

Abstract
The evaluation of possible interactions between chemical compounds and antitarget proteins is an important task of research and development process. Here we describe the development and validation of QSAR models for the prediction of antitarget end-points, created on the basis of Multilevel and Quantitative Neighborhoods of Atoms descriptors and self-consistent regression. Data on 4000 chemical compounds interacting with 18 antitarget proteins (13 receptors, 2 enzymes and 3 transporters) were used to model thirty two sets of end-points (IC50, Ki and Kact). Each set was randomly divided into training and test sets in a ratio of 80% to 20%, respectively. The test sets were used for external validation of QSAR models created on the basis of the training sets. The coverage of prediction for all test sets exceeded 95% and for half of the test sets it was 100%. The accuracy of prediction for 29 of the end-points, based on the external test sets was typically in the range of R2test = 0.6–0.9; three tests sets had a lower R2test values, specifically 0.55 – 0.6. The proposed approach showed a reasonable accuracy of prediction for 91% of the antitarget end-points and high coverage for all external test sets. On the basis of the created models we have developed a freely available on-line service for in silico prediction of 32 antitarget end-points: http://www.pharmaexpert.ru/GUSAR/antitargets.html.
Keywords: QSAR, antitargets, QNA, prediction, SCR, side effects, drug design
INTRODUCTION
The process of drug development is time-consuming and cost-intensive. Several years are required for lead identification, optimization, in vitro and in vivo testing before the first clinical trials are started. Preapproval costs of a new drug exceed US $800 million [1]. It is well known that about 90% of drug-candidates fail in the first phase of clinical trials [2].
Approximately 10% of new chemical entities (NCEs) show serious adverse drug reactions (ADRs) after their introduction into medical practice. More than 17 drugs were withdrawn from the market during the period from 1996 to 2006, because they had shown serious adverse drug reactions [3]. For example, Amineptine (launched in 1978) is an indirect dopamine agonist, which selectively inhibits dopamine uptake and induces its release, with additional stimulation of the adrenergic system. However, microcystic, macrocystic acne and hepatotoxicity were observed as common side effects [4]. Also, Duract (bromfenac sodium) was launched in 1997 for the treatment of acute pain as a non-steroidal antiinflammatory drug (NSAID). The drug was withdrawn after postmarketing reports of severe hepatic failure that led to four deaths and eight liver-transplants [5]. Another drug, Vioxx, was launched in 1999 for the treatment of pain and inflammation as an NSAID. In 2004, a long-term study of Vioxx in patients with increased risk of colon polyps was halted because of increased cardiovascular risk. Thus, Vioxx was withdrawn from the market in 2004 [6].
Interactions with some enzymes, receptors and channels have been identified as molecular mechanisms for certain side effects observed in the development of candidates or marketed drugs, and are named antitargets [3]. Several antitargets were previously considered as drug targets. Currently, it is thought that the benefit of action on these targets exceeds their side effects. For example, the main target of the well-known antidepressant and serotonin uptake inhibitor Prozac (Fluoxetine) is the sodium-dependent serotonin transporter that is considered as a withdrawn target in DrugBank. Most common ADRs (e.g. hepatic toxicity, hematologic toxicity and cardiovascular toxicity) are caused by drug action on antitargets (off-targets). Alpha-1A adrenergic antagonists may cause orthostatic hypotension, dizziness and fainting spells. D2 dopaminergic antagonists may cause extrapyramidal syndrome; M1 muscarinic antagonists may cause attention/memory deficits [7]. Antitarget-mediated side effects may risk the further development of promising clinical candidates.
Therefore, to avoid potential interactions of drugs with antitargets, specific studies for their detection should ideally be conducted before the pharmaceutical is launched. For this purpose, several different computational approaches have been proposed. 3D pharmacophore models rationalizing the affinity of several different chemical series have been described for alpha-1A, 5-HT 2A and D2 receptors [8]. The ligand–protein inverse docking approach was successfully used to predict the potential toxicity and side effects related to protein targets of small molecules [9]. Different 2D and 3D QSAR techniques have also been applied for prediction of antitarget activities [10–14]. However, until now there was no computational approach that allowed prediction of the profiles of antitarget effects for chemical compounds with reasonable accuracy (Q2model > 0.6 and R2test > 0.6) and speed (~ 1 compound per second).
Earlier, we have shown that GUSAR software based on Multilevel and Quantitative Neighbourhoods of Atoms (MNA, QNA) descriptors [15,16] and the self-consistent regression (SCR) algorithm [17,18], may successfully be applied for multiple QSAR tasks [15]. In this work, we have applied the GUSAR program to model the interactions between drug-like organic compounds and antitargets including three types of proteins: receptors, enzymes and transporters. A freely available on-line service for the quantitative prediction of antitarget interaction profiles was developed using the models derived.
MATERIALS AND METHODS
Data sets
Data on the chemical structures and quantitative end-point values (50% inhibitory concentration - IC50inhibition constant - Ki and activation constant - Kact) for approximately 4000 chemical compounds interacting with 18 antitarget proteins were collected from different literature sources. An example of chemical structures with end-point values is presented in Table 1. The InChI keys for all chemicals and their activities used in this work are available in the supplementary material S1.
Table 1.
Data set structural information.
| Structure | Activity | End- point |
-Log10(End- point), mol/L |
|---|---|---|---|
 |
5-hydroxytryptamine 1B receptor antagonist | Ki | 7 |
| 5-hydroxytryptamine 2A receptor antagonist | Ki | 6.45 | |
| alpha 1a adrenergic receptor antagonist | Ki | 7.95 | |
 |
5-hydroxytryptamine 2C receptor antagonist | IC50 | 6.3 |
| 5-hydroxytryptamine 2A receptor antagonist | IC50 | 5 | |
 |
alpha 1a adrenergic receptor antagonist | Ki | 9.7 |
| alpha 1b adrenergic receptor antagonist | Ki | 6.59 | |
 |
alpha-2A adrenergic receptor antagonist | Ki | 6.36 |
| alpha 1b adrenergic receptor antagonist | Ki | 6.66 | |
| alpha 1a adrenergic receptor antagonist | Ki | 7.55 | |
| IC50 | 8 | ||
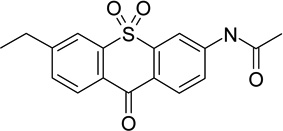 |
amine oxidase [flavin-containing] A inhibitor | ||
| IC50 | 7.66 | ||
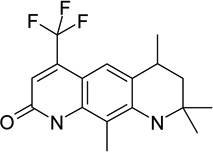 |
androgen receptor antagonist | ||
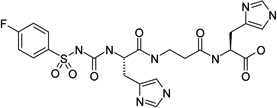 |
carbonic anhydrase I activator | Kact | 7.52 |
| carbonic anhydrase II activator | Kact | 6.3 | |
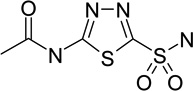 |
carbonic anhydrase I inhibitor | Ki | 6.05 |
| carbonic anhydrase II inhibitor | Ki | 7.92 | |
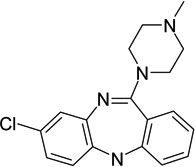 |
d(1 A) dopamine receptor antagonist | Ki | 6.38 |
| d3 dopamine receptor antagonist | Ki | 6.01 | |
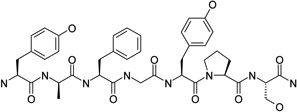 |
mu-type opioid receptor antagonist | IC50 | 8.43 |
| delta-type opioid receptor antagonist | Ki | 6.72 | |
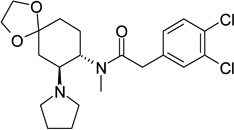 |
mu-type opioid receptor antagonist | Ki | 6.15 |
| kappa-type opioid receptor antagonist | Ki | 8.82 | |
| delta-type opioid receptor antagonist | Ki | 5.79 | |
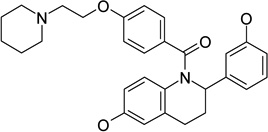 |
estrogen receptor antagonist | IC50 | 6.23 |
| estrogen receptor antagonist | Ki | 7.43 | |
| IC50 | 6.59 | ||
 |
sodium- and chloride-dependent GABA transporter 1 antagonist |
||
 |
sodium-dependent dopamine transporter antagonist |
IC50 | 8.66 |
| sodium-dependent serotonin transporter antagonist |
Ki | 8.61 |
Based on the “Withdrawn Drug Targets” and “Withdrawn Enzymes” represented in DrugBank (http://www.drugbank.ca/downloads), the list of antitarget proteins includes thirteen receptors, two enzymes and three transporters. A brief description of the data sets is given below.
The ranges of end-point values for the sets of compounds interacting with each receptor are presented in Table 2.
Table 2.
Receptor data sets.
| Activity Name | End-point | Mina | Maxb |
|---|---|---|---|
| 5-hydroxytryptamine 1B receptor antagonist | IC50 | 2.8 | 9.3 |
| 5-hydroxytryptamine 1B receptor antagonist | Ki | 3.8 | 9.7 |
| 5-hydroxytryptamine 2A receptor antagonist | IC50 | 3.1 | 10.3 |
| 5-hydroxytryptamine 2A receptor antagonist | Ki | 4.3 | 10.4 |
| 5-hydroxytryptamine 2C receptor antagonist | IC50 | 2.9 | 9.0 |
| 5-hydroxytryptamine 2C receptor antagonist | Ki | 3.4 | 10.7 |
| alpha-1 A adrenergic receptor antagonist | IC50 | 4.0 | 9.9 |
| alpha-1 A adrenergic receptor antagonist | Ki | 3.6 | 11.0 |
| alpha-1B adrenergic receptor antagonist | Ki | 4.3 | 10.0 |
| alpha-2A adrenergic receptor antagonist | IC50 | 3.2 | 9.3 |
| alpha-2A adrenergic receptor antagonist | Ki | 2.4 | 9.9 |
| androgen receptor antagonist | IC50 | 4.2 | 10 |
| d(1 A) dopamine receptor antagonist | IC50 | 3.9 | 9.1 |
| d(1 A) dopamine receptor antagonist | Ki | 4.1 | 10.1 |
| d3 dopamine receptor antagonist | Ki | 4.2 | 10.7 |
| delta-type opioid receptor antagonist | Ki | 4.3 | 12.0 |
| estrogen receptor antagonist | IC50 | 1.7 | 11.0 |
| estrogen receptor antagonist | Ki | 3.0 | 12.7 |
| kappa-type opioid receptor antagonist | Ki | 3.1 | 11.4 |
| mu-type opioid receptor antagonist | IC50 | 3.6 | 10.5 |
| mu-type opioid receptor antagonist | Ki | 3.4 | 12.3 |
Minimal value of -Log10(End-point) values in the set, mol/L.
Maximal value of -Log10(End-point) values in the set, mol/L.
Table 2 shows that the range of the modeled values for compounds from the receptor data sets exceeds 5 logarithmic units. This is a good prerequisite for the creation of accurate, robust and predictive QSAR models. The minimal values for all cases are less than 5 –log10(mol/L), which means that the created models can be used to assess both active and inactive molecules.
Thirteen receptors belong to the hydroxytryptamine, adrenergic, androgen, dopamine, opioid and estrogen families. The main adverse effects of antagonists to 5-hydroxytryptamine receptors are sickness, emesis, diarrhea, sleeplessness and anxiety [19]. Adrenergic receptor antagonists may cause orthostatic hypotension, reflex tachycardia, insomnia, nasal congestion, tachycardia and palpitation [20]. Antagonism to androgen receptors may lead to virilization, gynecomastia, hepatic pelioza and hepatoma [20]. The main undesirable effects of dopamine receptor antagonists are palpitations, ectopic rhythm, tachycardia, retrosternal pain, hypertension, vasoconstriction, shortness of breath and headache [7]. Antagonism to opioid receptors may cause sickness, emesis, respiratory depression, and sedation [21]. Adverse effects related to the action of estrogen receptor antagonists are depression, headache, obesity, sickness, hot flashes and puffiness [7].
The ranges of end-point values for the sets of compounds interacting with enzymes (amine oxidase and carbonic anhydrase) and transporters (GABA, dopamine and serotonin transporters) are represented in Table 3 and Table 4, respectively.
Table 3.
Enzyme data sets
| Activity Name | End-point | Mina | Maxb |
|---|---|---|---|
| amine oxidase [flavin-containing] A inhibitor | IC50 | 3.0 | 9.0 |
| amine oxidase [flavin-containing] A inhibitor | Ki | 3.5 | 9.5 |
| carbonic anhydrase II activator | Kact | 2.4 | 10.0 |
| carbonic anhydrase I activator | Kact | 1.5 | 10.9 |
| carbonic anhydrase I inhibitor | Ki | 0.4 | 9.4 |
| carbonic anhydrase II inhibitor | Ki | 4.4 | 9.7 |
Minimal value of -Log10(End-point) values in the set, mol/L.
Maximal value of -Log10(End-point) values in the set, mol/L.
Table 4.
Transporter data sets
| Activity Name | End-point | Mina | Maxb |
|---|---|---|---|
| sodium- and chloride-dependent GABA transporter 1 antagonist | IC50 | 3.1 | 7.3 |
| sodium-dependent dopamine transporter antagonist | IC50 | 3.0 | 10.5 |
| sodium-dependent dopamine transporter antagonist | Ki | 3.3 | 9.5 |
| sodium-dependent serotonin transporter antagonist | IC50 | 2.4 | 9.8 |
| sodium-dependent serotonin transporter antagonist | Ki | 3.4 | 11.1 |
Minimal value of -Log10(End-point) values in the set, mol/L.
Maximal value of -Log10(End-point) values in the set, mol/L.
Table 3 and Table 4 show that the ranges of the modeled values for compounds from the enzyme and transporter data sets exceed 4 logarithmic units. The minimal values for all cases are less than 5 –log10(mol/L), thus the created models can be used for the evaluation of both active and inactive molecules.
The main adverse effects of amine oxidase (MAO A) inhibitors are blood pressure lability, bradycardia, chorea, convulsions, delirium, diarrhea, hepatotoxicity and drowsiness. Interaction of ligands with carbonic anhydrase may be the cause of alopecia (hair loss), anaphylaxis, aplastic anemia, anxiety, bone marrow suppression, chronic fatigue syndrome, depression and renal tubular acidosis [20]. Neurotoxicity is the main adverse effect of GABA and dopamine transporters blockers [22]. Adverse effects caused by the serotonin transporter blockers are acute respiratory distress syndrome (ARDS), agitation, akathisia, constipation, diarrhea, drowsiness, emesis, glaucoma, headache, hemorrhage, hypomania, ischemic colitis, parkinsonism, myoclonus, QT interval prolongation and tremor [7].
The structural overlap was analyzed between the 32 dataset endpoints. The results are presented in the supplementary material S2. It was found that all end-points have some structural overlap except for the “sodium- and chloride-dependent GABA transporter 1 antagonists”. The major cases of overlap were found between receptor end-points, belonging to the hydroxytryptamine, adrenergic and dopamine families. More than one thousand compounds in common were found among the drug-like molecules acting on the different types of opioid receptors. In contrast to the receptor families, a small number of overlapping structures was found for the enzymes.
Each set was randomly divided into training and test sets according to the ratio 80%:20%, respectively. The training set was used to create the QSAR models and the test set was used to assess the external predictive accuracy. QSAR models were developed using Multilevel and Quantitative Neighbourhoods of Atoms (MNA, QNA) descriptors [15, 16] and the self-consistent regression (SCR) algorithm [17, 18].
QSAR modeling on the basis of QNA descriptors
QSAR modeling on the basis of QNA descriptors has previously been implemented in the software program GUSAR [15]. Reasonable results obtained by GUSAR modeling for different biological endpoints [15] prompted this study to investigate the utility of the method for modeling the interactions of chemical structures with sets of antitargets. A more detailed explanation of our approach is presented in the supplementary material S3. It is briefly described below.
The calculation of QNA descriptors is based on the connectivity matrix (C), and also, on the standard values of ionization potential (IP) and electron affinity (EA) of atoms in a molecule [15]. The main important feature of QNA descriptors is that they represent a molecule as a set of the P and Q values, or, in other words, as a “constellation” in a two-dimensional QNA descriptors’ space. The P and Q values can be considered as corresponding partial atomic hardness and electronegativity.
For any given atom ithe QNA descriptors are calculated as follows:
with .
The estimation of a target property for a chemical compound is calculated as a mean value of a function of the P and Q values for the atoms in a molecule in QNA descriptors space. We proposed the use of two-dimensional Chebyshev polynomials for approximating the function of P and Q values. So, the independent regression variables are calculated as average values of particular two-dimensional Chebyshev polynomials of P and Q values for the atoms in a molecule.
QNA descriptors and their polynomial transformations do not provide information on the shape and volume of a molecule. Since this type of information can be important for determining structure-activity relationships, these parameters were calculated separately and added to the variables already obtained from the Chebyshev polynomials. The topological length of the molecule is the maximal distance, calculated as the number of bonds between any two atoms (including hydrogen). The volume of a molecule is estimated as the sum of each atom’s volume.
The number of initial variables for QSAR modeling depends on the number of compounds in the training set and corresponds to the number of Chebyshev polynomials plus the number of the first, second and third power of the values of topological length and volume of a molecule. If the number of compounds in the training set varies from 100 to 2000, then the number of initial variables equals one-half of the number of compounds in the training set.
The GUSAR algorithm uses three randomly selected parameters to generate different QSAR models based on QNA descriptors: (a) calculation of the QNA descriptors for either all atoms or for only the atoms in a molecule with two or more immediate neighbors; (b) adjustment of the connectivity matrix coefficient (c) adjustment of the parameters of the Chebyshev polynomials. The detailed algorithm is described in the supplementary material S3. The final QSAR model is the consensus of several different QNA-based models built in this way.
QSAR modeling on the basis of biological activity profile prediction using MNA descriptors
GUSAR enables QSAR models to be derived based on predicted biological activity profiles of compounds. Each compound is represented as a list of MNA descriptors, which are used as input parameters [16–18] for predicting biological activity profiles in the PASS (Prediction of Activity Spectra for Substances) software program. The PASS algorithm is based on a Bayesian approach and is used to calculate this profile. A detailed description of the PASS algorithm is presented in the supplementary material S3.
GUSAR incorporates PASS version 10.1, which predicts 4130 types of biological activity with a mean prediction accuracy of about 95%. The list of predictable biological activities includes 501 pharmacotherapeutic effects, (e.g., Antihypertensive, Hepatoprotectant, Nootropic, etc.), 3295 mechanisms of action, (e.g., 5 Hydroxytryptamine antagonist, Acetylcholine M1 receptor agonist, Cyclooxygenase inhibitor, etc.), 57 adverse & toxic effects (e.g., Carcinogenic, Mutagenic, Hematotoxic, etc.), 199 metabolic terms (e.g., CYP1A inducer, CYP1A1 inhibitor, CYP3A4 substrate, etc.) 49 transporter proteins (e.g., P-glycoprotein 3 inhibitor, Nucleoside transporters inhibitors) and 29 activities related to gene expression (e.g., TH expression enhancer, TNF expression inhibitor, VEGF expression inhibitor). The results of a PASS prediction are given as a list of biological activities, for which the difference between the probability that a compound is active (Pa) and that it is inactive (Pi) is calculated.
To obtain different QSAR models, the Pa-Pi values for the activities, randomly selected from the total list of predicted biological activities, were input as independent variables for the regression analysis. Similar to the QSAR analysis with QNA descriptors, topological length and volume of molecules were added as variables to the biological activity profile; the number of initial variables for creating regression models was also selected depending on the number of compounds in the training set.
Self-consistent regression
GUSAR uses self-consistent regression (SCR) for building (Q)SAR models. SCR is based on the regularized least-squares method described in [15, 17]. Unlike stepwise regression and other methods of combinatorial search, the initial SCR model includes all regressors. The basic purpose of the SCR method is to remove the variables, which poorly describe the appropriate target value [15, 17]. The final number of variables in the QSAR equation, selected after the self-consistent regression procedure, is significantly less compared to the initial number of variables. Nevertheless, the final model contains a set of variables that correctly represents the existing relationship.
Nearest neighbor correction
It is well known that the use of both global and local models for non-congeneric sets improves the quality of QSAR models [23]. We used the experimental data on the three nearest neighbors (NN) to each input compound, to correct the prediction values obtained from the regression model. The correction value is estimated by taking an average of three values from the training set that are the most similar to the compound under prediction. The similarity of each pair of compounds is estimated as Pearson’s coefficient calculated in the space of the independent variables obtained after SCR. The mean experimental value obtained for the three nearest neighbor compounds from the training set is averaged with the predicted value of the test compound.
Applicability domain
The average similarity to the three nearest neighbor compounds in the training set was also used for the assessment of applicability domain (AD) of the model. If the average similarity exceeds the threshold, then the chemical compound under prediction is considered to be in the AD of the model and vice-versa. The higher the value selected for the threshold, the closer in similarity compounds must be to fall in the AD of the model. In this study a threshold for the AD equal to 0.7 was used.
Consensus modeling
The final predicted value for each end-point is estimated by including a weighted average of the predicted values from the set of QSAR models (for predictions that are within their respective applicability domains). The value obtained from each model is weighted by the similarity value calculated for the estimation of its applicability domain. This algorithm combines the results of QSAR modelling on the basis of QNA descriptors and on the basis of PASS-predicted biological activity profiles.
Interpretation of results
Typically, the affinity of pharmaceutical agent to the drug target should exceed the affinity to off-targets for at least one to two orders of magnitude. The medium affinity of current small molecule drugs to drug targets is about 16 nM, ranging from 16 mM to 1.6 pM [24]. Therefore, GUSAR prediction of interaction with antitarget(s) should be carefully considered in each individual case taking into account the predicted/measured affinity of the analyzed compound to the drug target. Particular attention should be paid to the compounds, for which predicted affinity of interaction with three or more antitargets exceeded 1 µM.
RESULTS AND DISCUSSION
QSAR modeling and validation of antitarget end-points
The initial data for each antitarget end-point was randomly divided into the training and external test sets in a ratio of 80% and 20%, respectively. External test sets were used for the assessment of predictivity in the obtained QSAR models. The number of compounds in the training and external test sets is shown in Table 5. Four antitarget end-points (Ki) include more than 1000 chemical compounds in the training set and more than 250 compounds in the external test set: 5-hydroxytryptamine 2A receptor antagonists, alpha1a adrenergic receptor antagonist, delta-type opioid receptor antagonists and mu-type opioid receptor antagonist. Thirty of the thirty two end-points (~94%) include more than 100 compounds in the training set. Therefore, only two training sets contain a small number of compounds: “sodium- and chloride-dependent GABA transporter 1 antagonist” with IC50 values and “amine oxidase [flavin-containing] A inhibitor” with Ki values (75 and 60 compounds, respectively).
Table 5.
Characteristics of QSAR models for the antitarget sets.
| Activity Name | End- point |
Number of compounds Training set / Test set |
Number of models |
R2 training set |
Q2 training set |
Average R2of test set during L10%Out |
R2 test set |
Coverage,% |
|---|---|---|---|---|---|---|---|---|
| 5-hydroxytryptamine 1B receptor antagonist |
IC50 | 297 / 74 | 8 | 0.83 | 0.79 | 0.63±0.02 | 0.67 | 100.0 |
| 5-hydroxytryptamine 1B receptor antagonist |
Ki | 266 / 66 | 7 | 0.73 | 0.66 | 0.55±0.06 | 0.72 | 100.0 |
| 5-hydroxytryptamine 2A receptor antagonist |
IC50 | 555/143 | 13 | 0.83 | 0.78 | 0.74±0.02 | 0.71 | 98.6 |
| 5-hydroxytryptamine 2A receptor antagonist |
Ki | 1010/252 | 13 | 0.72 | 0.65 | 0.58±0.05 | 0.59 | 99.6 |
| 5-hydroxytryptamine 2C receptor antagonist |
IC50 | 128/32 | 18 | 0.77 | 0.73 | 0.66±0.03 | 0.58 | 100.0 |
| 5-hydroxytryptamine 2C receptor antagonist |
Ki | 487/121 | 14 | 0.74 | 0.66 | 0.53±0.02 | 0.62 | 99.2 |
| alpha1a adrenergic receptor antagonist |
IC50 | 438/111 | 16 | 0.79 | 0.73 | 0.58±0.02 | 0.72 | 98.2 |
| alpha1a adrenergic receptor antagonist |
Ki | 1366/344 | 5 | 0.83 | 0.79 | 0.75±0.02 | 0.80 | 97.0 |
| alpha1b adrenergic receptor antagonist |
Ki | 410/102 | 17 | 0.73 | 0.66 | 0.54±0.03 | 0.63 | 100.0 |
| alpha-2A adrenergic receptor antagonist |
IC50 | 109/27 | 16 | 0.88 | 0.84 | 0.77±0.04 | 0.75 | 100.0 |
| alpha-2A adrenergic receptor antagonist |
Ki | 525/131 | 17 | 0.84 | 0.79 | 0.68±0.03 | 0.77 | 99.2 |
| amine oxidase [flavin- containing] A inhibitor |
IC50 | 286/71 | 9 | 0.80 | 0.75 | 0.58±0.02 | 0.72 | 100.0 |
| amine oxidase [flavin-containing] A inhibitor | Ki | 60/15 | 5 | 0.73 | 0.62 | 0.57±0.06 | 0.64 | 100.0 |
| androgen receptor antagonist |
IC50 | 116/29 | 8 | 0.79 | 0.73 | 0.68±0.06 | 0.67 | 100.0 |
| carbonic anhydrase II activator |
Kact | 104/26 | 20 | 0.92 | 0.90 | 0.76±0.04 | 0.91 | 100.0 |
| carbonic anhydrase I activator |
Kact | 108/27 | 12 | 0.98 | 0.97 | 0.92±0.01 | 0.93 | 100.0 |
| carbonic anhydrase I inhibitor |
Ki | 935/234 | 11 | 0.91 | 0.86 | 0.73±0.02 | 0.86 | 98.3 |
| carbonic anhydrase II inhibitor |
Ki | 866/217 | 7 | 0.87 | 0.79 | 0.74±0.04 | 0.76 | 98.6 |
| d(1A) dopamine receptor antagonist |
IC50 | 126/31 | 11 | 0.76 | 0.72 | 0.68±0.03 | 0.80 | 100.0 |
| d(1A) dopamine receptor antagonist |
Ki | 291/73 | 10 | 0.72 | 0.66 | 0.53±0.02 | 0.57 | 100.0 |
| d3 dopamine receptor antagonist |
Ki | 822 / 206 | 9 | 0.73 | 0.66 | 0.59±0.02 | 0.62 | 98.0 |
| delta-type opioid receptor antagonist |
Ki | 1044/261 | 16 | 0.75 | 0.70 | 0.60±0.04 | 0.65 | 98.5 |
| estrogen receptor antagonist | IC50 | 402 / 100 | 4 | 0.66 | 0.61 | 0.66±0.05 | 0.70 | 97.0 |
| estrogen receptor antagonist |
Ki | 255/68 | 13 | 0.76 | 0.71 | 0.66±0.04 | 0.70 | 100.0 |
| kappa-type opioid receptor antagonist |
Ki | 884/221 | 7 | 0.74 | 0.67 | 0.59±0.04 | 0.65 | 100.0 |
| mu-type opioid receptor antagonist |
IC50 | 545/136 | 7 | 0.67 | 0.61 | 0.60±0.03 | 0.70 | 97.8 |
| mu-type opioid receptor antagonist |
Ki | 1354/338 | 4 | 0.69 | 0.62 | 0.60±0.02 | 0.60 | 96.7 |
| sodium- and chloride- dependent GABA transporter 1 antagonist |
IC50 | 75/19 | 10 | 0.9 | 0.86 | 0.80±0.03 | 0.89 | 100.0 |
| sodium-dependent dopamine transporter antagonist |
IC50 | 920 / 230 | 5 | 0.7 | 0.65 | 0.65±0.04 | 0.67 | 98.3 |
| sodium-dependent dopamine transporter antagonist |
Ki | 655/164 | 7 | 0.77 | 0.69 | 0.59±0.04 | 0.64 | 100.0 |
| sodium-dependent serotonin transporter antagonist |
IC50 | 796 / 199 | 7 | 0.8 | 0.75 | 0.67±0.02 | 0.69 | 97.5 |
| sodium-dependent serotonin transporter antagonist |
Ki | 823 / 206 | 2 | 0.72 | 0.65 | 0.62±0.01 | 0.61 | 95.6 |
For each training set, forty models based on MNA descriptors and forty models based on QNA descriptors were created. A leave-10%-out cross-validation procedure was performed 20 times for each model, and the results were used to select the most predictive models. The average R2 value and the standard deviation of R2 values calculated for the test sets during the L-10%-out procedure are presented in Table 5. From the full set of 80 models, we selected only those models that satisfied the following conditions: a value of Q2 exceeding 0.6 and an R2 value from the leave-10%-out cross validation procedure exceeding 0.5. Thus, over 10 models were selected for sixteen of the end-points (50%) and only the “sodium-dependent serotonin transporter antagonist” activity was represented by two models. The selected models were used for consensus predictions on the external test set obtained for each antitarget end-point, taking into account the applicability domain of these models.
Table 5 shows the results of the consensus predictions on the external test sets for each antitarget endpoint. Sixteen test sets (50%) were predicted with 100% coverage, and the remaining test sets were predicted with coverage exceeding 95%. QSAR models obtained for twenty nine of the antitarget endpoints (91%) showed good statistical criteria for the external test sets. The accuracy of prediction for these end-points in external tests was in the range of 0.6–0.9. Thus, two activities (6%) were predicted with an accuracy higher than 0.9 – Kact of “carbonic anhydrase I activator” and “carbonic anhydrase II activator”. Plots of observed versus predicted values for these activities are shown in Figures 1 and 2. Six activities (19%) were predicted with an accuracy higher than 0.8 and sixteen (50%) with an accuracy higher than 0.7. Plots of observed versus predicted values for Ki of “alpha1a adrenergic receptor antagonist” and “carbonic anhydrase II inhibitor” are shown in Figures 3 and 4. Only three antitarget end-points – Ki of “5-hydroxytryptamine 2A receptor antagonist”, IC50 of “5-hydroxytryptamine 2C receptor antagonist” and Ki of “d(1A) dopamine receptor antagonist” showed not so high but reasonable values for accuracy exceeding 0.5.
Figure 1.

Carbonic anhydrase I activator test set, observed versus predicted Log10(Kact) values.
Figure 2.

Carbonic anhydrase II activator test set, observed versus predicted Log10(Kact) values.
Figure 3.

Alpha-1a adrenergic receptor antagonist test set, observed versus predicted Log10(Ki) values.
Figure 4.

Carbonic anhydrase I inhibitor test set, observed versus predicted Log10(Ki) values.
Some points in Figures 1–4 can be considered to be outliers; nevertheless, the number of outliers (~10–15) is negligible in comparison to the total number of points. In general, Figures 1–4 show that there are no any visible artefacts in the predicted values.
These results show that MNA and QNA descriptors with Self-Consistent Regression can be successfully used for the development of accurate and predictive QSAR models of antitarget effects.
On-line service for quantitative prediction of antitarget interaction profiles for chemical compounds
On the basis of our created QSAR models we have developed a freely available on-line service for the simultaneous prediction of thirty two antitarget end-points available at: http://www.pharmaexpert.ru/GUSAR/Antitargets/. It includes an on-line chemical editor (ChemAxon Marvin Sketch) [25] for drawing the studied structure. This service provides a reasonable computational speed (about 2 compounds per second for the simultaneous prediction of 32 antitarget end-points). The assessment of the prediction results for the 32 antitarget endpoints may done in the same way as an assessment for in vitro experimental assays. We consider that 1 µM (“6” in the units used on the website: -Log10(Value), mol/L) is a minimal cut-off value for any end-point. This means that a compound predicted to have an IC50 or Ki of less than 1 µM, is active against that undesirable target (antitarget). The significance of these predicted values depends on the therapeutic dose of the drug and the magnitude of the interaction between the drug and its target. However, the probability of adverse drug reactions may increase with the number of interactions with antitargets. For statistical reasons, any compound interacting with three or more antitargets may be considered potentially risky for further drug development and should be filtered out at the early stage of studies.
An example of prediction results for the antibiotic agent Temafloxacin (Omniflox) obtained with this service is represented in Figure 5. Temafloxacin was marketed in the United States from February to June 1992. During the three months of its use, the FDA received 50 reports of multiple side effects including three cases of death [6]. It was shown that Temafloxacin may produce hemolytic anemia and other blood cell abnormalities, kidney dysfunction and liver dysfunction [26]. Hemolytic anemia and blood cell abnormalities may be related to the inhibition of carbonic anhydrase. The cause of kidney dysfunction may be the interaction of Temafloxacin with the dopamine receptor and inhibition of the carbonic anhydrase. Liver dysfunction may be induced by action on the androgen receptor.
Figure 5.

The results of the antitarget interaction profile prediction for Temafloxacin using our on-line service.
Figure 5 shows that for these modeled antitarget end-points (carbonic anhydrase, dopamine and androgen receptors), Temafloxacin falls in the applicability domain and is predicted to be active (Ki and IC50 exceed 6 units in –log10(mol/L)). Thus, the GUSAR predictions correspond to the multiple side effects which were found during Temafloxacin usage in medical practice.
The web service also provides the total number of targets for which the input compound has been predicted to be active. This can be useful for selection and prioritization of compounds during the drug discovery process. A particular compound can be considered as a potential source of adverse drug reactions if interactions with three or more antitargets are predicted and exceed the cut-off value (1 µM). Compounds for which antitargets are not predicted can be selected for further development as potential drugs. In addition, the service can help medical chemists determine on which targets (molecular mechanism of toxicity) a particular compound should be tested experimentally, to avoid ADR.
We have applied the web-service to fourteen known drugs which have been withdrawn from the market, to estimate the number of antitargets they are predicted to interact with. In addition to the withdrawn drugs, seven currently marketed drugs were also analyzed to find out the difference in the number of antitargets. These prediction results are presented in Table 6. The results show that more than seven antitargets were predicted for each withdrawn drug, while less than three antitargets were predicted for each existing drug. Thus, our service can successfully be applied for the selection and prioritization of compounds during the drug discovery process.
Table 6.
Prediction results for withdrawn and marketed drugs.
| Drug Name | State | The number of predicted antitargets |
|---|---|---|
| Amineptine | withdrawn | 13 |
| Duract | withdrawn | 8 |
| Vioxx | withdrawn | 7 |
| Astemizole | withdrawn | 17 |
| Cerivastatin | withdrawn | 8 |
| Chlormezanone | withdrawn | 10 |
| Fenfluramine | withdrawn | 11 |
| Flosequinan | withdrawn | 11 |
| Glafenine | withdrawn | 14 |
| Grepafloxacin | withdrawn | 12 |
| Mibefradil | withdrawn | 16 |
| Rofecoxib | withdrawn | 7 |
| Troglitazone | withdrawn | 14 |
| Ximelagatran | withdrawn | 14 |
| Aspirin | marketed | 2 |
| Ibuprofen | marketed | 2 |
| Valtrex | marketed | 3 |
| Microzide | marketed | 3 |
| Neurontin | marketed | 3 |
| Enoxaparin | marketed | 2 |
| Lyrica | marketed | 2 |
CONCLUSIONS
We developed QSAR models for thirty two antitarget end-points based on MNA and QNA descriptors and self-consistent regression. These models showed good accuracy of prediction for 91% of the activities and high coverage of the external test sets for all end-points. Thus, the QSAR models could successfully be used for filtering out chemical compounds with a high probability of antitarget activity during the R&D process.
Our freely available on-line service for quantitative prediction of antitarget interaction profiles of chemical compounds is useful for researchers to increase the efficacy of finding drug-like leads with desirable pharmacological effects but without side effects and toxicity caused by interactions with antitargets.
Supplementary Material
ACKNOWLEDGEMENTS
The work is partially supported by RFBR grant No. 12-07-00597 (D.A.F., A.A.L.) and RFBR/NIH grant No. 12-04-91445/RUB1-31081-MO-12 (V.V.P.). Finally, two anonymous reviewers are thanked for many helpful suggestions to improve the manuscript.
ABBREVIATIONS LIST
- GUSAR
general unrestricted structure-activity relationships
- PASS
prediction of activity spectra for substances
- SCR
self-consistent regression
- QSAR
quantitative structure-activity relationships
- MNA
multilevel neighborhoods of atoms descriptors
- QNA
quantitative neighborhoods of atoms descriptors
Footnotes
ASSOCIATED CONTENT
Supporting Information
A detailed description of the QNA descriptors and the PASS algorithm, the chemical structures used in this work and an overlap analysis of the datasets are presented in the Supplementary Material. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
REFERENCES
- 1.European Federation of Pharmaceutical Industries and Associations. The Pharmaceutical Industry in Figures, EFPIA. 2010 [Google Scholar]
- 2.Czerepak E, Ryser S. Drug approvals and failures: implications for alliances. Nature reviews drug discovery. 2008;7:197–198. [Google Scholar]
- 3.Schuster D, Laggner C, Langer T. Why Drugs Fail – A Study on Side Effects in New Chemical Entities. In: Vaz RJ, Klabunde T, editors. Antitargets. KGaA, Wienheim: WILEY-VCH, Verlag GmbH & Co; 2008. pp. 3–22. [Google Scholar]
- 4.Larrey D, Berson A, Habersetzer F, Tinel M, Castot A, Babany G, Letteron P, Freneaux E, Loeper J, Dansette P, Pessyre D. Hepatology. 1989;10:168–173. doi: 10.1002/hep.1840100208. [DOI] [PubMed] [Google Scholar]
- 5.FDA Duract Volunterily Withdrawn. http://www.fda.gov/downloads/Safety/MedWatch/SafetyInformation/UCM189811.zip.
- 6.FDA. FDA News. FDA; 2004. FDA issues public health advisory on Vioxx as its manufacturer voluntary withdraws the product. [Google Scholar]
- 7.ELSEVIER. Meyler's Side Effects of Drugs Fourteenth Edition. An Encyclopedia of Adverse Reactions and Interactions. Hardbound: ELSEVIER; 2006. [Google Scholar]
- 8.Klabunde T, Evers A. Chembiochem: A European Journal of Chemical Biology. 2005;6:876–889. doi: 10.1002/cbic.200400369. [DOI] [PubMed] [Google Scholar]
- 9.Chen YZ, Ung CY. Journal of Molecular Graphics and Modelling. 2001;20:199–218. doi: 10.1016/s1093-3263(01)00109-7. [DOI] [PubMed] [Google Scholar]
- 10.Fanelli F, Menziani M, Carotti A, Benedetti PA, Benedetti De. Bioorganic and Medicinal Chemistry. 1994;2:195–211. doi: 10.1016/s0968-0896(00)82015-5. [DOI] [PubMed] [Google Scholar]
- 11.Lopez-Rodriguez M, Rosado M, Benhamu B, Morcillo M, Fernandez E, Schaper K. Journal of Medicinal Chemistry. 1997;40:1648–1656. doi: 10.1021/jm960744g. [DOI] [PubMed] [Google Scholar]
- 12.Vistoli G, Pedretti A, Villa L, Tesla B. Journal of Medicinal Chemistry. 2005;48:4947–4952. doi: 10.1021/jm0408969. [DOI] [PubMed] [Google Scholar]
- 13.Hegymegi-Barakonyi B, Eros D, Szántai-Kis C, Breza N, Bánhegyi P, Szabó G, Várkondi E, Peták I, Orfi L, Kéri G. Curr. Opin. Mol. Ther. 2009;11(3):308–321. [PubMed] [Google Scholar]
- 14.Matthews E, Frid A. Regul Toxicol Pharmacol. 2010;56(3):247–275. doi: 10.1016/j.yrtph.2009.11.006. [DOI] [PubMed] [Google Scholar]
- 15.Filimonov DA, Zakharov AV, Lagunin AA, Poroikov VV. SAR and QSAR Environ. Res. 2009;20(7–8):679–709. doi: 10.1080/10629360903438370. [DOI] [PubMed] [Google Scholar]
- 16.Lagunin AA, Zakharov AV, Filimonov DA, Poroikov VV. QSAR Modelling of Rat Acute Toxicity on the Basis of PASS Prediction. Molecular Informatics. 2011;30(2–3):241–250. doi: 10.1002/minf.201000151. [DOI] [PubMed] [Google Scholar]
- 17.Lagunin AA, Zakharov AV, Filimonov DA, Poroikov VV. SAR QSAR Environ Res. 2007;18(3–4):285–298. doi: 10.1080/10629360701304253. [DOI] [PubMed] [Google Scholar]
- 18.Sadym A, Lagunin A, Filimonov D, Poroikov V. SAR QSAR Env. Res. 2003;14:339–347. doi: 10.1080/10629360310001623935. [DOI] [PubMed] [Google Scholar]
- 19.Maurer I, Volz H. Arzneimittelforschung. 2001;51(10):785–792. doi: 10.1055/s-0031-1300116. [DOI] [PubMed] [Google Scholar]
- 20.Rang HP, Dale MM, Ritter JM. Pharmacology. 4th Edition. Churchill Livingstone: 1999. [Google Scholar]
- 21.Gutstein HB, Akil H. In: Goodman & Gilman's The Pharmacological Basis of Therapeutics Eleventh edition. Brunton LL, Lazo JS, Parker KL, editors. The McGraw-Hill Companies; 2006. pp. 547–590. [Google Scholar]
- 22.Maus M, Glowinski J, Premont J. J Neurochem. 2002;82(4):763–773. doi: 10.1046/j.1471-4159.2002.01011.x. [DOI] [PubMed] [Google Scholar]
- 23.Lei B, Xi L, Li J, Liu H, Yao X. Anal Chim Acta. 2009;644(1–2):17–24. doi: 10.1016/j.aca.2009.04.019. [DOI] [PubMed] [Google Scholar]
- 24.Overington JP, Al-Lazikani B, Hopkins AL. Nat. Rev. Drug Discov. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 25. http://www.chemaxon.com/marvin/help/index.html. [Google Scholar]
- 26.FDA. Omniflox recalled - antibiotic. FDA Consumer; 1992. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.