropathol Exp Neurol. 69:1–15 [DOI] [PubMed] [Google Scholar]
- Penninger JM, Irie-Sasaki J, Sasaki T, Oliveira-dos-Santos AJ. 2001. CD45: new jobs for an old acquaintance. Nat Immunol. 2:389–396 [DOI] [PubMed] [Google Scholar]
- Petrova TV, Bono P, Holnthoner W, Chesnes J, Pytowski B, Sihto H, Laakkonen P, Heikkilä P, Joensuu H, Alitalo K. 2008. VEGFR-3 expression is restricted to blood and lymphatic vessels in solid tumors. Cancer Cell. 13:554–556 [DOI] [PubMed] [Google Scholar]
- Polfliet MM, van de, Veerdonk F, Döpp EA, van Kesteren-Hendrikx EM, van Rooijen N, Dijkstra CD, van den Berg TK. 2002. The role of perivascular and meningeal macrophages in experimental allergic encephalomyelitis. J Neuroimmunol. 122:1–8 [DOI] [PubMed] [Google Scholar]
- Proescholdt MA, Jacobson S, Tresser N, Oldfield EH, Merrill MJ. 2002. Vascular endothelial growth factor is expressed in multiple sclerosis plaques and can induce inflammatory lesions in experimental allergic encephalomyelitis rats. J Neuropathol Exp Neurol. 61:914–925 [DOI] [PubMed] [Google Scholar]
- Raivich G, Banati R. 2004. Brain microglia and blood-derived macrophages: molecular profiles and functional roles in multiple sclerosis and animal models of autoimmune demyelinating disease. Brain Res Brain Res Rev. 46:261–281 [DOI] [PubMed] [Google Scholar]
- Saria A, Lundberg JM. 1983. Evans blue fluorescence: quantitative and morphological evaluation of vascular permeability in animal tissues. J Neurosci Methods. 8:41–49 [DOI] [PubMed] [Google Scholar]
- Sasaki M, Lankford KL, Brown RJ, Ruddle NH, Kocsis JD. 2010. Focal experimental autoimmune encephalomyelitis in the Lewis rat induced by immunization with myelin oligodendrocyte glycoprotein and intraspinal injection of vascular endothelial growth factor. Glia. 58:1523–1531 [DOI] [PubMed] [Google Scholar]
- Seabrook TJ, Littlewood-Evans A, Brinkmann V, Pöllinger B, Schnell C, Hiestand PC. 2010. Angiogenesis is present in experimental autoimmune encephalomyelitis and pro-angiogenic factors are increased in multiple sclerosis lesions. J Neuroinflammation. 22:7–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin YJ, Choi JS, Choi JY, Hou Y, Cha JH, Chun MH, Lee MY. 2010. Induction of vascular endothelial growth factor receptor-3 mRNA in glial cells following focal cerebral ischemia in rats. J Neuroimmunol. 229:81–90 [DOI] [PubMed] [Google Scholar]
- Shin YJ, Choi JS, Lee JY, Choi JY, Cha JH, Chun MH, Lee MY. 2008. Differential regulation of vascular endothelial growth factor–C and its receptor in the rat hippocampus following transient forebrain ischemia. Acta Neuropathol. 116:517–527 [DOI] [PubMed] [Google Scholar]
- Skobe M, Hamberg LM, Hawighorst T, Schirner M, Wolf GL, Alitalo K, Detmar M. 2001. Concurrent induction of lymphangiogenesis, angiogenesis, and macrophage recruitment by vascular endothelial growth factor–C in melanoma. Am J Pathol. 159:893–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavin A, Kelly-Modis L, Labadia M, Ryan K, Brown ML. 2010. Pathogenic mechanisms and experimental models of multiple sclerosis. Autoimmunity. 43:504–513 [DOI] [PubMed] [Google Scholar]
- Stepanova OI, Krylov AV, Lioudyno VI, Kisseleva EP. 2007. Gene expression for VEGF-A, VEGF-C, and their receptors in murine lymphocytes and macrophages. Biochemistry. 72:1194–1198 [DOI] [PubMed] [Google Scholar]
- Strbian D, Durukan A, Pitkonen M, Marinkovic I, Tatlisumak E, Pedrono E, Abo-Ramadan U, Tatlisumak T. 2008. The blood-brain barrier is continuously open for several weeks following transient focal cerebral ischemia. Neuroscience. 153:175–181 [DOI] [PubMed] [Google Scholar]
- Su JJ, Osoegawa M, Matsuoka T, Minohara M, Tanaka M, Ishizu T, Mihara F, Taniwaki T, Kira J. 2006. Upregulation of vascular growth factors in multiple sclerosis: correlation with MRI findings. J Neurol Sci. 243:21–30 [DOI] [PubMed] [Google Scholar]
- Tham E, Gielen AW, Khademi M, Martin C, Piehl F. 2006. Decreased expression of VEGF-A in rat experimental autoimmune encephalomyelitis and in cerebrospinal fluid mononuclear cells from patients with multiple sclerosis. Scand J Immunol. 64:609–622 [DOI] [PubMed] [Google Scholar]
- Toft-Hansen H, Füchtbauer L, Owens T. 2011. Inhibition of reactive astrocytosis in established experimental autoimmune encephalomyelitis favors infiltration by myeloid cells over T cells and enhances severity of disease. Glia. 59:166–176 [DOI] [PubMed] [Google Scholar]
- Voskuhl RR, Peterson RS, Song B, Ao Y, Morales LB, Tiwari-Woodruff S, Sofroniew MV. 2009. Reactive astrocytes form scar-like perivascular barriers to leukocytes during adaptive immune inflammation of the CNS. J Neurosci. 29:11511–11522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Ohlsson M, Warner EA, Loo KK, Hoang TX, Voskuhl RR, Havton LA. 2008. Glial reactions and degeneration of myelinated processes in spinal cord gray matter in chronic experimental autoimmune encephalomyelitis. Neuroscience. 156:586–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu CS, Hu XQ, Xiong ZJ, Lu ZQ, Zhou GY, Wang DJ. 2008. Adenoviral delivery of soluble VEGF receptor 1 (sFlt-1) inhibits experimental autoimmune encephalomyelitis in dark Agouti (DA) rats. Life Sci. 83:404–412 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.