

Abstract
Intermicrobial binding plays an important role in the ecology of the oral cavity because it represents one mechanism by which specific bacteria colonize dental plaque. The formation of “corncobs”, a morphologically distinct microbial unit composed of Streptococcus crista and Fusobacterium nucleatum, is a highly specific binding interaction that depends on the presence of polar tufts of fimbriae on the streptococci. We have used a genetic approach to examine the role of streptococcal cell surface components involved in the binding of S. crista to F. nucleatum. Such binding may be an important component of corncob formation. A method for the genetic transformation of S. crista was used to transfer the broad host range transposon, Tn916, into the bacteria. Cells were grown to early log phase in brain heart infusion broth containing 10% fetal calf serum. The competent cells were mixed with purified DNA from pDL916, a plasmid construct consisting of Tn916 and the streptococcal/Escherichia coli shuttle vector pDL278. Over 300 transformants were screened for a reduction in binding to F. nucleatum. Five of the transformants showed a change in binding ranging from 59% to 29% of the positive control values. Southern blots revealed that the binding-deficient transformants contained the Tn916 element integrated into one of 4 different sites in the chromosome. The transposon, integrated into 4 different sites, appeared to be stable in the absence of selective pressure. Based on these findings, it appears that some strains of S. crista are naturally competent and that insertional inactivation methods can be used to facilitate the study of binding receptors in this group of oral streptococci.
Keywords: Streptococcus crista, Fusobacterium nucleatum, corncob, transposon, transformation, dental plaque
Dental plaque is a complex microbial ecological system in which the temporal changes from the initial pioneer community to the final climax community (20) follow a recognized bacteriological pattern. Among the major pioneer populations associated with these changes are species in the sanguis group of streptococci. This group is of interest because it contains some species with higher affinity for salivary pellicle and some that bind to other bacteria. These distinctive activities provide a biological basis for the formation of the plaque community by allowing incoming bacteria to fix to the tooth surface and grow to form the mature climax community. In the absence of these binding capacities, the bacteria would be dislodged by the mechanical forces in the mouth. Without some form of physical removal, these communities eventually induce the inflammatory changes associated with some types of periodontal disease. One of the major efforts for control of this disease is directed at removal and/or control of dental plaque formation and maturation (11). Such control could be achieved with agents that block various phases of the attachment process. It is generally believed that the adhesins responsible for the attachment process are located in the fimbriae of the streptococci.
One of the most unusual fimbrial arrangements is that found in the streptococci associated with “corncob” formation. This is a distinctive two-cell microbial community that resembles an ear of corn and is a prominent feature of dental plaque. The corncob consists of a central filamentous organism surrounded by streptococci bound to the filament by a polar tuft of fimbriae. In previous studies (15, 16), we have shown that the central filaments could be either the gram-positive, aerobic species Corynebacterium matruchotii (formerly Bacterionema) or the gram-negative, anaerobic bacterium, Fusobacterium nucleatum. The species designation of the streptococci associated with corncobs was recently changed from Streptococcus sanguis to Streptococcus crista (9). The relative simplicity of this community as well as its distinctive morphology are properties that make the complex an ideal model for the study of both the binding process as well as the possible communication that may take place between the partners in this bi-cellular complex. We have concentrated on the fusobacterial corncobs because this model might be archetypal of mechanisms associated with the conversion of the pioneer plaque community to the climax community dominated by anaerobic bacteria. Our initial studies have focused on the adhesins of S. crista. We hypothesized that if the synthesis or assembly of the polar fimbriae of S. crista were altered then corncob formation would be disrupted. Theoretically, such disruption could be accomplished by using a transposon to direct the insertional inactivation of genes associated with adhesin synthesis.
Tn916 is a conjugative transposon that is able to insert at different sites in a range of bacterial species (2, 5). The insertion of Tn916 into a gene inactivates it. Gawron-Burke & Clewell (7) have suggested that these transposable elements would be useful for the targeting of genes and their subsequent cloning. This strategy has been used to identify a number of genes in gram-positive bacteria (1, 4, 27). The simplest method for introducing Tn916 into a bacterium is by conjugation. However, this method was unsuccessful with S. crista. Therefore, we have concentrated on developing a transformation system to insert Tn916 into S. crista. Among the sanguis group of streptococci, only Streptococcus gordonii Challis has been transformed at a high efficiency, using either plasmid or chromosomal DNA (25). The objectives of our study were to develop a similar transformation system for S. crista and to obtain mutants of the bacterium that are deficient in binding to F. nucleatum.
Material and methods
Strains, plasmids and culturing conditions
S. crista CC5A and F. nucleatum ATCC 10953 have been described previously (16). S. crista PSH1a, PSH1b, CR3, CR311 and CH3410 were obtained from P. Handley (University of Manchester, UK) and their general properties described (8, 9). The steptococci were grown in brain heart infusion broth (Difco Laboratories, St. Louis, MO) at 37°C for 18 h. One-millimeter aliquots were rapidly frozen in alcohol-dry ice and stored at −70°C as stock cultures for transformation experiments. F. nucleatum was grown anaerobically in brain heart infusion broth supplemented with 0.2% yeast extract and 0.05% L-cysteine. Escherichia coli JM109 (endA, recA1, gyrA96, thi, hsdR17, (rk−, mk+), relA1, supE44, Δ(lac-proAB), [F′, traD36, proAB, lacIqZΔM15], (λDE3) was grown in LB medium.
Plasmid pDL278 (Fig. 1) is a strepto-coccal/Escherichia coli shuttle vector and was obtained from G. Dunny (University of Minnesota). This plasmid contains an origin of replication (380-1 rep) and the spectinomycin resistance gene from pDL602 and the ori gene and multiple cloning site from pUC19 (6, 18, 26). Plasmid pAM120 (Fig. 1) was obtained from D. Clewell (University of Michigan). Plasmid pAM120 contains the EcoRI F′ fragment (containing the entire Tn916 sequence) cloned into the unique EcoRI site of pGL101 (7).
Fig. 1.

Correlation between competency and growth of strain CC5A. A 1/5000 dilution of an overnight culture of CC5A was incubated at 37°C Samples (0.1 ml) were removed at 1-h intervals, diluted and plated to determine total colony-forming units. A second aliquot (0.33ml) was transformed with1μg of pDL278 DNA. Colony-forming units, —○—; transformant number, —■—.
Transformation
Strain CC5A was streaked, from a frozen stock, onto brain heart infusion agar and grown at 37°C overnight. Several colonies were then used to inoculate 2.0 ml of brain heart infusion broth. This culture was incubated at 37°C overnight. A 5000-fold dilution of the overnight culture was made in brain heart infusion broth containing 10% fetal calf serum (HYCLONE, Logan, UT). After 4 h of growth, 1.0 μg of cesium chloride purified plasmid DNA was added to 0.33-ml aliquots of the culture. This process was repeated hourly for an additional 3–4 h. The transformation mixtures were incubated for 45 min at 37°C. Following incubation, the cells were spread on appropriate selective media and grown aerobically for 48 h.
Isolation of streptococcal DNA
Streptococcal DNA was isolated using a modification of the protocol described by Lunsford & Macrina (19). Briefly, cells from an overnight culture of CC5A were harvested by centrifugation at 5800 ×g for 5 min. Cells were washed twice with an equal volume of 10 mM Tris-HCl (pH 7.0) and resuspended in 1/4 volume of 20 mM Tris-HCl buffer. One-half volume of an aqueous solution of 24% polyethylene glycol (PEG 20 M) was added followed by the addition of 1/4 volume of 20 mM Tris-HCl (pH 7.0) containing 1000 units/ml of mutanolysin (Sigma Chemical Co., St. Louis, MO). The preparation was incubated for 1 h at 37°C. Spheroplasts were harvested by centrifugation at 5800 × g for 20 min. The spheroplasts were suspended in 1/10 volume of 1% sodium dodecyl sulfate (SDS) in 10 mM Tris-HCl (pH 8.0) containing 10 mM EDTA (pH 8.0) and 1 mg/ml proteinase K. This mixture was incubated for 1 h at 50 °C. An equal volume of buffer-saturated phenol was added. After vortexing, the emulsion was centrifuged at 10,000 × g for 15 min and the aqueous phase was collected. An equal volume of chloroform:isoamyl alcohol (24:1) was added, the mixture was centrifuged, and the aqueous phase collected. Two volumes of ethanol and 1/10 volume of sodium acetate were added to precipitate the DNA. After chilling to 0°C, the DNA was collected by centrifugation at 20,000 × g for 20 min and was resuspended in 10 mM Tris-HCl (pH 8.0) –1 mM EDTA (TE) buffer.
Recombinant DNA techniques
Restriction endonuclease digestion was carried out according to the manufacturer’s instructions. Plasmid DNA isolation, ligations and transformation of E. coli were performed as described (23). Probes were labeled with α[32P]dATP (6000 Ci/mmol; Amersham Corp., Arlington Heights, IL) using a random primer labeling kit (Boehringer Mannheim Biochemicals, Indianapolis, IN). Southern blots were prehybridized for 2 h at 42 °C and then hybridized overnight, at the same temperature, in 50% formamide. Usually 2× 106 cpm of probe DNA (specific activity 2×107 cpm/μg DNA) were used. Following hybridization the blots were washed in 2×SSC (1×SSC is 0.15 M sodium choloride and 0.015 M sodium citrate, pH 7.0) containing 0.1% SDS for 15 min at room temperature. This was followed by a second wash in 0.1×SSC–0.1% SDS for 1 h at 42 °C. The blots were exposed to NEN-DuPont Reflection™ Autoradiography film with one Reflection™ intensifying screen at −70°C.
Biotinylation of F. nucleatum
F. nucleatum was biotinylated as described previously (13). Briefly, bacteria were recovered by centrifugation, washed in buffered KCl (5 mM KCl, 2 mM K2PO4, 1 mM CaCl2, pH 6.0) and suspended in 0.1 M NaHCO3 (pH 8.1) at 1010 cells/ml. N-hydroxysuccinimidobiotin (Sigma Chemical Co.) was added to yield a final cell concentration of 1 mg/ml. After incubation for 3 h at room temperature the cells were recovered by centrifugation (10,000 × g, 10 minutes), washed twice and resuspended in the KCl buffer.
Colony lift assay for fusobacterial/streptococcal binding
Transformed streptococcal colonies were grown on brain heart infusion agar containing 10 μg/ml of tetracycline and were transferred to nitrocellulose paper by an overlay method. Binding of the bacteria to the nitrocellulose was stabilized by incubation at 37°C for 30 min and free sites on the filter were blocked with KCl buffer containing 0.1% Tween 20 (KCl-Tween). The blots were incubated, with gentle rocking, with 109 cells of biotinylated F. nucleatum for 1 h at room temperature (12, 14). The filter was washed 4 times with KCl-Tween and fusobacteria-streptococci binding visualized by developing the filter with streptavidin-alkaline phosphatase conjugate (Bio-Rad Laboratories, Rockville Centre, NY) and the chromogenic substrates 5-bromo-4-chloro-3-indolyl phosphate and nitro-blue tetrazolium (Bio-Rad Laboratories) according to the manufacturer’s instructions. Transformant colonies that failed to bind F. nucleatum were further tested in the quantitative binding assay.
Quantitative fusobacterial/streptococcal binding assay
Quantitative adherence of CC5A transformants to F. nucleatum was determined by a nitrocellulose blot assay described previously (14). Briefly, the fusobacteria were suspended in buffered KCl and 5×107 cells/well were deposited on nitrocellulose paper in a dot blot apparatus. The blot was washed 3 times with KCl-Tween and incubated for 2 h at room temperature with 8×107 cells of [3H]thymidine-labeled CC5A (suspended in KCl-Tween; specific activity 0.6–1 ×10−4 cpm/cell). The blot was washed to remove any unbound CC5A. The areas of the nitrocellulose filter that corresponded to the wells were punched out of the blot and the amount of bound streptococci determined in a liquid scintillation counter.
SDS-polyacrylamide gel electrophoresis and Western blotting
Bacteria were grown overnight in 10 ml of brain heart infusion broth, washed twice in 10 mM Tris-HCl (pH 8.0) and suspended in 0.5 ml of lysis buffer [2% SDS, 8 mM Tris-HCl (pH 6.8), 10% glycerol]. The samples were heated in a boiling water bath for 20 min, and 40 μl was applied to a 17.5% acrylamide gel. Following electrophoresis, the gel was stained with Coomassie blue. Samples were run on a duplicate gel and a Western blot was prepared as described (23). The blot was treated with anti-whole cell S. crista CC5A serum, made in rabbits, at a 1:500 dilution.
Results
The shuttle vector pDL278 was used to determine the optimal transformation conditions for S. crista CC5A. Selection of transformants was on brain heart infusion agar containing 1000 μg/ml spectinomycin. As shown in Fig. 1, maximum competence was achieved during log phase at approximately 5–6 h after the dilution of the culture. The narrow zone of competence was typical for this transformation. In 6 independent experiments, transformation frequencies ranged from 0.2×10−3 to 4.3×10−3 (Table 1). As shown in the dose-response curve in Fig. 2, the maximum number of transformants were obtained when 2 μg of DNA were used in the transformation mixture. The transformation appeared to be linear up to 1 μg of DNA.
Table 1.
Tranformation frequencies for S. crista CC5A
| Experiment | Number of transformants3 | Total number of bacteria (×105) | Transformation frequency15 (×10−3) |
|---|---|---|---|
| 1 | 540 | 1.3 | 4.2 |
| 2 | 265 | 2.2 | 1.2 |
| 3 | 10,400 | 23.9 | 4.3 |
| 4 | 460 | 8.4 | 0.6 |
| 5 | 131 | 8.3 | 0.2 |
| 6 | 2,105 | 12.0 | 1.8 |
Based on 0.33 ml of competent cell culture and 1 μg/ml of pDL278 DNA.
Transformation frequencies, for each experiment, were calculated from the peak value similar to that shown in Fig. 1.
Fig. 2.
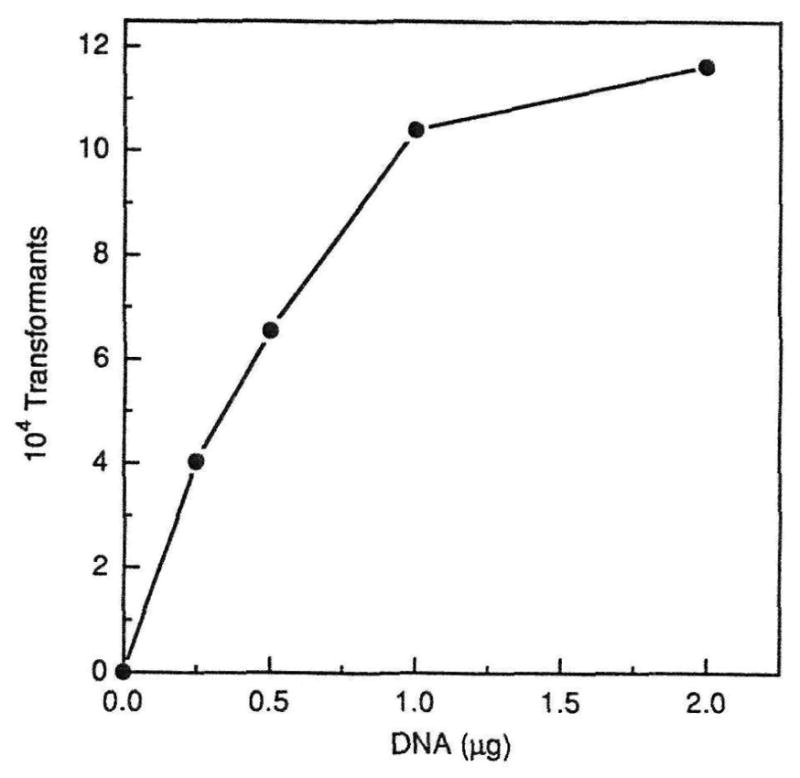
Effect of increasing amounts of DNA on transformation. Competent cells of CC5A were transformed with 0.25 to 2.0 μg of pDL278 DNA. The total number of transformants is plotted against DNA concentration.
A vector was constructed to introduce transposon Tn916 into the chromosome of CC5A. The EcoRI F′ (EcoRI::Tn916) fragment of pAM120 was extracted from an agarose gel and cloned into the unique EcoRI site of pDL278 to generate pDL916 (Fig. 3). The pDL916 construct contains the entire coding region of transposon Tn916 including the tetracycline resistance determinant, tetM. Plasmid DNA was extracted from E. coli, grown in the presence of 10 μg/ml tetracycline and purified on a cesium chloride-ethidium bromide density gradient. A 2.7-kb fragment was also found in the EcoRI digested gradient purified preparations in addition to the 19.1- and 6.6-kb fragments. This additional fragment has been noted in other constructs containing Tn916 (7) and derives from the excision of Tn916 from the EcoRI F′ fragment during replication in E. coli.
Fig. 3.

Schematic representation of the steps in the construction of the insertional mutagenesis vector pDL916. tet, tetracycline; bla, β-lactamase; spc, spectinomycin.
In addition to CC5A, we also attempted to transform S. crista strains PSH1a, PSH1b, CR3, CR311 and CH3410 with pDL916. Strains PSH1a and CC5A transformed with an optimum frequency of 3.5 ×10−6 and 1.1 ×10−6 per μg of DNA, respectively. The 4 other strains of S. crista were not transformable with the pDL916 construct.
The frequency of transformation of S. crista CC5A with the pDL916 construct was 2 orders of magnitude lower than that obtained with pDL278. A total of 330 tetracycline-resistant transformants were obtained. Thirteen percent of these were also spectinomycin-resistant, suggesting a low incidence of the plasmid. Nineteen of the 330 tetracycline-resistant transformants were randomly selected and their DNA digested with HincII and hybridized to the entire Tn916 sequence on a Southern blot. In all cases the tetracycline-resistant transformants contained Tn916 specific DNA.
The colony lift binding assay was then used to rapidly screen all of the transformants for a decrease in binding to F. nucleatum ATCC 10953. Approximately 20 colonies that showed a reduced chromogenic reaction on the colony lifts were assayed quantitatively for binding. As shown in Table 2, 5 of the transformants bound lower numbers of F. nucleatum than the control streptococci. The number of fusobacteria that bound to the 5 suspected mutants ranged from 59% to 29% of the positive control values. Two strains served as positive binding controls. These included the wild-type strain CC5A and one of the transformants (A24) that exhibited unaltered binding in the initial colony lift screening assay. Strain A24 bound to the fusobacteria nearly as well as CC5A (87.8% of control), suggesting that just the presence of the transposon in the bacterium had little effect on binding. Total protein extracts from the putative binding mutants and transformant A24 were compared with that of strain CC5A on SDS-polyacrylamide gels (data not shown). There were no apparent differences among the stained protein patterns on the gel. Western blots, using anti-strain CC5A whole cell serum, also showed no differences between the antigen patterns of the mutants, transformant A24 and the wild-type strain (data not shown).
Table 2.
Binding of S. crista CC5A mutants to F. nucleatum ATCC 10953
| Strain | Average number of cells bound (×107) | Standard deviation (×107) | na | Percent of control |
|---|---|---|---|---|
| CC5A | 4.1 | 0.40 | 6 | 100 |
| A24 | 3.6 | 0.60 | 12 | 87.8 |
| B15 | 1.2 | 0.50 | 9 | 29.3 |
| B40 | 1.7 | 1.00 | 9 | 41.5 |
| C29 | 1.7 | 0.30 | 6 | 41.5 |
| D36 | 2.4 | 0.60 | 6 | 58.5 |
| D38 | 1.9 | 0.90 | 5 | 46.3 |
Number of experiments performed.
Cesium chloride-purified chromosomal DNA was extracted from the 5 binding deficient mutants, control strain A24 and the wild-type strain CC5A. The DNA was digested with HincII and a Southern blot was prepared. The blot was hybridized with the HincII fragment C, which is the junction fragment that represents the left end of the transposon. Fragment C is 3.6 kb in length in pDL916 (Fig. 3). The results of the hybridization are shown in Fig. 4 and demonstrate that the HincII fragment C in each mutant and in the A24 control vary in size relative to the same fragment in pDL916. Mutants B40 and D38 showed similar hybridization patterns and, therefore, may have identical insertions.
Fig. 4.

Southern blot showing single insertions of Tn916 in binding-deficient transformants of CC5A. Chromosomal DNA, extracted from 5 tetracycline-resistant transformants that showed reduced binding to F. nucleatum, was digested with HincII and hybridized to the left end (fragment C; 3.6-kb) of Tn916. DNA from wild-type CC5A and a tetracycline-resistant transformant (A24) that showed no reduction in binding was also run on the blot. pDL916 DNA is shown in the first lane. The autoradiogram is shown.
It was noted that fragment C hybridized to several different bands in the pDL916 lane. As expected, the smallest hybridization-positive band was 3.6 kb. The appearance of the larger hybridization-positive bands, including a 9.8-kb species, is reproducible and may have resulted from the instability of Tn916 when replicated in E. coli. A 9.8-kb supercoiled species of Tn916 has been reported by Scott et al. (24).
When the same chromosomal DNA digests were hybridized to the EcoRI F′ fragment containing the entire Tn916 element, it was apparent that the internal HincII fragments A, B, F and G were present, at their expected sizes, in all of the binding deficient transformants (Fig. 5). Based on these data, it was concluded that no gross rearrangements of Tn916 occurred in these mutants. The hybridization-positive fragments, between fragments F and C in the mutant lanes on the blot (see small arrows), most likely represent the right junction fragment of the Tn916 element. This element is 0.8 kb in pDL916. As in the case of fragment C, each mutant, except B40 and D38, showed a distinct hybridization pattern for these fragments.
Fig. 5.
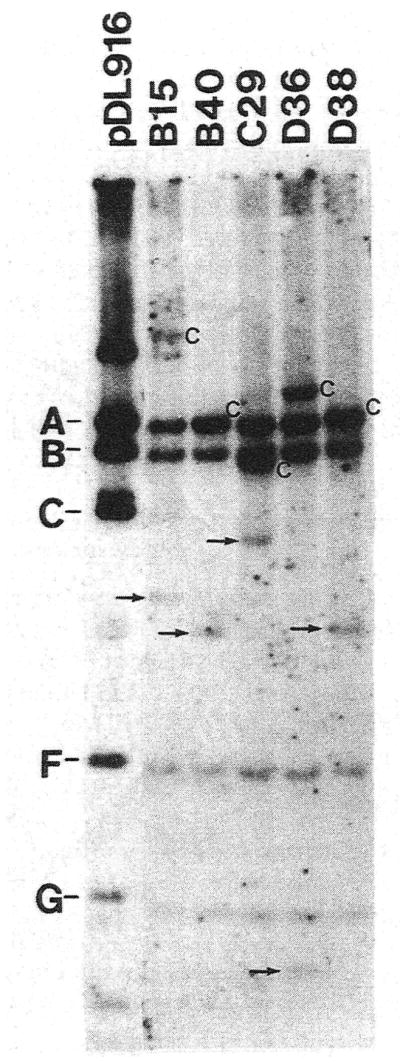
Autoradiogram of a Southern blot depicting the status of the inserted transposon in the binding-deficient mutants of CC5A. The entire Tn916 was hybridized to chromosomal DNA, digested with HincII, from the 5 binding mutants. pDL916 DNA, digested with HincII, is shown in the first lane. The upper case letters designate the HincII fragments of Tn916 (pDL916 lane). The left end junction fragment, in each of the transformant lanes, is marked by an uppercase C. The HincII fragments A and C overlap in transformant lanes B40 and D38. The small arrows mark additional hybridization-positive fragments thought to represent the right end junction fragment of the transposon.
Discussion
This is the first report of the transformation of S. crista. We found that at least 2 strains of S. crista (formally S. sanguis) were naturally competent. Previous studies of the sanguis group showed that only S. gordonii Challis (22) was naturally competent. The narrow growth window in which the cells develop competence mimics the phenomenon seen in S. gordonii (17) and Streptococcus pneumoniae (28). In both these cases, as in CC5A, transformation is associated with a brief time during exponential growth during which the cells are maximally competent.
Plasmid pAM120, a suicide vector containing Tn916, could not be used in these experiments because we were concerned about the possibility of introducing a β-lactamase gene into S. crista. The shuttle vector pDL278 was used because it gave reasonably high transformation frequencies in CC5A, it permitted the isolation of large amounts of plasmid DNA from E. coli, it had a selectable marker (spectinomycin resistance) other than ampicillin resistance and it contained a unique Eco-RI site for the cloning of the entire Tn916 element.
The large plasmid construct that was required to introduce Tn916 into the bacterium showed significantly lower transformation frequencies than those obtained with pDL278. This was attributed to the four-fold larger size of pDL916 (25.7 kb) compared with that of pDL278 (6.6 kb). Tn916 was very stable once inserted into the chromosome of S. crista even when selective pressure was removed. Tn9I6 could not be transferred, by conjugation, from the CC5A transformants back to wild-type S. crista. These results suggested that gene insertions can be obtained only during the initial transformation. This observation was similar to the finding of Bringel et al. (3) that Tn916 could not transpose in Lactococcus lactis due to the lack of a host fertility factor. We also found that new gene insertions could not be obtained in the existing transformants even if the selective pressure (tetracycline) was removed.
Despite the low transformation frequencies for pDL916, we obtained over 300 tetracycline-resistant transformants. Twenty putative mutants were identified by their reduced binding to biotinylated cells of F. nucleatum. Of these, 5 consistently showed reduced binding relative to the wild-type CC5A in a quantitative binding assay.
The molecular characterization of the transformants was complicated by the fact that pDL916 can replicate autonomously in the host because it contains the 380-1 replication origin (Fig. 3). To establish that the transformants contained chromosomal insertions, the plasmid DNA was removed, prior to further analysis, by cesium chloride-ethidium bromide gradient purification. All of the transformants that showed a reduction in binding contained a single Tn916 insertion in the chromosome, and the insertion site appeared to be unique in 4 of the 5 mutants. The transformants (see transformant A24) that had Tn916 insertions in other unique chromosomal sites showed no reduction in binding to the fusobacteria. Additional evidence that the transposon insertions were random was obtained by screening the transformants for loss of [β-N-acetyl glucosaminidase, an enzyme known to be present in S. crista (9). One of the 330 transformants showed loss of this activity (data not shown). The results of these experiments indicated that specific loci in S. crista were involved in the binding to F. nucleatum. Values for the 5 mutants ranged from 59% to 29% of the wild type. Only a partial reduction in binding was expected, because other studies of streptococcal adhesins have shown similar findings (12, 14, 16). This is attributed to the existence of multifactorial binding components or pleiotropic mutation effects (10, 21). Pleiotropic mutations may explain the high percentage of binding-deficient transformants obtained. However, no differences in total protein or immunoblot patterns between the binding-deficient transformants and wild-type strain were apparent.
In summary, transposon mutagenesis has been used to identify at least 4 types of mutants of S. crista CC5A that show reduced binding to F. nucleatum. In the course of developing this approach, we found that S. crista can be genetically transformed by standard methods. Future work will be directed towards the cloning and characterization of the mutated genes reported here.
Acknowledgments
We thank D. Clewell and G. Dunny for supplying plasmids and P. Handley for providing bacterial strains. The technical assistance of T Oswald is also greatly appreciated. This work was supported by USPHS grants DE03180 and DE09439 from the National Institutes of Health.
References
- 1.Bensing BA, Dunny GM. Cloning and molecular analysis of genes affecting expression of binding substance, the recipient-encoded receptor(s) mediating mating aggregate formation in Enterococcus faecalis. J Bacteriol. 1993;175:7421–7429. doi: 10.1128/jb.175.22.7421-7429.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bertram J, Strätz M, Dürre P. Natural transfer of conjugative transposon Tn916 between gram-positive and gram-negative bacteria. J Bacteriol. 1991;173:443–448. doi: 10.1128/jb.173.2.443-448.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bringel F, van Alstine GL, Scott JR. Transfer of Tn916 between Lactococcus lactis subsplactis strains is nontrans-positional: evidence for a chromosomal fertility function in strain MG1363. J Bacteriol. 1992;174:5840–5847. doi: 10.1128/jb.174.18.5840-5847.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caparon MG, Scott JR. Identification of a gene that regulates expression of M protein, the major virulence determinant of group A streptococcci. Proc Natl Acad Sci U S A. 1987;84:8677–8681. doi: 10.1073/pnas.84.23.8677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clewell DB, Flannagan SE. The conjugative transposons of gram-positive bacteria. In: Clewell D, editor. Bacterial conjugation. New York: Plenum Press; 1993. pp. 321–325. [Google Scholar]
- 6.Dunny GM, Lee LN, LeBlanc DJ. Improved electroporation and cloning vector system for gram-positive bacteria. Appl Environ Microbiol. 1991;57:1194–1201. doi: 10.1128/aem.57.4.1194-1201.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gawron-Burke C, Clewell DB. Regeneration of insertionally inactivated streptococcal DNA fragments after excision of transposon Tn916 in Escherichia coli: strategy for targeting and cloning of genes from gram-positive bacteria. J Bacteriol. 1984;159:214–221. doi: 10.1128/jb.159.1.214-221.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Handley P, Carter PL, Wyatt JE, Hesketh LM. Surface structures (peritrichous fibrils and tufts of fibrils) found on Streptococcus sanguis strains may be related to their ability to coaggregate with other oral genera. Infect Immun. 1985;47:217–227. doi: 10.1128/iai.47.1.217-227.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Handley P, Coykendall A, Beighton D, Hardie JM, Whiley RA. Streptococcus crista sp. nov., a viridans streptococcus with tufted fibrils, isolated from the human oral cavity and throat. Int J Syst Bacteriol. 1991;41:543–547. doi: 10.1099/00207713-41-4-543. [DOI] [PubMed] [Google Scholar]
- 10.Harrington DJ, Russell RRB. Multiple changes in cell wall antigens of isogenic mutants of Streptococcus mutans. J Bacteriol. 1993;175:5925–5933. doi: 10.1128/jb.175.18.5925-5933.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jenkins GN. Opening statement. In: Leach SA, editor. Dental plaque and surface interactions in the oral cavity. London: Information Retrieval; 1980. pp. 1–3. [Google Scholar]
- 12.Lamont RJ, Bevan CA, Gil S, Persson RE, Rosan B. Involvement of Porphyromonas gingivalis fimbriae in adherence to Streptococcus gordonii. Oral Microbiol Immunol. 1993;8:272–276. doi: 10.1111/j.1399-302x.1993.tb00573.x. [DOI] [PubMed] [Google Scholar]
- 13.Lamont RJ, Gil S, Demuth DR, Malamud D, Rosan B. Molecules of Streptococcus gordonii that bind to Porphyromonas gingivalis. Microbiology. doi: 10.1099/00221287-140-4-867. in press. [DOI] [PubMed] [Google Scholar]
- 14.Lamont RJ, Rosan B. Adherence of mutans streptococci to other oral bacteria. Infect Immun. 1990;58:1738–1743. doi: 10.1128/iai.58.6.1738-1743.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lancy P, Jr, Appelbaum B, Holt SC, Rosan B. Quantitative in vitro assay for “corncob” formation. Infect Immun. 1980;29:663–670. doi: 10.1128/iai.29.2.663-670.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lancy P, Jr, DiRienzo JM, Appelbaum B, Rosan B, Holt SC. Corncob formation between Fusobacterium nucleatum and Streptococcus sanguis. Infect Immun. 1983;40:303–309. doi: 10.1128/iai.40.1.303-309.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lawson JW, Gooder H. Growth and development of competence in the group H streptococci. J Bacteriol. 1970;102:820–825. doi: 10.1128/jb.102.3.820-825.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.LeBlanc DJ, Lee LN. Replicative functions of pVA380-1. In: Dunny GM, Cleary PP, McKay LL, editors. Genetics and molecular biology of streptococci, lactococci, and enterococci. Washington, DC: Am Soc Microbiol; 1991. pp. 235–239. [Google Scholar]
- 19.Lunsford RD, Macrina EL. Molecular cloning and characterization of scrB, the structural gene for the Streptococcus mutans phosphoenolpyruvate-dependent sucrose phosphotransferase system sucrose-6-phosphate hydrolase. J Bacteriol. 1986;166:426–434. doi: 10.1128/jb.166.2.426-434.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marsh P, Martin M. Oral microbiology. 3. New York: Chapman & Hall; 1992. Acquisition, adherence, distribution and functions of the oral microflora; pp. 56–60. [Google Scholar]
- 21.McNab R, Jenkinson HF. Gene disruption identifies a 290 kDa cell-surface polypeptide conferring hydrophobicity and coaggregation properties in Streptococcus gordonii. Mol Microbiol. 1992;6:2939–2949. doi: 10.1111/j.1365-2958.1992.tb01753.x. [DOI] [PubMed] [Google Scholar]
- 22.Pakula R, Fluder Z, Hulanicka E, Walczak W. Studies on transformation of streptococci. Bull Acad Polon Sci Ser Sci Biol. 1958;6:325–328. [Google Scholar]
- 23.Sambrook J, Fritch EF, Maniatis T. Molecular cloning: a laboratory manual. New York: Cold Spring Harbor; 1989. [Google Scholar]
- 24.Scott JR, Kirchman PA, Caparon MG. An intermediate in transposition of the conjugative transposon Tn916. Proc Natl Acad Sci U S A. 1988;85:4809–4813. doi: 10.1073/pnas.85.13.4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith HO, Danner DB, Deich RA. Genetic transformation. Annu Rev Biochem. 1981;50:41–68. doi: 10.1146/annurev.bi.50.070181.000353. [DOI] [PubMed] [Google Scholar]
- 26.Tao L, LeBlanc DJ, Ferretti JJ. Novel streptococcal-integration shuttle vectors for gene cloning and inactivation. Gene. 1992;120:105–110. doi: 10.1016/0378-1119(92)90016-i. [DOI] [PubMed] [Google Scholar]
- 27.Tao L, Tanzer JM, Kuramitsu HK, Das A. Identification of several rod loci and cloning of rod D locus of Streptococcus mutans. Gene. 1993;126:123–128. doi: 10.1016/0378-1119(93)90600-8. [DOI] [PubMed] [Google Scholar]
- 28.Tomasz A, Hotchkiss RD. Regulation of the transformability of pneumococcal cultures by macromolecular cell products. Proc Natl Acad Sci USA. 1964;51:480–487. doi: 10.1073/pnas.51.3.480. [DOI] [PMC free article] [PubMed] [Google Scholar]