

Abstract
Background
Drug–drug interactions (DDIs) are responsible for many serious adverse events; their detection is crucial for patient safety but is very challenging. Currently, the US Food and Drug Administration and pharmaceutical companies are showing great interest in the development of improved tools for identifying DDIs.
Methods
We present a new methodology applicable on a large scale that identifies novel DDIs based on molecular structural similarity to drugs involved in established DDIs. The underlying assumption is that if drug A and drug B interact to produce a specific biological effect, then drugs similar to drug A (or drug B) are likely to interact with drug B (or drug A) to produce the same effect. DrugBank was used as a resource for collecting 9454 established DDIs. The structural similarity of all pairs of drugs in DrugBank was computed to identify DDI candidates.
Results
The methodology was evaluated using as a gold standard the interactions retrieved from the initial DrugBank database. Results demonstrated an overall sensitivity of 0.68, specificity of 0.96, and precision of 0.26. Additionally, the methodology was also evaluated in an independent test using the Micromedex/Drugdex database.
Conclusion
The proposed methodology is simple, efficient, allows the investigation of large numbers of drugs, and helps highlight the etiology of DDI. A database of 58 403 predicted DDIs with structural evidence is provided as an open resource for investigators seeking to analyze DDIs.
Keywords: Drug-drug interaction, adverse drug event, structure similarity, molecular fingerprints, QSAR, molecular modeling, drug design, automated learning, statistical analysis of large datasets, discovery, and text and data mining methods
Introduction
Adverse drug events are a serious problem worldwide. In the US, they result in many injuries and deaths each year,1–3 costing millions of dollars per hospital annually and billions overall,4 5 and lead to increased hospital care.6–8 Drug–drug interactions (DDIs) are an important patient safety problem and have been reported to cause up to 30% of patient adverse events9 10 resulting in warning notices for or the withdrawal of many drugs from the market. The safety and efficacy profile of a drug can be altered significantly by the co-administration of other drugs. Multiple drug combinations in therapy are common11 and increase the risk of adverse events since concomitant drugs can share pharmacological or metabolic pathways. In extreme cases, some drugs have caused death due to the heightened adverse effect of the drug affected by the interaction. For example, cerivastatin, a drug withdrawn from the US market, caused 31 cases of fatal rhabdomyolysis prior to June 2001; the combination cerivastatin–gemfibrozil was implicated in 12 of the 31 deaths.12 Gemfibrozil causes increased blood levels of the statin resulting in a higher risk of myopathy and rhabdomyolysis.
The development of tools to predict DDIs is important in the drug development process and in postmarketing surveillance in order to detect new drug combinations that should be contraindicated. Indeed, there is currently strong interest among regulatory authorities, such as the US Food and Drug Administration,13 and pharmaceutical companies in developing better tools for assessing drug interactions.14
The concept that similar molecules result in similar biological properties has been employed over the years by medicinal chemists.15–18 Methodologies such as QSAR/QSPR (Quantitative Structure-Activity Relationship or Quantitative Structure-Property Relationship), frequently used in computer aided drug design, are very helpful to establish relationships between the structures of molecules and their corresponding biological activity or other biological properties.16 19 Molecular fingerprint-based modeling has also been applied successfully to the identification of molecules structurally similar to those with a selected property.20 21 The same idea can be expanded to explain DDIs based on their structural similarities. In previous work, the similarity concept was used to develop interesting approaches comparing biological targets through the chemical similarity of their ligands.22 23
In this article we present a large-scale method for DDI discovery and prediction that uses molecular structure similarity information derived from fingerprint-based modeling. Identifying new DDIs using structural similarity is based on the basic idea that if drug A interacts with drug B, and drug C is structurally similar to A, then C should also interact with B (the argument also follows if A is replaced with B). Hence, by combining knowledge of known interactions with structural similarity it is possible to identify new interactions. As an example, it has been reported in the medical literature24 and in Micromedex25 that simvastatin, a drug that reduces levels of cholesterol by inhibiting the enzyme HMG-CoA reductase, can interact with fluconazole, a triazole antifungal drug, resulting in increased risk of myopathy or rhabdomyolysis. The methodology presented in this paper suggests new interactions by exploiting the concept that drugs similar to simvastatin can also interact with fluconazole and cause a similar effect as described above. At the same time, drugs similar to fluconazole can interact with simvastatin causing the same mentioned effect (figure 1 illustrates this with another example). We have created a database of 58 403 new predicted interactions (not mentioned in DrugBank) for approved and experimental drugs, and have made this data resource publically available (see online supplementary tables S1–S3), which can be used by itself or in combination with other methods to identify possible candidates and improve DDI detection.
Figure 1.

Overview of the construction of an interaction similarity model. Employing a list of known drug–drug interactions from DrugBank (step 1), structural similarity computation was carried out using molecular fingerprints (step 2) and a new list of predicted interactions based on structural similarity was generated (step 3).
Methods
DrugBank database
A total of 6624 drugs and 9454 DDIs mentioned in DrugBank V.3.0 were used in this work.26 Drugs with more than one active ingredient, such as oxtriphylline, aminophylline, or colesevelam, and proteins and peptidic drugs were not included because molecular fingerprints are not appropriate descriptors for these types of molecules.
DrugBank DDI database
Drugs included in the DrugBank database were searched for possible interactions using the Interax Interaction Search engine on the DrugBank website,26 27 and duplicate DDIs from the database were eliminated. Interaction information was available for 928 drugs, resulting in a set of 9454 unique DDIs represented as follows: drug A, the description of the effect, and drug B, as shown in figure 1. The effect of the interaction associated with drug pairs was included in our analysis (eg, the DrugBank entry for the DDI tramadol–nefazodone is: increased risk of serotonin syndrome). To prepare for the calculation of DDI detection, the spreadsheet with the set of known DDIs was then transformed into a binary matrix M1 (with 928 rows and 928 columns) where a matrix cell value of 1 represented a known interaction between a pair of drugs and a value of 0 represented no interaction.
Molecular structure similarity analysis
Structural similarity was identified in three steps:
Collecting and processing drug structures: Information on the structures of the compounds in DrugBank was downloaded from the website along with the SMILE code (a chemical notation representing a chemical structure in linear textual form). The molecular structures were preprocessed using the Wash module implemented in MOE software,28 disconnecting group I metals in simple salts and retaining only the largest molecular fragment. The protonation state was considered neutral and explicit hydrogens were added. This step is a common process necessary to prepare the molecules for the next modeling process.
Structural representation: BIT_MACCS (MACCS Structural Keys Bit packed) fingerprints were calculated for all molecules included in the study.28 29 Different molecular fingerprints have been published but the basic technique is to represent a molecule as a bit vector that codes the presence or absence of structural features where each feature is assigned a specific bit position. For example, some structural features in the BIT_MACCS fingerprint for the molecule C6H5-C(O)-NH2 are: bit 84 (NH2, amine group), bit 154 (C=O, carbonyl group), bit 162 (aromatic, C6H5), and bit 163 (six member ring, C6H5).28 29
Similarity measures, computation, and data representation: Different measures are used to compare similarity between two molecular fingerprints. In this study, the molecular fingerprints were compared using the widely applied Tanimoto coefficient (TC).29 30 The TC can span values between 0 and 1, where 0 means ‘maximum dissimilarity’ and 1 means ‘maximum similarity.’ The TC between two fingerprint representations A and B is defined as the number of features present in the intersection of both fingerprints A and B divided by the number of features present in the union of both fingerprints:
Using the Fingerprint Cluster module and the sim_matrix2txt.svl script in MOE,28 a similarity matrix M2 was constructed to capture the TC measure of similarity between pairs of drugs in DrugBank (the matrix cell value represented the TC between pairs of drugs).
Predicting new DDIs
From a technical standpoint, efficiently predicting all new DDIs reduces to matrix multiplication of the matrices M1, which consists of the established interactions, and M2, which consists of the similarity matrix (see step 3a in figure 2). Values in the diagonals of all the matrices are 0 because the interaction of a drug with itself is not considered. However, the same interaction can be generated at different times based on similarities obtained from different pairs, and therefore only the maximum value in the array is retained for each entry, so that the predicted interaction with the highest TC value only is considered (step 3b in figure 2). As an example, the predicted interaction voriconazole–triazolam, which increases the effect of the benzodiazepine, can be generated from the interaction voriconazole–alprazolam (the TC between triazolam and alprazolam is 0.98) or from the interaction voriconazole–midazolam (the TC between triazolam and midazolam is 0.91). In this case, the interaction associated with the highest TC value is used, and the prevailing source for the interaction voriconazole–triazolam is the interaction voriconazole–alprazolam. A symmetric transformation is carried out to obtain the final M3 matrix (step 3c in figure 2), considering the highest value for each pair of drugs (note that the matrix in 3b of figure 2 is not a symmetric matrix). In the example of figure 2, interactions 1–2 and 2–3 from M1 are retrieved in M3 with a TC>0.75. Interaction 1–4 is retrieved by the model with a low score (TC=0.3). The model also predicts the new interaction 3–4 (TC=0.9).
Figure 2.

Generating a drug–drug interactions (DDI) similarity model through combination of the DrugBank interaction database and molecular fingerprint-based modeling. In step 1, interaction matrix M1 is created where the interactions in DrugBank are represented as ‘1’. In step 2, the similarity matrix M2 is created based on the Tanimoto coefficient values. In step 3, M1×M2 is performed, the maximum value for each entry is retained, and the final matrix, M3, is formed based on a symmetry-based transformation (retrieved interactions from M1 when TC>0.75 are represented in red, new predicted interactions with TC>0.75 are represented in blue, and non-retrieved interactions from M1 when TC>0.75 are represented in green). Values in the diagonal of all the matrices are 0 because the interaction of a drug with itself is not considered.
Once the final list of possible interactions is generated from M3, the interactions are associated with the corresponding row in the initial spreadsheet containing the effect of the interaction so that the effect of the interaction can also be captured. The list of interactions predicted by the model with TC>0.75 is given in online supplementary table S1 for the initial 928 DrugBank drugs used to construct the model. The same methodology was applied to the other drugs in DrugBank for which no interaction information was found in the Interax Interaction Search module,27 generating a database of new interactions for 5696 approved, nutraceutical, and experimental drugs (see online supplementary tables S2 and S3).
Evaluation
The performance of the model was evaluated by comparing the predicted interactions based on our methodology when using different TC cut-off values with the established interactions in the initial DrugBank database. The interactions in the DrugBank database were retrieved by the method based on maximum similarity with other drug interaction pairs. The overall performance is summarized using the measures of sensitivity, specificity, precision, and enrichment factors. A receiver operating characteristic (ROC) curve has been generated for more accurate interpretation of model performance. A second evaluation by an external source other than DrugBank was also carried out for the 50 most frequently sold drugs in 2009,31 and the performance of the method was assessed using Micromedex/Drugdex databases as a gold standard to establish the number of correct predictions.
Results
Analysis of model performance using the DrugBank database
A total of 9454 DDIs were obtained from DrugBank which were associated with 928 drugs. Similarity information using molecular fingerprint-based modeling was computed for all 928 drugs and integrated into the system as described in the Methods section to develop the final model. Different cut-offs of similarity values of the TC were used to estimate sensitivity, specificity, precision, and an enrichment factor for the model. Based on a TC>0.85, the model detected 4335 of the 9454 known interactions in the DrugBank database. It was highly unlikely that our system identified this set of 4335 interactions by chance (p<0.0001, one-sided Fisher's exact test). A random methodology considering the same number of possible cases (430 128 possible interactions) and the same number of true positive cases (4335) and false positive cases (6792) as predicted by our model, is capable of selecting only 245 known interactions (true positives), whereas our method identified over 17-fold more interactions. Table 1 shows the performance of our model using different cut-off values for the TC. An ROC curve containing all the possible interactions generated by the model has been plotted in figure 3 and shows an area under the curve of 0.92.
Table 1.
Comparison of model performance on the DrugBank database using different cut-off values for the Tanimoto coefficient
| Model performance on the DrugBank database | ||||
| Tanimoto coefficient >0.85 | ||||
| TP | FP | Total (TP+FP) | Sensitivity | Precision |
| 4335 | 6792 | 11127 | 0.46 | 0.39 |
| FN | TN | Total (FN+TN) | Specificity | Enrichment factor |
| 5119 | 413882 | 419001 | 0.98 | 17.73 |
| Tanimoto coefficient >0.80 | ||||
| 5560 | 10018 | 15578 | 0.59 | 0.36 |
| FN | TN | Total (FN+TN) | Specificity | Enrichment factor |
| 3894 | 410656 | 414550 | 0.98 | 16.24 |
| Tanimoto coefficient >0.75 | ||||
| 6442 | 18366 | 24808 | 0.68 | 0.26 |
| FN | TN | Total (FN+TN) | Specificity | Enrichment factor |
| 3012 | 402308 | 405320 | 0.96 | 11.81 |
FN, false negatives; FP, false positives; Precision, TP/(TP+FP); Sensitivity, TP/(TP+FN); Specificity, TN/(TN+FP); TN, true negatives; TP, true positives.
Figure 3.
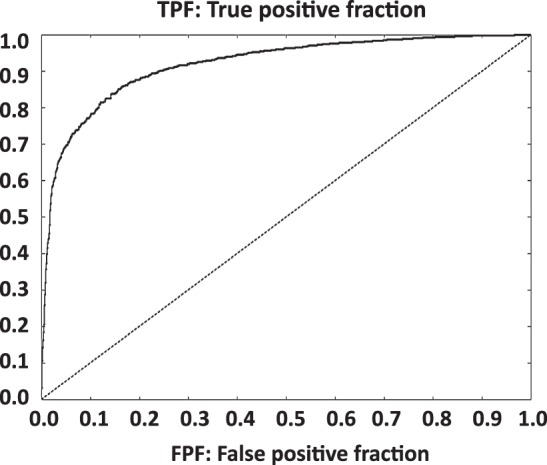
Receiver operating characteristic (ROC) curve showing the performance of the interaction model on the DrugBank database (430 128 possible interactions were computed). The area under the curve is 0.92.
A sensitivity analysis through cross-validation was carried out by dividing the database randomly into two sets: a training set and a test set. Three evaluations were performed by moving 15%, 30%, and 45%, respectively, of the initial interactions to the test set, and by constructing the model with the remaining DrugBank interactions. Sensitivity and specificity values were calculated for the three training and test sets and showed metrics very close to the initial results using TC>0.75 (sensitivity was 0.64, 0.61, and 0.55; specificity was 0.96, 0.97, and 0.97 for the three models, respectively; see online supplementary table S4 for more details). The robustness and the stability of the final model were barely affected by the division of two sets.
Prediction of the effect produced by the DDI
Another feature of the model is its ability to detect the biological effect produced by the DDI. As an example, an interaction could produce an effect based on alterations in the bioavailability of one of the drugs due to both drugs being metabolized by the same enzymes or due to competition for the same transporter protein. In order to verify whether the model is also capable of predicting the effect produced by the DDI, a random selection of DrugBank interactions was reviewed manually to determine the degree of precision of the predicted biological effect. Out of 100 interactions selected using a TC cut-off value of 0.85, the effect produced by the drug combination was correctly predicted in 99 interactions where the effect was the same as that originally specified in DrugBank. Using other cut-offs, that is, 0.85≥TC>0.80 and 0.80≥TC>0.75, the model correctly predicted the effect in 96% and 91% of the evaluated interactions, respectively (see online supplementary table S5 for more details). However, in future predictions the nature of the predicted interactions should be carefully analyzed, especially when the TC is lower and the pharmacological class of the drugs detected as structurally similar is different. For this reason, for values of TC<0.85, appropriate pharmacological knowledge to correctly interpret the effect of the interaction predicted would be beneficial.
Evaluation in Micromedex/Drugdex
In the second part of the evaluation, interactions for the 50 most frequent commercial drugs (consisting of 44 unique generic drugs) sold in 2009 were searched in the Micromedex/Drugdex database. Table 2 provides details of the sources of the drug information as well as the results (see also figure 4). Specifically, table 2A gives the number of interactions specified in DrugBank and in Micromedex/Drugdex; table 2B gives the number of predicted interactions and the number of interactions correctly predicted by our model; and table 2C gives the sensitivity, specificity, precision, and enrichment factor for the three different TC cut-off values. A total of 1760 interactions were associated with the drugs specified in Micromedex, and the model predicted 548 interactions with a TC>0.75 (31% correct classification) and 348 interactions with a TC>0.85 (20% correct classification). Detailed results are given in table 2 and online supplementary tables S6 and S7. It was highly unlikely that our model identified 348 true interactions by chance (p<0.0001, one-sided Fisher's exact test). A random method considering 63 932 possible interactions (interactions generated between 1454 drugs from DrugBank and the 44 most frequently sold drugs in 2009) and randomly selecting 1141 positive cases (the same as the model when TC>0.85) would detect 31 interactions described in the Micromedex database (1.78% correct classification).
Table 2.
Model performance predicting the interactions for the 50 most frequently sold drugs in 2009 based on the Micromedex/Drugdex database as a gold standard and using different cut-off values for the Tanimoto coefficient
| A | ||
| Interactions described in DrugBank | 773 | |
| Interactions described in Micromedex/Drugdex | 2323 | |
| Interactions described in Micromedex with DrugBank drugs* | 1760 | |
| B | |||
| Tanimoto coefficient | >0.85 | >0.80 | >0.75 |
| Interactions predicted by the model† | 1141 | 1591 | 2565 |
| Number of correct interactions predicted by the model | 348 | 444 | 548 |
| Number of correct interactions predicted by the model and described in DrugBank | 212 | 252 | 269 |
| Number of correct interactions predicted by the model and not described in DrugBank (ie, new for DrugBank) | 136 | 192 | 279 |
| C | ||||
| Model performance (Tanimoto coefficient >0.85) | ||||
| TP | FP | Total (TP+FP) | Sensitivity | Precision |
| 348 | 793 | 1141 | 0.20 | 0.30 |
| FN | TN | Total (FN+TN) | Specificity | Enrichment factor |
| 1412 | 61379 | 62791 | 0.99 | 11.08 |
| Model performance (Tanimoto coefficient >0.80) | ||||
| TP | FP | Total (TP+FP) | Sensitivity | Precision |
| 444 | 1147 | 1591 | 0.25 | 0.28 |
| FN | TN | Total (FN+TN) | Specificity | Enrichment factor |
| 1316 | 61025 | 62341 | 0.98 | 10.14 |
| Model performance (Tanimoto coefficient >0.75) | ||||
| TP | FP | Total (TP+FP) | Sensitivity | Precision |
| 548 | 2017 | 2565 | 0.31 | 0.21 |
| FN | TN | Total (FN+TN) | Specificity | Enrichment factor |
| 1212 | 60155 | 61367 | 0.97 | 7.76 |
The evaluation is carried out using the interactions described in Micromedex/Drugdex when the drugs implicated in the interaction are also present in DrugBank and the molecular structure is also available so the likelihood of the interaction can be computed.
Predicted interactions using the 1454 drugs from DrugBank (approved and nutraceuticals).
FN, false negatives; FP, false positives; Precision, TP/(TP+FP); Sensitivity, TP/(TP+FN); Specificity, TN/(TN+FP); TN, true negatives; TP, true positives.
Figure 4.
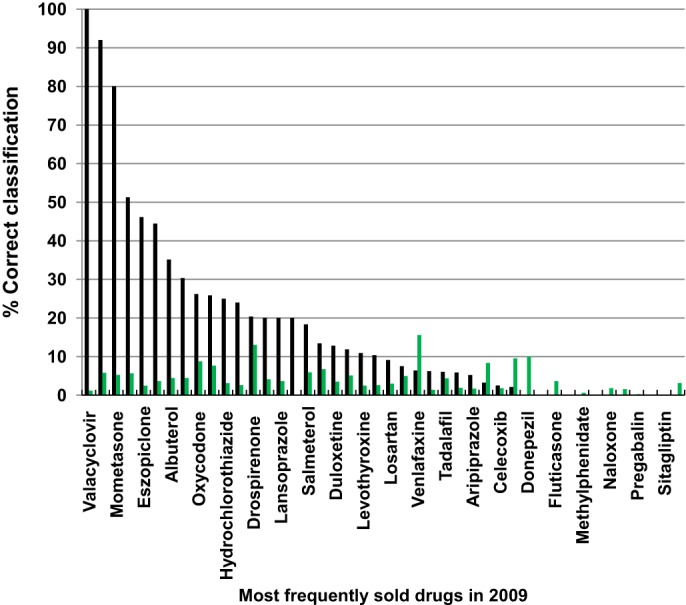
Percentage of correct classifications for a random set of interactions described in Micromedex/Drugdex for the 50 most frequently sold drugs in 2009 (44 generic names) using the similarity interaction model (TC>0.75) and a random set of drug interactions (more details are provided in online supplementary tables S6 and S7). Drugs are sorted according to the percentage classified correctly by the model. Only interactions described in Micromedex/Drugdex but not in the DrugBank database are taken into account. % Correct classification (Micromedex/Drugdex interactions not described previously in DrugBank and correctly predicted by the model). % Correct classification (random set of interactions).
The results identify interesting drug interactions belonging to two categories. The nature of the system permits the identification of drugs belonging to pharmacological classes different from those of the drugs implicated in the interaction (eg, drug A and a similar drug C do not belong to the same pharmacological class but each interact with drug B), which occurs more frequently as the TC value decreases. However, the method is more likely to identify interactions between drugs with similar pharmacological profiles. The information provided by the model in this case is more obvious but still could be very useful to researchers, particularly those without a strong background in pharmacology.
The interaction examples shown below were predicted by our model and not described in DrugBank, but were described in Micromedex/Drugdex with different levels of documentation, from ‘the existence of the interaction was clearly established through controlled studies’ to ‘limited documentation but pharmacological knowledge lead clinicians to recognize the possible interaction’.
Examples of different pharmacological classes
Several interaction examples predicted for the 50 most frequently sold drugs in 2009 showed that the DDI similarity model can detect drugs that belong to different pharmacological classes but have similar structural features (see table 3). An example of an interaction correctly predicted by the model according to the Micromedex/Drugdex database is aripiprazole–nefazodone. Concomitant use of these drugs can cause increased concentration of aripiprazole. Our model detected this interaction because the interaction aripiprazole–itraconazole is described in DrugBank, where the result is that itraconazole increases the effect of aripiprazole. According to our analysis, itraconazole shows some structural features similar to nefazodone (TC=0.82), although both molecules have different pharmacological profiles (itraconazole is an antifungal and nefazodone is an antidepressant).
Table 3.
Examples of new predictions for the top 50 drugs not described in DrugBank but detected by our method and confirmed through Micromedex/Drugdex
|


Another example of an interaction found by our methodology is mometasone and different protease inhibitors used in HIV therapy, such as indinavir, nelfinavir, ritonavir, and saquinavir, possibly increasing the effect and toxicity of mometasone (see table 3). Mometasone is similar to fusidic acid (TC=0.77), and it is established in DrugBank that fusidic acid can interact with protease inhibitors. The possible interaction mometasone–protease inhibitors is described in Micomedex/Drugdex and may cause increased mometasone plasma concentrations due to inhibition of CYP3A4-mediated mometasone metabolism by the antiretroviral drugs.
Buprenorphine, an opioid analgesic, has been found to share some structural similarity with vinblastine, an antineoplastic agent used for the treatment of different types of cancer (TC=0.76). The model correctly predicts, based on Micromedex/Drugdex, that buprenorphine can interact with different protease inhibitors (atazanavir, darunavir, indinavir, ritonavir, and saquinavir), with the antifungal ketoconazole and with the macrolide antibiotic erythromycin, causing decreased metabolism of buprenorphine and increased drug plasma concentrations (see table 3).
Different interactions predicted by our model and described in Micromedex/Drugdex have been found for venlafaxine, an antidepressant of the serotonin–norepinephrine reuptake inhibitor (SNRI) class. According to our fingerprint-based model, tramadol was found to be similar to venlafaxine with TC=0.93. Therefore, venlafaxine was predicted to interact with cimetidine, clozapine, haloperidol, and dextroamphetamine, producing different plasma concentrations of the drugs implicated in the interaction (see table 3).
The possibility of finding drugs belonging to different classes increases as the TC value decreases, which is interesting. However, since the similarity is lower, the risk of incorrect predictions is higher. For this reason, we considered a cut-off value of 0.75 for the TC appropriate since similarity is still remarkable and many different classes of related drugs can be identified.
Examples of the same pharmacological classes
Although the DDI model can associate drugs which have different pharmacological profiles but are structurally similar, some of the predicted interactions can identify a drug belonging to the same pharmacological class of one of the drugs implicated in the known interaction. The DrugBank database describes the interactions acetophenazine–cisapride and acetophenazine–terfenadine as resulting in an increased risk of cardiotoxicity and arrhythmias. Our model detects that acetophenazine, a first generation antipsychotic of the phenothiazine class, is similar to quetiapine, a second generation antipsychotic, with TC=0.78. Quetiapine is predicted to have the same interactions, which were confirmed in Micromedex/Drugdex. Other examples of predictions validated in Micromedex are the reduction in hydrochlorotiazide absorption due to concomitant use of colestipol, fenofibrate may increase the anticoagulant effect of phenprocoumon with risk of excessive bleeding, and buprenorphine can interact with different opioids resulting in precipitation of withdrawal symptoms (see table 3 and online supplementary tables for more details).
Discussion
Different types of models for predicting DDIs have been recently published.9 32–34 However, the majority of the in silico approaches to predicting drug interactions have focused on the integration of in vitro data to generate models for the in vivo prediction of drug interactions.33 These models mainly try to predict possible metabolic interactions, especially interactions related to CYP enzymes. Nevertheless, there are many examples of drugs that follow other metabolic routes. There are also many DDIs due to similar distribution profiles of the investigated drugs. The importance of some mechanisms, as interaction with transporters, has been recognized later.14
We propose a large-scale method based on identifying molecular similarity to analyze multiple types of drug interactions caused by the inhibition of metabolizing enzymes, transporters, or even the pharmacological targets. The model described in this article can exploit experimental knowledge to identify the possible causes of the interaction. The system allows the researcher to monitor the data and the model's predictions preserve the nature of the original DDI that generates the outcome, which is very useful for examining the effect and the type of interaction predicted. Indeed, we reviewed 300 randomly selected interactions and have shown that the system can predict the effect of the interactions in more than 90% of cases when TC>0.75 (see online supplementary table S5).
The model potentiates a visible pattern in the DrugBank database (similar drugs have similar interactions) by detecting drugs similar to the drugs implicated in the interactions described previously. Therefore, one limitation of this study is that the performance of the model depends on the comprehensiveness of the information in the original interaction database. This method was applied to the interactions and drugs only specified in DrugBank, but the addition of other sources of established DDIs, such as those mentioned in drug labels, could be taken into account to generate the final model.
An additional issue is that 2D similarity fingerprints were used, which have some limitations in describing the molecular structure. The 3D structure is a very important component in the interaction drug–receptor and is a better representation of the molecules.35 36 However, although the information provided by 2D methods is more limited than the 3D information, the 2D methods still offer good results and are much simpler and require less computational effort, avoiding important problems such as the selection of bioactive conformations and the calculation and superimposition of the 3D structure of all the drugs implicated in the study. Different 2D molecular fingerprints could also be used in the development of this type of model.37 Nevertheless, in the current study, BIT_MACCS fingerprints were calculated because they are simple and have offered good results for recognizing similar molecules in large databases.21 38 39
Although the similarity model provides valuable information associated with the initial interactions, a more reliable and complex system could be implemented through the integration of structural similarity measures and knowledge in pharmacological databases containing information about possible targets and metabolizing or transporter enzymes. This method could also be combined with other methodologies using different types of information, such as the Food and Drug Administration's Adverse Event Reporting System,40 which was created to provide postmarketing drug safety information, or the use of clinical data in electronic health records.41 An extensive database of annotated possible drug interactions predicted by our model for the drugs in DrugBank (approved and experimental drugs) is provided in online supplementary tables S1–S3). This database is a valuable source of information on drug interactions that is available for download and can be used by itself or in combination with other methods to filter out possible candidates and improve DDI detection.
Several DDIs highlighted by our methodology were not known and consequently were considered false positives in our evaluation. However, it is possible that some of these drugs actually do interact but have not yet been identified. Therefore, it is possible that the false positive rate is lower than we estimated.
Conclusion
The results presented in this study demonstrate the usefulness of the proposed drug–drug interaction methodology as a promising approach for in silico prediction of drug interactions and their effects. The method described in this article is very simple, efficient, applicable to large-scale investigation and helps highlight the etiology of DDIs (see table 3). In this study, the application of structure similarity information to drug interaction knowledge as specified in DrugBank led to retrieval of the majority of known interactions, showing a sensitivity of 0.68 when the specificity was 0.96. A set of interactions not described in the literature but with strong supporting evidence according to our model has been constructed for further analysis. Experimental drugs were also evaluated by the model and ranked according to interaction probability. The database of 58 403 new predicted DDIs provided in this study could be useful for further study of possible candidates, and is available for download (online supplementary tables S1–S3). This database could be used as a powerful pharmacovigilance tool by itself or combined with other methods, such as the Food and Drug Administration's Adverse Event Reporting System or electronic health records, to facilitate drug safety by selecting candidates with a strong possibility of interacting in the human body.
Supplementary Material
Footnotes
Contributors: SV and CF conceived and designed the study; SV, RH, EU, LS, RR, and CF suggested data and analysis tools; SV performed and analyzed the data; and SV, RH, EU, LS, RR, and CF wrote the paper.
Funding: This work was supported by grants R01 LM010016 (CF), R01 LM010016-0S1 (CF), R01 LM010016-0S2 (CF), R01 LM008635 (CF), and 1R01LM010140-01 (RR) from the National Library of Medicine, ‘Plan Galego de Investigación, Innovación e Crecemento 2011-2015 (I2C)’, the European Social Fund (ESF), and the Angeles Alvariño program from Xunta de Galicia (Spain).
Competing interests: None.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1. Classen DC, Pestotnik SL, Evans RS, et al. Adverse drug events in hospitalized patients. Excess length of stay, extra costs, and attributable mortality. JAMA 1997;277:301–6 [PubMed] [Google Scholar]
- 2. Cullen DJ, Sweitzer BJ, Bates DW, et al. Preventable adverse drug events in hospitalized patients: a comparative study of intensive care and general care units. Crit Care Med 1997;25:1289–97 [DOI] [PubMed] [Google Scholar]
- 3. Cullen DJ, Bates DW, Small SD, et al. The incident reporting system does not detect adverse drug events: a problem for quality improvement. Jt Comm J Qual Improv 1995;21:541–8 [DOI] [PubMed] [Google Scholar]
- 4. Bates DW, Spell N, Cullen DJ, et al. The costs of adverse drug events in hospitalized patients. Adverse Drug Events Prevention Study Group. JAMA 1997;277:307–11 [PubMed] [Google Scholar]
- 5. Bates DW, Cullen DJ, Laird N, et al. Incidence of adverse drug events and potential adverse drug events-implications for prevention. ADE Prevention Study Group. JAMA 1995;274:29–34 [PubMed] [Google Scholar]
- 6. Mjorndal T, Boman MD, Hagg S, et al. Adverse drug reactions as a cause for admissions to a department of internal medicine. Pharmacoepidemiol Drug Saf 2002;11:65–72 [DOI] [PubMed] [Google Scholar]
- 7. Schneeweiss S, Hasford J, Gottler M, et al. Admissions caused by adverse drug events to internal medicine and emergency departments in hospitals: a longitudinal population-based study. Eur J Clin Pharmacol 2002;58:285–91 [DOI] [PubMed] [Google Scholar]
- 8. Hohl CM, Nosyk B, Kuramoto L, et al. Outcomes of emergency department patients presenting with adverse drug events. Ann Emerg Med 2011;58:270–9.e4 [DOI] [PubMed] [Google Scholar]
- 9. Tatonetti NP, Fernald GH, Altman RB. A novel signal detection algorithm for identifying hidden drug-drug interactions in adverse event reports. J Am Med Inform Assoc. Published Online First: 2011. doi:10.1136/amiajnl-2011-000214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pirmohamed M. Ml O: Drug Interactions of Clinical Importance. London: Chapman & Hall, 1998 [Google Scholar]
- 11. Harpaz R, Chase HS, Friedman C. Mining multi-item drug adverse effect associations in spontaneous reporting systems. BMC Bioinformatics 2010;11(Suppl 9):S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Staffa JA, Chang J, Green L. Cerivastatin and reports of fatal rhabdomyolysis. N Engl J Med 2002;346:539–40 [DOI] [PubMed] [Google Scholar]
- 13.http://www.fda.gov/
- 14. Bjornsson TD, Callaghan JT, Einolf HJ, et al. The conduct of in vitro and in vivo drug-drug interaction studies: a Pharmaceutical Research and Manufacturers of America (PhRMA) perspective. Drug Metab Dispos 2003;31:815–32 [DOI] [PubMed] [Google Scholar]
- 15. Martin YC, Kofron JL, Traphagen LM. Do structurally similar molecules have similar biological activity? J Med Chem 2002;45:4350–8 [DOI] [PubMed] [Google Scholar]
- 16. Vilar S, Santana L, Uriarte E. Probabilistic neural network model for the in silico evaluation of anti-HIV activity and mechanism of action. J Med Chem 2006;49:1118–24 [DOI] [PubMed] [Google Scholar]
- 17. Vilar S, Cozza G, Moro S. Medicinal chemistry and the molecular operating environment (MOE): application of QSAR and molecular docking to drug discovery. Curr Top Med Chem 2008;8:1555–72 [DOI] [PubMed] [Google Scholar]
- 18. Gedeck P, Lewis RA. Exploiting QSAR models in lead optimization. Curr Opin Drug Discov Devel 2008;11:569–75 [PubMed] [Google Scholar]
- 19. Winkler DA. The role of quantitative structure–activity relationships (QSAR) in biomolecular discovery. Brief Bioinform 2002;3:73–86 [DOI] [PubMed] [Google Scholar]
- 20. Costanzi S, Vilar S, Micozzi D, et al. Delineation of the molecular mechanisms of nucleoside recognition by cytidine deaminase through virtual screening. Chem Med Chem 2011;6:1452–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ewing T, Baber JC, Feher M. Novel 2D fingerprints for ligand-based virtual screening. J Chem Inf Model 2006;46:2423–31 [DOI] [PubMed] [Google Scholar]
- 22. Hert J, Keiser MJ, Irwin JJ, et al. Quantifying the relationships among drug classes. J Chem Inf Model 2008;48:755–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Keiser MJ, Roth BL, Armbruster BN, et al. Relating protein pharmacology by ligand chemistry. Nat Biotechnol 2007;25:197–206 [DOI] [PubMed] [Google Scholar]
- 24. Molden E, Skovlund E, Braathen P. Risk management of simvastatin or atorvastatin interactions with CYP3A4 inhibitors. Drug Saf 2008;31:587–96 [DOI] [PubMed] [Google Scholar]
- 25. Micromedex ® Healthcare Series [Internet database] Greenwood Village, Colo: Thomson Reuters (Healthcare) Inc. Thomson Reuters; USA: Updated periodically. 2011 [Google Scholar]
- 26. DrugBank Database, Version 3.0. http://www.drugbank.ca/ (accessed Apr 2011).
- 27. http://www.drugbank.ca/interax/drug_lookup (accessed Apr 2011).
- 28. MOE, Version 2011.10; Chemical Computing Group, Inc. http://www.chemcomp.com
- 29.MACCS Keys; MDL Information Systems I, 14600 Catalina Street, San Leandro, CA 94577.
- 30.http://www.daylight.com/dayhtml/doc/theory/theory.finger.html
- 31.http://www.drugs.com/top200_units.html
- 32. Dickins M, Van de Waterbeemd H. Simulation models for drug disposition and drug interactions. Drug Discov Today Biosilico 2004;2:38–45 [Google Scholar]
- 33. Zhang L, Zhang YC, Zhao P, et al. Predicting drug-drug interactions: an FDA perspective. AAPS J 2009;11:300–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tatonetti NP, Denny JC, Murphy SN, et al. Detecting drug interactions from adverse-event reports: interaction between paroxetine and pravastatin increases blood glucose levels. Clin Pharmacol Ther 2011;90:133–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vilar S, Karpiak J, Costanzi S. Ligand and structure-based models for the prediction of ligand-receptor affinities and virtual screenings: development and application to the beta(2)-adrenergic receptor. J Comput Chem 2010;31:707–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Engel S, Skoumbourdis AP, Childress J, et al. A virtual screen for diverse ligands: discovery of selective G protein-coupled receptor antagonists. J Am Chem Soc 2008;130:5115–23 [DOI] [PubMed] [Google Scholar]
- 37. Steffen A, Kogej T, Tyrchan C, et al. Comparison of molecular fingerprint methods on the basis of biological profile data. J Chem Inf Model 2009;49:338–47 [DOI] [PubMed] [Google Scholar]
- 38. Durant JL, Leland BA, Henry DR, et al. Reoptimization of MDL keys for use in drug discovery. J Chem Inf Comput Sci 2002;42:1273–80 [DOI] [PubMed] [Google Scholar]
- 39. Vilar S, Harpaz R, Chase HS, et al. Facilitating adverse drug event detection in pharmacovigilance databases using molecular structure similarity: application to rhabdomyolysis. J Am Med Inform Assoc 2011;18(Suppl 1):i73–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Adverse Event Reporting System. http://www.fda.gov/cder/aers/default.htm
- 41. Wang X, Hripcsak G, Markatou M, et al. Active computerized pharmacovigilance using natural language processing, statistics, and electronic health records: a feasibility study. J Am Med Inform Assoc 2009;16:328–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.