

Abstract
Species in the filamentous fungal genus Aspergillus display a wide diversity of lifestyles and are of great importance to humans. The decoding of genome sequences from a dozen species that vary widely in their degree of evolutionary affinity has galvanized studies of the function and evolution of the Aspergillus genome in clinical, industrial, and agricultural environments. Here, we synthesize recent key findings that shed light on the architecture of the Aspergillus genome, on the molecular foundations of the genus’ astounding dexterity and diversity in secondary metabolism, and on the genetic underpinnings of virulence in Aspergillus fumigatus, one of the most lethal fungal pathogens. Many of these insights dramatically expand our knowledge of fungal and microbial eukaryote genome evolution and function and argue that Aspergillus constitutes a superb model clade for the study of functional and comparative genomics.
Keywords: sexual reproduction, pathogenesis, A. fumigatus, development, genome structure, horizontal gene transfer, genetic structure
Aspergillus: the ‘Dr. Jekyll and Mr. Hyde’ genus of fungi
There is probably no genus better suited than Aspergillus, an important and efficient saprophytic genus found in diverse environments, to illustrate how inextricably intertwined fungi are with human affairs. Its Mr. Hyde personality is exemplified by species such as Aspergillus fumigatus, responsible for the highest number of deaths from fungi and the second highest number of human infections from fungi [1]; Aspergillus flavus, the opportunistic but very destructive agricultural pest that contaminates several crops with the potent carcinogen aflatoxin, causing major crop yield losses and a few deaths per year [2]; or Aspergillus sydowii, the opportunistic pathogen of Caribbean gorgonian coral communities, whose recent outbreak of infection threatens the collapse of this fragile ecosystem [3]. In contrast, no species better illustrate its Dr. Jekyll side than Aspergillus niger, a biotechnological ‘cell factory’ widely used in the food industry [4, 5], Aspergillus nidulans, an important model for eukaryotic genetics and cell biology [6], or the several Aspergillus species that drive production of beverages and sauces in the Far East; among others, Aspergillus oryzae is used in the making of sake [7], Aspergillus sojae of soy sauce [8], and Aspergillus kawachii in the brewing of the spirit shochu [9].
First described nearly three hundred years ago by the priest and botanist Antonio Micheli, Aspergillus got its name from the resemblance of its asexual spore-forming structure to the aspergillum, an instrument used to disperse holy water in some Christian liturgical services. Aspergillus is thus the name that describes the asexual cycle of the fungus. Because the phenotypic diversity of the sexual fruiting bodies is greater, ten different genera describe the sexual cycles of Aspergillus species [10] (Figure 1). For example, A. nidulans and A. fumigatus describe the asexual cycles of these species, whereas Emericella nidulans and Neosartorya fumigata their sexual counterparts. Most commonly, species in the genus are referred to as Aspergillus species, which is quite practical given that only a third of Aspergillus species known to have a sexual cycle [10]. In their classic 1965 treatise on the genus, Raper and Fennell recognized 132 species [11], but the systematic application of a polyphasic approach that uses morphological, physiological, and molecular data to identify and classify new species, including several cryptic ones, has resulted in the present circumscription of more than 250 species [12]. The pace of discovery of new species continues unabated with approximately 50 new species having been described this century [12]. This genomics-enabled systematic revision of the Aspergillus taxonomy has dramatically influenced the design and application of molecular techniques to identify medically important Aspergillus [13], but also aided the identification of new clinically relevant species [14].
Figure 1. Evolutionary relationship, relevance and content of available and in progress Aspergillus genomes.
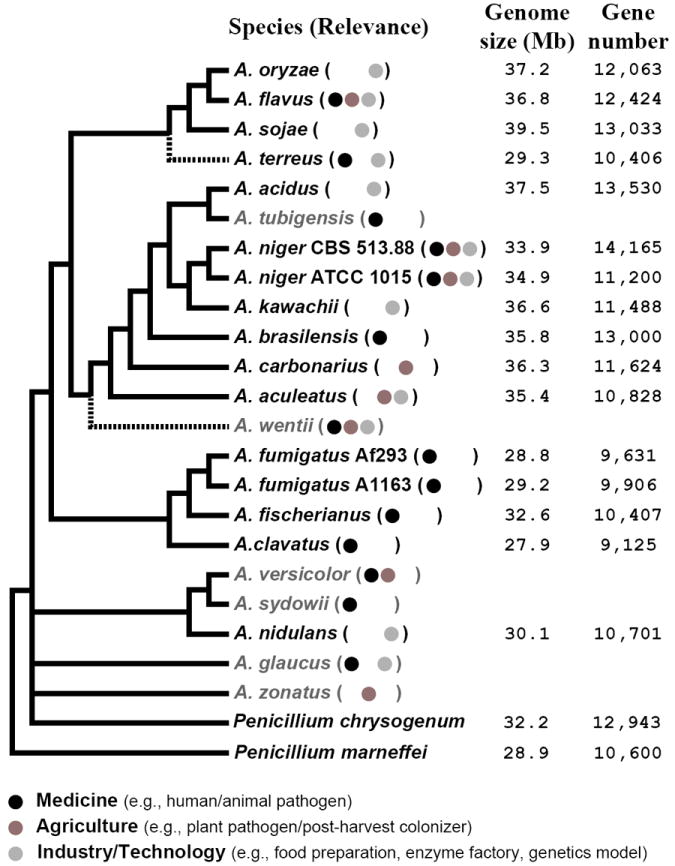
The phylogeny of Aspergillus genomes was synthesized from the phylogenies described by Houbraken and Samson [37], Geiser and co-workers [35], Peterson [36], and Rokas and Galagan [6]. Dotted line branches indicate uncertainty about their placement on the phylogeny. Genome size and gene number data are from the published genome analyses [4, 8, 9, 15-18, 42, 94]; for unpublished genomes, these values were obtained from the JGI genome portal [41] (A. acidus, A. aculeatus, A. brasiliensis, A. carbonarius, and A. tubigensis) or the Broad Institute’s Aspergillus Comparative Database (A. flavus and A. terreus).
The early decoding of the genomes from some of the species currently available [4, 15-18], their widely varying degree of evolutionary affinity [6, 18, 19], as well as the advent of novel molecular [e.g., 20, 21, 22] and computational [e.g., 23, 24-26] tools, have dramatically accelerated Aspergillus ‘-omics’ research and the pace of discovery in genome-wide functional and evolutionary studies [e.g., 7, 27, 28-33]. Here, we describe the current status of genomics research on Aspergillus and synthesize recent key findings in three key areas, namely genome architecture, secondary metabolism and virulence, that not only dramatically expand our understanding of the function and evolution of the Aspergillus genome, but also argue that Aspergillus represents a model clade for the study of eukaryote comparative functional genomics.
A cornucopia of genomes and lifestyles
With the genomes from 14 species already publicly available (Figure 1), Aspergillus is the most genome sequenced-rich fungal genus, surpassing even the genome sequenced-rich Saccharomyces and Candida yeasts [34]. Aspergillus is likely to continue holding on to this distinction because the U. S. Department of Energy Joint Genome Institute (JGI), as part of its 2011 community sequencing proposal mechanism, approved a multi-investigator proposal and is currently sequencing the genomes of eight additional species (Figure 1), including the coral pathogen A. sydowii and the xerophile Aspergillus glaucus. The panel of currently available genomes includes a good sample of the diversity of the fungi comprising Aspergillus; the model organism A. nidulans, the ‘cell factory’ A. niger, the human pathogens A. fumigatus and Aspergillus terreus, the human pathogen and agricultural pest A. flavus, as well as the fermenters A. oryzae, A. sojae and A. kawachii. Importantly, there appears to be no association between lifestyle and evolutionary affinity. For example, A. oryzae is a domesticated ecotype of A. flavus and their genomes share 99.5% identity, yet the first is used in the making of several traditional Far Eastern sauces and beverages and has a Generally Regarded as Safe label by the United States Department of Agriculture, whereas the second is a destructive agricultural pest and potent mycotoxin producer [7, 19]. Similarly, the top three most common human pathogens, A. fumigatus, A. flavus and A. terreus, do not group together in the Aspergillus family tree and all possess relatives that rarely, if ever, infect humans (Figure 1) [35-37]. This lack of association between lifestyle and evolutionary affinity is probably because many of the traits render fungi into potent pathogens, agricultural pests, or cell factories are likely features that are generally associated with the saprophytic lifestyle and selected for survival in conditions independent of their current roles in pathogenesis, pestilence, or biotechnology.
Although no database contains all 14 available Aspergillus genomes, most are available from several, including AspGD [25], FungiDB [38], CADRE [39], and the Aspergillus Comparative Database (http://www.broadinstitute.org/annotation/genome/aspergillus_group/), with the rest available from GenBank [40] or the JGI genome portal [41]. Unfortunately, the annotations of some species, such as A. sojae [8], have not yet been made publicly available effectively stymieing easy access to some of the data for inclusion in ‘-omics’ studies. Furthermore, the gene models for the 14 currently available genomes have been constructed using several different algorithms and different standards of analysis, which is problematic because the process of whole genome annotation is highly sensitive to the algorithms and assumptions used in constructing these gene models. Take, for example, the genomes of two A. niger isolates; isolate CBS 513.88 is reported to contain 14,165 genes and isolate ATCC 1015 to contain 11,200 genes, but further analysis suggests that less than one third of the ~3,000 gene differential between the two annotations is real [42]. This lack of consistency and uniformity in the annotation of Aspergillus genomes significantly reduces the utility and value of the data. For gene-centered studies, elucidation of whether the annotation differences observed at a particular locus across genomes are real is non-trivial, whereas for genome-wide studies, gene number overestimation or underestimation makes studies that fundamentally rely on accurate gene counts, such as examination of gene family evolution or of genome size differences, vulnerable to annotation bias.
Although the availability and quality of Aspergillus genome data is a long-standing problem that is unlikely to disappear soon, the advent of next-generation sequencing technologies (NGSTs) have ameliorated another problem [21], namely the ability to generate genomic or other high-throughput sequencing data from any Aspergillus species. Besides the use of NGSTs to sequence the genomes of some of the species shown in Figure 1, such as A. kawachii [9] and A. sojae [8], NGSTs have been used to sequence the genomes of additional isolates from already sequenced species [7], whereas NGST applications such as RNA-Seq [43], have been employed to characterize the structure and variation of the Aspergillus transcriptome [7, 27, 30, 44, 45]. For example, in the most thorough application of these technologies in the genus Aspergillus to date, Gibbons and co-workers sequenced the genomes of seven A. oryzae and seven A. flavus isolates as well as three of the transcriptomes of each species [7].
The content and genetic structure of the Aspergillus genome
The genus Aspergillus is characterized by remarkable genome sequence diversity; using proteome divergence as a yardstick, Aspergillus is as diverse as our own phylum, the Vertebrates, whereas the ‘very close’ relatives A. fumigatus and Aspergillus fischerianus are as divergent as humans and mice [6, 18, 19]. In contrast to several other fungal lineages where genome structure within and between species is quite plastic [46, 47], and contrary to what would be expected based on the degree of sequence diversity in the genus, the structure of the Aspergillus genomes appears rather stable (Figure 1). All Aspergillus genomes sequenced so far have eight chromosomes, ranging in size between 28-40 Mb, and appear to have similar characteristics, although karyotype analyses suggest that natural populations of several of these species harbor chromosomal variants [see Ref. 48 and references therein].
The apparent lack of genome plasticity does not mean that the Aspergillus genome is devoid of conundrums. One question that has attracted considerable interest is why the genomes of species like A. oryzae and A. flavus are ~20% bigger and substantially more gene rich than those of A. nidulans and A. fumigatus. Several potential explanations could account for the difference, including genome duplication, segmental duplication, as well as massive horizontal gene transfer (HGT). However, an in-depth comparison of the A. oryzae, A. nidulans and A. flavus genomes by Khaldi and coworkers did not find support for any of these explanations [49], suggesting that several different mechanisms, each acting in a piecemeal fashion, likely account for the difference. Nevertheless, both this study as well as a few others have provided several examples indicating that the Aspergillus genome has been sculpted by HGT (Figure 2), serving both as the donor lineage [42, 50] as well as the recipient [42, 49, 51-54].
Figure 2. The genomic and functional footprint of horizontal gene transfer to and from Aspergillus.
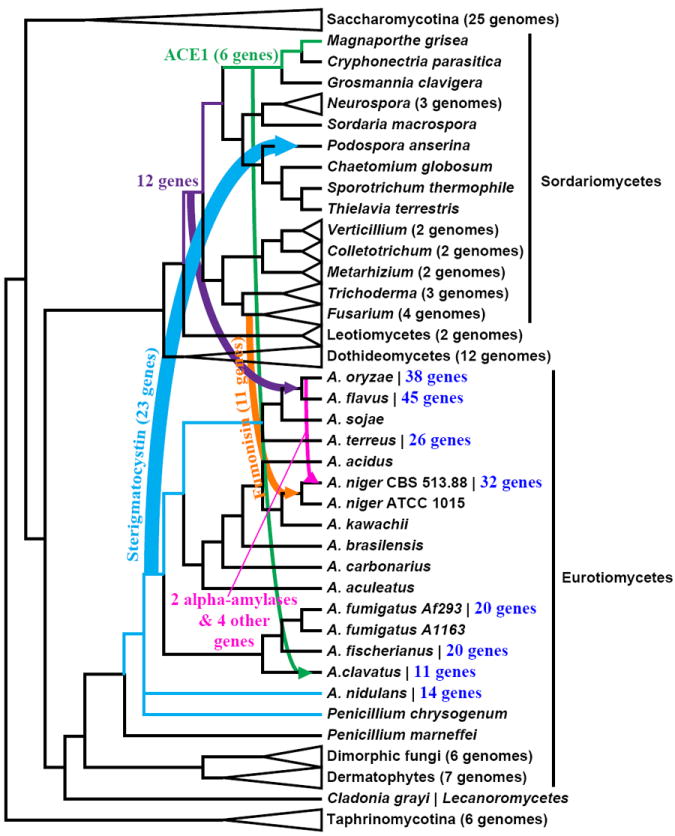
The fungal species phylogeny was synthesized from the literature [6, 35, 36, 95, 96]. Arrows illustrate key examples of horizontal gene transfer (HGT) of single genes as well as gene clusters and the direction of transfer, when known, between fungi [42, 49-51, 53]; arrow colors correspond to different HGT events, whereas arrow thickness corresponds to the number of genes transferred. The numbers of Aspergillus genes inferred to have been acquired via HGT from prokaryotes [52], when known, are shown in blue color font next to each species name. Concept in figure adapted from [97].
Early analysis of the Aspergillus genome focused considerably on whether all species have a sexual cycle [55], greatly contributing to what has been termed the ‘fungal sexual revolution’ [56]. This revolution encompasses not only the demonstration of sex in a few, previously thought to be asexual, species [57-59], but also the realization that experiments, such as the ability of the mating genes to regulate expression of downstream genes in a mating type-specific manner [60], suggest that most, if not all, asexual fungal species have cryptic sexual cycles yet to be discovered. In A. fumigatus, perhaps the most celebrated case of fungal sex cycle discovery [59], mixing and matching of pairs has revealed considerable variation in fertility [61], opening the door to understanding why the sexual cycle has been so elusive.
Another major, and perhaps more complex, question whose investigation has been dramatically enhanced by the availability of genomes is whether Aspergillus populations are genetically differentiated, and the implication of population structure for their lifestyles. Global surveys from a variety of species show lack of genetic differentiation [3, 62, 63]; however, the presence of distinct lineages in phylogenetic analyses of such cosmopolitan species, which are usually interpreted to represent cryptic species [64], could also be interpreted as evidence for the existence of genetically distinct populations within species. Nevertheless, local examinations often identify considerable levels of differentiation and the existence of genetically distinct populations. For example, despite the lack of differentiation of A. fumigatus isolates across the globe [62, 64], a recent analysis of 255 Dutch isolates using data from 20 molecular markers identified five distinct populations [65]. Interestingly, all multidrug-resistant isolates nest within a single, predominantly asexual, population, suggesting that both genetic differentiation and reproductive mode are in uencing the dynamics of drug resistance patterns in natural A. fumigatus populations [65]. Similarly, the apparent lack of structure in A. flavus isolates from around the globe [63], contrasts with the existence and long-term (~10,000 years) maintenance of three genetically distinct sympatric populations [66]. Intriguingly, Olarte and co-workers recently showed that crosses of isolates from distinct A. flavus populations not only interbreed in the laboratory, but also recombine and convert nonaflatoxigenic isolates into aflatoxin-producing ones [67]. Whether and how these lab-based findings impact the efficacy of nonaflatoxigenic biocontrol strains aimed to competitively exclude their aflatoxin-producing relatives in the field is a major, yet unanswered, riddle [68].
Secondary metabolites: the drugs behind the lifestyles
The cholesterol-reducing drug lovastatin, the antibiotic penicillin, as well as the potent mycotoxins aflatoxin and gliotoxin are just a tiny sample of the pharmacopoeia encoded in and manufactured by the Aspergillus genome. One is tempted to think that this bewildering diversity of small organic molecules, also known as secondary metabolites (SMs), is a direct byproduct of the enormous genomic diversity of Aspergillus. Remarkably, the levels of variation in both the number and the identity of SM pathways, which most often are physically linked or clustered on the chromosome, far exceed those observed in the rest of the genome. For example, although A. fumigatus, A. fischerianus and Aspergillus clavatus share ~80% of their genes [18, 69], only 30% of their SM genes are conserved across all three species [18].
The presence of SM gene clusters is highly variable even within species. Genome-wide comparison of two A. fumigatus isolates identified a putative SM gene cluster that was uniquely present in only one of the isolates [18], whereas a similar analysis between two A. niger isolates revealed unique putative SM gene clusters in both of their genomes [42]. Furthermore, a recent population genomic survey of A. flavus and A. oryzae lead to the discovery of a SM locus occupied by two different gene clusters. The two gene clusters differ in gene number, gene content and evolutionary history, with A. flavus isolates being polymorphic for the two gene cluster ‘alleles’ [7]. Although much less in known about SM production, the available data suggests that the dramatic variation observed within and between Aspergillus species extends beyond differences in genome sequence; for example, a comparison of small molecule chemistry between A. oryzae and A. flavus, two species that are ~99.5% identical at the nucleotide level, suggests that they have quite dissimilar metabolite profiles [70].
Commonalities in the genome architecture of SM gene clusters, such as the presence of an essential biosynthetic ‘backbone’ gene as well as ‘decorating’ ones that encode for proteins involved in modification, transport and regulation of SM, have led to the recent development of powerful computational algorithms that predict SM gene clusters in genomes [23, 71]. Application of these algorithms on Aspergillus genomes has identified surprisingly large numbers of putative SM gene clusters; for example, although only the gene clusters responsible for the synthesis of aflatoxin, cyclopiazonic acid and aflatrem were previously known to be produced by A. flavus, computational analysis of its genome predicts the presence of 55 putative SM gene clusters [23, 72].
Another characteristic of SM gene clusters that is useful in predicting novel clusters is that the expression of their genes is regulated in a coordinated fashion. For instance, comparison of the expression patterns of A. nidulans strains whose laeA gene copy, a global regulator of SM as well as of numerous non-SM genes [73], was either knocked out or overexpressed identified a novel five-gene SM cluster that is responsible for the synthesis of terrequinone, a class of compounds for which no gene cluster had been previously described [74]. Extending this strategy, a recent examination of the transcriptome profile of A. fumigatus grown in two different growth conditions showed that changes in gene expression were not randomly distributed across the genome; rather, they tended to reside within genomic neighborhoods largely composed of gene sets containing many of the hallmarks characteristic of SM gene clusters [27]. Given the importance of SMs to humans, the emerging consensus that there are many more putative SM gene clusters than previously indicated is perhaps one of the most significant, and potentially far-reaching, discoveries of the decade-long exploration of the Aspergillus genome.
The advent of Aspergillus genomes has also augmented studies on understanding the regulation of SM gene clusters. One of the most interesting recent developments is the discovery that the velvet family of proteins, together with the global regulator laeA [73], form a complex that links and coordinates SM production with morphological differentiation [75, 76], which is in turn activated when a highly conserved signal transduction module receives the appropriate external environmental signals (Figure 3) [77]. This coupling of SM with development presumably evolved because the protection offered by the deposition of SMs into the spores is vital to propagation [76]. In line with this hypothesis, A. nidulans mutants deficient in the production of SMs are less toxic to their insect predators than the wild-type [78]. However, SMs are not only important in predator avoidance; evidence that certain Aspergillus SM gene clusters are activated only when physically interacting with other microbes [79], that SMs provide a competitive advantage [80], as well as the discovery of self-protection genes nested within others [81], suggest that SMs are also likely to be critical in interactions between Aspergillus and other microbes.
Figure 3. A model of the genetic mechanisms underpinning the coordination of secondary metabolism and development in Aspergillus.
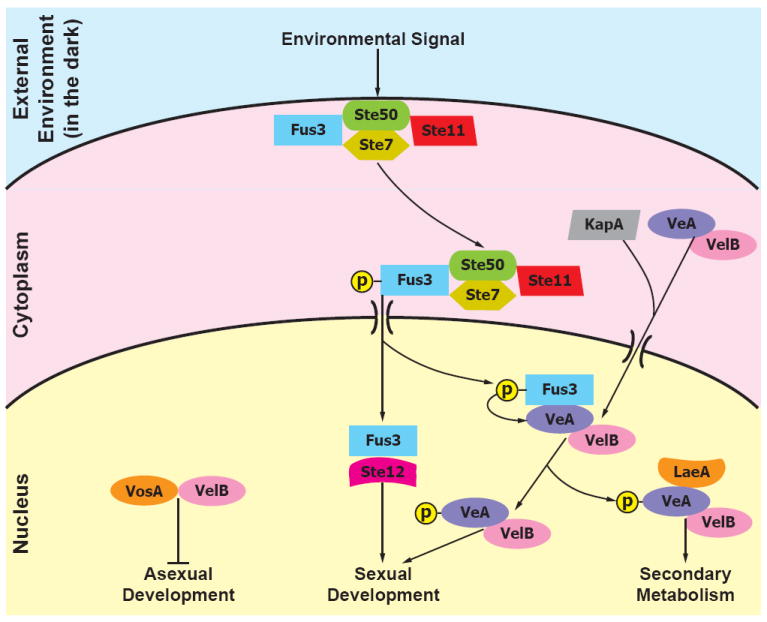
Upon receiving the appropriate environmental signals (e.g., darkness), the Ste50–Ste11–Ste7–Fus3 complex leaves the internal side of cell membrane where it is attached partly through the action of the Ste50 protein and migrates to the nuclear envelope. There Fus3 is phosphorylated and enters the nucleus where it interacts with Ste12, forming a complex that is necessary for sexual development, as well as phosphorylates the VeA protein, which is also transferred to the nucleus as part of the VeA–VelB dimer through the aid of the α-importin KapA in dark conditions. Once in the nucleus, the VelB–VeA dimer activates sexual development as well as interacts with the global regulator LaeA to activate secondary metabolism. VelB also interacts with VosA, also a member of the velvet protein family together with VeA and VelB, to repress asexual development [76, 77].
Additional support for the hypothesis that SMs are critical components of fungal–microbial interactions comes from a recent study aimed at identifying the molecular signature of domestication in A. oryzae [7], one of the two fungi used in the making of sake in the last few millennia in the Far East. During sake making, A. oryzae is responsible for breaking down rice starch into simpler sugars, a process that occurs, to a large degree, in parallel with the conversion of sugars to alcohol by the brewer’s yeast Saccharomyces cerevisiae. In contrast to its wild relative A. flavus, the entire SM profile of A. oryzae is dramatically downregulated when grown on rice, including the gene clusters responsible for the synthesis of the mycotoxins aflatoxin and cyclopiazonic acid [7]. Because aflatoxin, and presumably other SMs as well, is genotoxic to S. cerevisiae [82] and its presence during fermentation would affect yeast survival and, consequently, sake making, the domestication process may have converted A. oryzae into a microbe that is ‘friendly’ to its other microbial co-inhabitants.
The inner workings of a pathogen
More than two dozen Aspergillus species are capable of causing opportunistic infections in humans with compromised immune systems, which are collectively known as aspergillosis [14, 83]. Aspergillosis is typically acquired following inhalation of the very small, aerially dispersed, asexual spores produced by Aspergillus fungi [84]. Aspergillosis covers a remarkably wide spectrum of diseases that encompasses chronic diseases, such as chronic fibrosing aspergillosis, which occurs in individuals with normal immune systems but who have preexisting structure lung diseases (e.g., tuberculosis), rapidly progressing and acute infections of immunocompromised individuals, such as acute invasive aspergillosis, as well as allergies, such as sinusitis (Figure 4) [84]. This diversity of host-pathogen interactions provides a potentially extremely fertile ground for high-throughput comparative studies that simultaneously examine the in vivo transcriptome or proteome profiles of pathogen and host.
Figure 4. Aspergillosis encompasses a diverse spectrum of diseases due to interactions between Aspergillus and the human host.
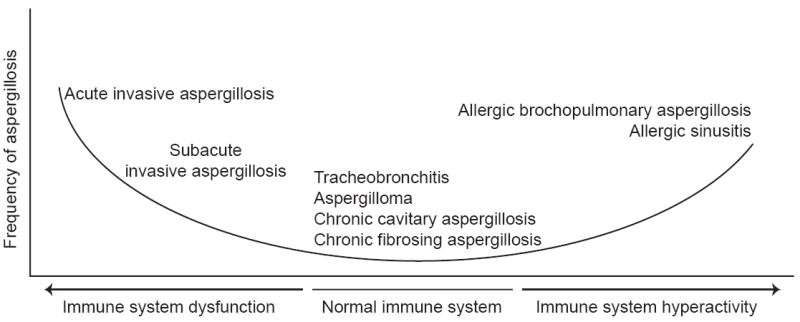
In compromised immune systems, the risk of developing invasive aspergillosis positively correlates with dysfunction. In contrast, the risk of allergic aspergillosis increases in individuals with hyperactive immune systems. The figure was modified and reproduced from a concept developed by David Denning, with his permission.
By far the leading cause of aspergillosis is A. fumigatus, the fungal opportunistic pathogen with the biggest impact on human pathogenesis; it is responsible for the highest number of deaths and for the second highest number of infections, behind only Candida albicans [83]. This dominance of A. fumigatus is likely due to ecological traits, such as the high prevalence and buoyancy of its spores in the environment [85], as well as genetic ones, such as the ability to grow well at 37°C and the coating of its spores with a hydrophobin that renders them immunologically inert [86]. In the aftermath of the decoding of the A. fumigatus genome and its close relatives [17, 18], a number of studies have greatly increased our understanding of its genetic makeup and how it interacts with the human host to produce a wide range of aspergillosis diseases.
A broad comparison of A. fumigatus with the rarely pathogenic A. fischerianus and A. clavatus reveals extensive conservation [18]. For example, all of the 45 known and predicted A. fumigatus allergens appear to be conserved across all Aspergillus genomes, suggesting that differences in gene content between species are unlikely to explain why A. fumigatus is a major contributor to diseases such as allergic bronchopulmonary aspergillosis. Nevertheless, patterns of allergen gene expression in response to oxidative stress may differ between species [87]. Despite the broad conservation of gene content, approximately 8.5% of genes appear to be present only in A. fumigatus, and lacking or absent from A. fischerianus and A. clavatus [18].
One of the most striking findings of the analysis of A. fumigatus lineage-specific genes is that they are much smaller than genes that are conserved across Aspergillus [18]. Although small proteins have received a great deal of attention in the study of fungal plant pathogens [88], understanding their involvement in A. fumigatus pathogenesis, remains an important, yet virtually unexplored topic. Part of the reason for this neglect may be because genome annotation pipelines often exclude small gene models due to lack of independent evidence supporting their validity. For example, the original annotation of the A. fumigatus Af293 genome did not include transcripts smaller than 150 base pairs [17], whereas the current annotation excluded only species-specific transcripts that fall below the same length threshold [18]. However, recent work suggests that small transcripts and proteins may play important roles in the infection process. For example, during conidial dormancy, the first stage of infection at which spores are inhaled and adhere to host tissue [89], the A. fumigatus proteome profile is dominated by small, lineage specific proteins whose function is mostly unknown [90]. Transcriptome studies in the same species also reveal an abundance of small transcripts; although some of these transcripts have significant similarity to sequences from other species most of them appear to be uniquely present in A. fumigatus [43]. Similarly, sequencing of non-coding transcripts shorter than 500 base pairs in A. fumigatus identified a few dozen non-coding RNAs, several of which appear to also be developmentally regulated [91]. In the next few years, aided by RNA-Seq [43] and high-throughput proteomics approaches [22], we predict that the identification and functional characterization of these small transcripts and proteins will become an indispensable part of the study of Aspergillus development and pathogenicity.
Notwithstanding our knowledge gap on small transcripts and proteins, a considerable amount of effort has been devoted on obtaining the transcriptome and proteome profile of in vitro and in vivo aspergillosis models [27, 28, 32]. For example, two recent studies describe the transcriptome and proteome profile of an in vitro model of the A. fumigatus biofilm [27, 28], the dense network of hyphae embedded in an extracellular matrix made up of proteins, monosaccharides, polysaccharides and secondary metabolites that A. fumigatus grows as when in aerial colony conditions [92, 93]. In agreement with the morphology and presumed function of the biofilm, these studies revealed extensive upregulation of gene sets encoding for structural and adhesive components of the extracellular matrix, for drug resistance, as well as for SM [27, 28].
Remarkably, transcriptome analysis of an in vivo murine model of invasive aspergillosis infection [32] shared several commonalities with the biofilm studies, including upregulation of SM gene clusters. For example, pseurotin, a SM produced by the fumitremorgin gene cluster, was upregulated both in vivo [32] and in vitro [27, 28]. Finally, examination of the physical location of differentially regulated genes along the A. fumigatus chromosomes suggests that they are not randomly distributed. In vitro [27] and in vivo [32] studies argue that genes located near chromosome ends are much more likely to be upregulated during biofilm growth and infection, arguing that the increased diversity in genome structure near A. fumigatus telomeres may be key to its pathobiology.
Concluding remarks
Less than a decade after the release of the first publicly available Aspergillus genomes, the study of the function and evolution of the genome of this broadly important genus has already begun yielding remarkable novel insights into genome architecture [18, 42, 50], sexual reproduction [56, 59, 61], population biology [7, 65-67], secondary metabolism and development [76], and virulence mechanisms [32, 86]. The emerging synergy between the substantial and ever-expanding bodies of knowledge on Aspergillus genomics, natural history, systematics, molecular genetics and development, natural products chemistry, human, animal and plant disease, and biotechnology, coupled with the remarkable phenotypic versatility present in the genus make it ideal for addressing fundamental questions across several levels of biological organization. However, several novel challenges and big questions remain unaddressed. Understanding why the sexual cycle is cryptic in so many species, what are the molecular mechanisms that drive the production of such diverse chemistry, or what is the Achilles’ heel(s) of A. fumigatus during human infections, are just a small sample of major questions that await answers. Antonio Micheli would have been happy to know that study of the organism he discovered nearly three centuries ago continues to sprinkle us with new insights.
Acknowledgments
We thank David Denning for allowing us to develop and reproduce a modified version of Figure 4. J.G.G. was funded by the Graduate Program in Biological Sciences at Vanderbilt University and the National Institute of Allergy and Infectious Diseases, National Institutes of Health (NIH, NIAID: F31AI091343-01). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIAID or the NIH. Research in A.R.’s lab is supported by the Searle Scholars Program and the National Science Foundation (DEB-0844968).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pfaller MA, et al. Invasive fungal pathogens: Current epidemiological trends. Clin Infect Dis. 2006;43:S3–S14. [Google Scholar]
- 2.Amaike S, Keller NP. Aspergillus flavus Annu Rev Phytopathol. 2011;49:107–133. doi: 10.1146/annurev-phyto-072910-095221. [DOI] [PubMed] [Google Scholar]
- 3.Rypien KL, et al. Globally panmictic population structure in the opportunistic fungal pathogen Aspergillus sydowii. Mol Ecol. 2008;17:4068–4078. doi: 10.1111/j.1365-294X.2008.03894.x. [DOI] [PubMed] [Google Scholar]
- 4.Pel HJ, et al. Genome sequencing and analysis of the versatile cell factory Aspergillus niger CBS 513.88. Nat Biotechnol. 2007;25:221–231. doi: 10.1038/nbt1282. [DOI] [PubMed] [Google Scholar]
- 5.Machida M, et al. Genomics of Aspergillus oryzae: learning from the history of Koji mold and exploration of its future. DNA Res. 2008;15:173–183. doi: 10.1093/dnares/dsn020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rokas A, Galagan JE. The Aspergillus nidulans genome and a comparative analysis of genome evolution in Aspergillus. In: Goldman GH, Osmani SA, editors. The Aspergilli: Genomics, Medical Applications, Biotechnology, and Research Methods. CRC Press; 2008. pp. 43–55. [Google Scholar]
- 7.Gibbons JG, et al. The evolutionary imprint of domestication on genome variation and function of the filamentous fungus Aspergillus oryzae. Curr Biol. 2012;22:1403–1409. doi: 10.1016/j.cub.2012.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sato A, et al. Draft genome sequencing and comparative analysis of Aspergillus sojae NBRC4239. DNA Res. 2011;18:165–176. doi: 10.1093/dnares/dsr009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Futagami T, et al. Genome sequence of the white koji mold Aspergillus kawachii IFO 4308, used for brewing the Japanese distilled spirit shochu. Eukaryot Cell. 2011;10:1586–1587. doi: 10.1128/EC.05224-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Geiser DM. Sexual structures in Aspergillus: morphology, importance and genomics. Med Mycol. 2009;47(Suppl 1):S21–26. doi: 10.1080/13693780802139859. [DOI] [PubMed] [Google Scholar]
- 11.Raper KB, Fennell DI. The Genus Aspergillus. Williams & Wilkins; 1965. [Google Scholar]
- 12.Geiser DM, et al. The current status of species recognition and identification in Aspergillus. Stud Mycol. 2007;59:1–10. doi: 10.3114/sim.2007.59.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klaassen CHW, Osherov N. Aspergillus strain typing in the genomics era. Stud Mycol. 2007:47–51. doi: 10.3114/sim.2007.59.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Balajee SA, et al. Molecular identification of Aspergillus species collected for the Transplant-Associated Infection Surveillance Network. J Clin Microbiol. 2009;47:3138–3141. doi: 10.1128/JCM.01070-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galagan JE, et al. Sequencing of Aspergillus nidulans and comparative analysis with A. fumigatus and A. oryzae. Nature. 2005;438:1105–1115. doi: 10.1038/nature04341. [DOI] [PubMed] [Google Scholar]
- 16.Machida M, et al. Genome sequencing and analysis of Aspergillus oryzae. Nature. 2005;438:1157–1161. doi: 10.1038/nature04300. [DOI] [PubMed] [Google Scholar]
- 17.Nierman WC, et al. Genomic sequence of the pathogenic and allergenic filamentous fungus Aspergillus fumigatus. Nature. 2005;438:1151–1156. doi: 10.1038/nature04332. [DOI] [PubMed] [Google Scholar]
- 18.Fedorova ND, et al. Genomic islands in the pathogenic filamentous fungus Aspergillus fumigatus. PLoS Genet. 2008;4:e1000046. doi: 10.1371/journal.pgen.1000046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rokas A, et al. What can comparative genomics tell us about species concepts in the genus Aspergillus? Stud Mycol. 2007;59:11–17. doi: 10.3114/sim.2007.59.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuck U, Hoff B. New tools for the genetic manipulation of filamentous fungi. Appl Microbiol Biotechnol. 2010;86:51–62. doi: 10.1007/s00253-009-2416-7. [DOI] [PubMed] [Google Scholar]
- 21.Rokas A, Abbot P. Harnessing genomics for evolutionary insights. Trends Ecol Evol. 2009;24:192–200. doi: 10.1016/j.tree.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 22.Doyle S. Fungal proteomics: from identification to function. FEMS Microbiol Lett. 2011;321:1–9. doi: 10.1111/j.1574-6968.2011.02292.x. [DOI] [PubMed] [Google Scholar]
- 23.Khaldi N, et al. SMURF: Genomic mapping of fungal secondary metabolite clusters. Fungal Genet Biol. 2010;47:736–741. doi: 10.1016/j.fgb.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tuckwell D, et al. A public resource for metabolic pathway mapping of Aspergillus fumigatus Af293. Med Mycol. 2011;49(Suppl 1):S114–119. doi: 10.3109/13693786.2010.490243. [DOI] [PubMed] [Google Scholar]
- 25.Arnaud MB, et al. The Aspergillus Genome Database (AspGD): recent developments in comprehensive multispecies curation, comparative genomics and community resources. Nucleic Acids Res. 2012;40:D653–659. doi: 10.1093/nar/gkr875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nitsche BM, et al. New resources for functional analysis of omics data for the genus Aspergillus. BMC Genomics. 2011;12:486. doi: 10.1186/1471-2164-12-486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gibbons JG, et al. Global transcriptome changes underlying colony growth in the opportunistic human pathogen Aspergillus fumigatus. Eukaryot Cell. 2012;11:68–78. doi: 10.1128/EC.05102-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bruns S, et al. Functional genomic profiling of Aspergillus fumigatus biofilm reveals enhanced production of the mycotoxin gliotoxin. Proteomics. 2010;10:3097–3107. doi: 10.1002/pmic.201000129. [DOI] [PubMed] [Google Scholar]
- 29.Andersen MR, et al. A trispecies Aspergillus microarray: comparative transcriptomics of three Aspergillus species. Proc Natl Acad Sci USA. 2008;105:4387–4392. doi: 10.1073/pnas.0709964105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang B, et al. Survey of the transcriptome of Aspergillus oryzae via massively parallel mRNA sequencing. Nucleic Acids Res. 2010;38:5075–5087. doi: 10.1093/nar/gkq256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liang Y, et al. Analysis of extracellular proteins of Aspergillus oryzae grown on soy sauce koji. Biosci Biotechnol Biochem. 2009;73:192–195. doi: 10.1271/bbb.80500. [DOI] [PubMed] [Google Scholar]
- 32.McDonagh A, et al. Sub-telomere directed gene expression during initiation of invasive aspergillosis. PLoS Pathog. 2008;4:e1000154. doi: 10.1371/journal.ppat.1000154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reese BN, et al. Gene expression profile and response to maize kernels by Aspergillus flavus. Phytopathology. 2011;101:797–804. doi: 10.1094/PHYTO-09-10-0261. [DOI] [PubMed] [Google Scholar]
- 34.Dujon B. Yeast evolutionary genomics. Nat Rev Genet. 2010;11:512–524. doi: 10.1038/nrg2811. [DOI] [PubMed] [Google Scholar]
- 35.Geiser DM, et al. A review of molecular phylogenetics in Aspergillus, and prospects for a robust genus-wide phylogeny. In: Varga J, Samson RA, editors. Aspergillus in the Genomics Era. Wageningen Academic Publishers; 2008. pp. 17–32. [Google Scholar]
- 36.Peterson SW. Phylogenetic analysis of Aspergillus species using DNA sequences from four loci. Mycologia. 2008;100:205–226. doi: 10.3852/mycologia.100.2.205. [DOI] [PubMed] [Google Scholar]
- 37.Houbraken J, Samson RA. Phylogeny of Penicillium and the segregation of Trichocomaceae into three families. Stud Mycol. 2011;70:1–51. doi: 10.3114/sim.2011.70.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stajich JE, et al. FungiDB: an integrated functional genomics database for fungi. Nucleic Acids Res. 2012;40:D675–681. doi: 10.1093/nar/gkr918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mabey Gilsenan J, et al. CADRE: the Central Aspergillus Data REpository 2012. Nucleic Acids Res. 2012;40:D660–666. doi: 10.1093/nar/gkr971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Benson DA, et al. GenBank. Nucleic Acids Res. 2009;37:D26–31. doi: 10.1093/nar/gkn723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grigoriev IV, et al. The genome portal of the Department of Energy Joint Genome Institute. Nucleic Acids Res. 2012;40:D26–32. doi: 10.1093/nar/gkr947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Andersen MR, et al. Comparative genomics of citric-acid-producing Aspergillus niger ATCC 1015 versus enzyme-producing CBS 513.88. Genome Res. 2011;21:885–897. doi: 10.1101/gr.112169.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rokas A, et al. The diverse applications of RNA-Seq for functional genomics studies in Aspergillus fumigatus. Ann N Y Acad Sci. 2012 doi: 10.1111/j.1749-6632.2012.06755.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu JJ, et al. Tight control of mycotoxin biosynthesis gene expression in Aspergillus flavus by temperature as revealed by RNA-Seq. FEMS Microbiol Lett. 2011;322:145–149. doi: 10.1111/j.1574-6968.2011.02345.x. [DOI] [PubMed] [Google Scholar]
- 45.Delmas S, et al. Uncovering the genome-wide transcriptional responses of the filamentous fungus Aspergillus niger to lignocellulose using RNA sequencing. PLoS Genet. 2012;8:e1002875. doi: 10.1371/journal.pgen.1002875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Selmecki A, et al. Genomic plasticity of the human fungal pathogen Candida albicans. Eukaryot Cell. 2010;9:991–1008. doi: 10.1128/EC.00060-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ma LJ, et al. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature. 2010;464:367–373. doi: 10.1038/nature08850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Geiser DM, et al. Wild chromosomal variants in Aspergillus nidulans. Curr Genet. 1996;29:293–300. doi: 10.1007/BF02221561. [DOI] [PubMed] [Google Scholar]
- 49.Khaldi N, Wolfe KH. Elusive origins of the extra genes in Aspergillus oryzae. PLoS One. 2008;3:e3036. doi: 10.1371/journal.pone.0003036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Slot JC, Rokas A. Horizontal transfer of a large and highly toxic secondary metabolic gene cluster between fungi. Curr Biol. 2011;21:134–139. doi: 10.1016/j.cub.2010.12.020. [DOI] [PubMed] [Google Scholar]
- 51.Khaldi N, et al. Evidence for horizontal transfer of a secondary metabolite gene cluster between fungi. Genome Biol. 2008;9:R18. doi: 10.1186/gb-2008-9-1-r18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marcet-Houben M, Gabaldon T. Acquisition of prokaryotic genes by fungal genomes. Trends Genet. 2010;26:5–8. doi: 10.1016/j.tig.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 53.Khaldi N, Wolfe KH. Evolutionary origins of the fumonisin secondary metabolite gene cluster in Fusarium verticillioides and Aspergillus niger. Int J Evol Biol. 2011 doi: 10.4061/2011/423821. 423821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mallet LV, et al. Whole genome evaluation of horizontal transfers in the pathogenic fungus Aspergillus fumigatus. BMC Genomics. 2010;11:171. doi: 10.1186/1471-2164-11-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Scazzocchio C. Aspergillus genomes: secret sex and the secrets of sex. Trends Genet. 2006;22:521–525. doi: 10.1016/j.tig.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 56.Dyer PS, O’Gorman CM. A fungal sexual revolution: Aspergillus and Penicillium show the way. Curr Opin Microbiol. 2011;14:649–654. doi: 10.1016/j.mib.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 57.Horn BW, et al. Sexual reproduction and recombination in the aflatoxin-producing fungus Aspergillus parasiticus. Fungal Genet Biol. 2009;46:169–175. doi: 10.1016/j.fgb.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 58.Horn BW, et al. Sexual reproduction in aflatoxin-producing Aspergillus nomius. Mycologia. 2011;103:174–183. doi: 10.3852/10-115. [DOI] [PubMed] [Google Scholar]
- 59.O’Gorman CM, et al. Discovery of a sexual cycle in the opportunistic fungal pathogen Aspergillus fumigatus. Nature. 2009;457:471–474. doi: 10.1038/nature07528. [DOI] [PubMed] [Google Scholar]
- 60.Wada R, et al. Presence and functionality of mating type genes in the supposedly asexual filamentous fungus Aspergillus oryzae. Appl Environ Microbiol. 2012;78:2819–2829. doi: 10.1128/AEM.07034-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sugui JA, et al. Identification and characterization of an Aspergillus fumigatus “supermater” pair. MBio. 2011;2:e00234–00211. doi: 10.1128/mBio.00234-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rydholm C, et al. Low genetic variation and no detectable population structure in Aspergillus fumigatus compared to closely related Neosartorya species. Eukaryot Cell. 2006;5:650–657. doi: 10.1128/EC.5.4.650-657.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ramirez-Camejo LA, et al. Phylogeography of the cosmopolitan fungus Aspergillus flavus: is everything everywhere? Fungal Biol. 2012;116:452–463. doi: 10.1016/j.funbio.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 64.Pringle A, et al. Cryptic speciation in the cosmopolitan and clonal human pathogenic fungus Aspergillus fumigatus. Evolution. 2005;59:1886–1899. [PubMed] [Google Scholar]
- 65.Klaassen CH, et al. Evidence for genetic differentiation and variable recombination rates among Dutch populations of the opportunistic human pathogen Aspergillus fumigatus. Mol Ecol. 2012;21:57–70. doi: 10.1111/j.1365-294X.2011.05364.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Grubisha LC, Cotty PJ. Genetic isolation among sympatric vegetative compatibility groups of the aflatoxin-producing fungus Aspergillus flavus. Mol Ecol. 2010;19:269–280. doi: 10.1111/j.1365-294X.2009.04467.x. [DOI] [PubMed] [Google Scholar]
- 67.Olarte RA, et al. Effect of sexual recombination on population diversity in aflatoxin production by Aspergillus flavus and evidence for cryptic heterokaryosis. Mol Ecol. 2012;21:1453–1476. doi: 10.1111/j.1365-294X.2011.05398.x. [DOI] [PubMed] [Google Scholar]
- 68.Fisher MC, Henk DA. Sex, drugs and recombination: the wild life of Aspergillus. Mol Ecol. 2012;21:1305–1306. doi: 10.1111/j.1365-294X.2012.05506.x. [DOI] [PubMed] [Google Scholar]
- 69.Gibbons JG, Rokas A. Comparative and functional characterization of intragenic tandem repeats in 10 Aspergillus genomes. Mol Biol Evol. 2009;26:591–602. doi: 10.1093/molbev/msn277. [DOI] [PubMed] [Google Scholar]
- 70.Rank C, et al. Comparative Chemistry of Aspergillus oryzae (RIB40) and A. flavus (NRRL 3357) Metabolites. 2012;2:39–56. doi: 10.3390/metabo2010039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Medema MH, et al. antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011;39:W339–346. doi: 10.1093/nar/gkr466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Georgianna DR, et al. Beyond aflatoxin: four distinct expression patterns and functional roles associated with Aspergillus flavus secondary metabolism gene clusters. Mol Plant Pathol. 2010;11:213–226. doi: 10.1111/j.1364-3703.2009.00594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Perrin RM, et al. Transcriptional regulation of chemical diversity in Aspergillus fumigatus by LaeA. PLoS Pathog. 2007;3:e50. doi: 10.1371/journal.ppat.0030050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bok JW, et al. Genomic mining for Aspergillus natural products. Chem Biol. 2006;13:31–37. doi: 10.1016/j.chembiol.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 75.Calvo AM. The VeA regulatory system and its role in morphological and chemical development in fungi. Fungal Genet Biol. 2008;45:1053–1061. doi: 10.1016/j.fgb.2008.03.014. [DOI] [PubMed] [Google Scholar]
- 76.Bayram O, Braus GH. Coordination of secondary metabolism and development in fungi: the velvet family of regulatory proteins. FEMS Microbiol Rev. 2012;36:1–24. doi: 10.1111/j.1574-6976.2011.00285.x. [DOI] [PubMed] [Google Scholar]
- 77.Bayram O, et al. The Aspergillus nidulans MAPK module AnSte11-Ste50-Ste7-Fus3 controls development and secondary metabolism. PLoS Genet. 2012;8:e1002816. doi: 10.1371/journal.pgen.1002816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rohlfs M, et al. Secondary chemicals protect mould from fungivory. Biol Lett. 2007;3:523–525. doi: 10.1098/rsbl.2007.0338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schroeckh V, et al. Intimate bacterial-fungal interaction triggers biosynthesis of archetypal polyketides in Aspergillus nidulans. Proc Natl Acad Sci USA. 2009;106:14558–14563. doi: 10.1073/pnas.0901870106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Losada L, et al. Effect of competition on the production and activity of secondary metabolites in Aspergillus species. Med Mycol. 2009;47(Suppl 1):S88–96. doi: 10.1080/13693780802409542. [DOI] [PubMed] [Google Scholar]
- 81.Schrettl M, et al. Self-protection against gliotoxin--a component of the gliotoxin biosynthetic cluster, GliT, completely protects Aspergillus fumigatus against exogenous gliotoxin. PLoS Pathog. 2010;6:e1000952. doi: 10.1371/journal.ppat.1000952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Keller-Seitz MU, et al. Transcriptional response of yeast to aflatoxin B1: recombinational repair involving RAD51 and RAD1. Mol Biol Cell. 2004;15:4321–4336. doi: 10.1091/mbc.E04-05-0375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Latge JP, Steinbach WJ, editors. Aspergillus fumigatus and Aspergillosis. ASM Press; 2009. [Google Scholar]
- 84.Segal BH. Aspergillosis. N Engl J Med. 2009;360:1870–1884. doi: 10.1056/NEJMra0808853. [DOI] [PubMed] [Google Scholar]
- 85.O’Gorman CM. Airborne Aspergillus fumigatus conidia: a risk factor for aspergillosis. Fungal Biol Rev. 2011;25:151–157. [Google Scholar]
- 86.Aimanianda V, et al. Surface hydrophobin prevents immune recognition of airborne fungal spores. Nature. 2009;460:1117–1121. doi: 10.1038/nature08264. [DOI] [PubMed] [Google Scholar]
- 87.Fraczek MG, et al. Aspergillus fumigatus allergen expression is coordinately regulated in response to hydrogen peroxide and cyclic AMP. Clin Mol Allergy. 2010;8:15. doi: 10.1186/1476-7961-8-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Stergiopoulos I, de Wit PJ. Fungal effector proteins. Annu Rev Phytopathol. 2009;47:233–263. doi: 10.1146/annurev.phyto.112408.132637. [DOI] [PubMed] [Google Scholar]
- 89.Sheppard DC. Molecular mechanism of Aspergillus fumigatus adherence to host constituents. Curr Opin Microbiol. 2011;14:375–379. doi: 10.1016/j.mib.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Suh MJ, et al. Development stage-specific proteomic profiling uncovers small, lineage specific proteins most abundant in the Aspergillus fumigatus conidial proteome. Proteome Sci. 2012;10:30. doi: 10.1186/1477-5956-10-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jochl C, et al. Small ncRNA transcriptome analysis from Aspergillus fumigatus suggests a novel mechanism for regulation of protein synthesis. Nucleic Acids Res. 2008;36:2677–2689. doi: 10.1093/nar/gkn123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Loussert C, et al. In vivo biofilm composition of Aspergillus fumigatus. Cell Microbiol. 2010;12:405–410. doi: 10.1111/j.1462-5822.2009.01409.x. [DOI] [PubMed] [Google Scholar]
- 93.Beauvais A, et al. An extracellular matrix glues together the aerial-grown hyphae of Aspergillus fumigatus. Cell Microbiol. 2007;9:1588–1600. doi: 10.1111/j.1462-5822.2007.00895.x. [DOI] [PubMed] [Google Scholar]
- 94.van den Berg MA, et al. Genome sequencing and analysis of the filamentous fungus Penicillium chrysogenum. Nat Biotechnol. 2008;26:1161–1168. doi: 10.1038/nbt.1498. [DOI] [PubMed] [Google Scholar]
- 95.James TY, et al. Reconstructing the early evolution of Fungi using a six-gene phylogeny. Nature. 2006;443:818–822. doi: 10.1038/nature05110. [DOI] [PubMed] [Google Scholar]
- 96.Geiser DM, et al. Eurotiomycetes: Eurotiomycetidae and Chaetothyriomycetidae. Mycologia. 2006;98:1053–1064. doi: 10.3852/mycologia.98.6.1053. [DOI] [PubMed] [Google Scholar]
- 97.Richards TA. Genome evolution: horizontal movements in the Fungi. Current Biology. 2011;21:R166–R168. doi: 10.1016/j.cub.2011.01.028. [DOI] [PubMed] [Google Scholar]