

Abstract
Activation of arterial smooth muscle alpha1-adrenergic receptors results in vasoconstriction, as well as a secondary release of nitric oxide and slow vasodilation, presumably through gap junction communication from smooth muscle to endothelium. We hypothesized that this slow vasodilation is due to activation of eNOS through phosphorylation at Ser1179 and dephosphorylation at Thr495. Phosphorylation was measured by western blot using mouse mesenteric arteries that were cannulated and pressurized (75 mmHg) and treated either by 1) 5 min of phenylephrine superfusion (10−5 M) (PE5), 2) 15 minutes of phenylephrine (PE15), 3) 15 min phenylephrine followed by acetylcholine (10−4 M) (PE+ACh), or 4) 20 min time control with no treatment (NT) [4–5 arteries pooled per treatment per blot; 5 blots performed]. These treatments allowed correlation between vasomotor changes, namely maximal constriction (PE5), slow vasodilation (PE15), and maximal dilation (PE+ACh), and relative phosphorylation changes. Phosphorylation of eNOS at Ser1179 was increased relative to NT by more than 2-fold at PE5 and remained similarly increased at PE15 and PE+ACh. Phosphorylation of eNOS at Thr495 was less in all treatments relative to NT, but not significantly. Treatment with L-NAME (10−4 M) or endothelial denudation indicated that the slow dilation in response to phenylephrine was completely due to nitric oxide synthase and was endothelial dependent. These results indicate that eNOS phosphorylation at Ser1179 occurs before the slow dilation and is not actively involved in this vasodilation or dilation to acetylcholine, but may play a permissive role in eNOS activation by other mechanisms. It is not yet known what mechanism is responsible for Ser1179 phosphorylation with phenylephrine stimulation.
Keywords: mouse, mesenteric, myoendothelial junctions, vasodilation, phenylephrine, acetylcholine
1. Introduction
Endothelial nitric oxide synthase (eNOS), which produces the gas nitric oxide, is an important enzyme for cardiovascular health as it is essential for maintenance of normal blood pressure (Shesely et al, 1996), for preventing atherosclerosis (Kuhlencordt et al, 2001), for angiogenesis (Murohara et al, 1998; Yu et al, 2005), and for vasodilation and remodeling in large arteries (Brandes et al, 2000; Rudic et al, 1998). It is activated by shear stress, hypoxia, and endothelial agonists, such as bradykinin, estrogen, and VEGF (Sessa, 2004). Its catalytic activity is regulated by intracellular calcium, phosphorylation, dimerization, and interaction with other proteins (Rafikov et al, 2011). Phosphorylation changes occur at several sites and stimulation of cultured endothelial cells with agonists induces increased eNOS phosphorylation at Ser1179 and desphosphorylation at Thr495 (human sequence nomenclature) (Harris et al, 2001; Rafikov et al, 2011; Sessa, 2004). Phosphorylation at Ser1179 seems to be a critical event because in cultured endothelial cells, alteration of Ser1179 to alanine results in decreased production of nitric oxide (Dimmeler et al, 1999; Fulton et al, 1999; Scotland et al, 2002), and conversion to aspartate (mimicking phosphorylation) causes maximal enzymatic activity of the enzyme and independence from increases in calcium concentration (Dimmeler et al, 1999). Moreover, in intact carotid arteries, transgenic alteration of amino acid 1179 from serine to alanine or adenoviral reconstitution of eNOS knockout mice to Ala1179 impairs acetycholine-induced vasodilation (Atochin et al, 2007; Scotland et al, 2002). It is unclear, however, how and at what sites eNOS is phosphorylated in intact arteries in response to agonists and over time and how this correlates with vasomotor responses.
Generation of nitric oxide is also stimulated in some vessels during vasoconstriction by activation of sympathetic nerves (Boric et al, 1999) or by the alpha1-adrenergic receptor agonist, phenylephrine (Dora et al, 1997; Tuttle & Falcone, 2001). Dora et al. (Dora et al, 1997) were the first to show that with stimulation of hamster arterioles with phenylephrine lead to an increase in endothelial cell calcium and subsequent generation of nitric oxide and attenuation of constriction. The rise in endothelial cell calcium provided evidence for gap junction communication from smooth muscle to endothelial cells (myoendothelial gap junction communication), because phenylephrine is not known to have direct effects on endothelial cells. This phenomenon has also been observed in rat skeletal muscle arterioles (Tuttle & Falcone, 2001) and in mouse mesenteric arteries (Nausch et al, 2012). In this latter study, it was recently shown that in mouse mesenteric arteries, smooth muscle alpha1-adrenergic receptor stimulation with phenylephrine results in IP3-mediated release of Ca++ from the endothelial sarcoplasmic reticulum, presumably due to diffusion of IP3 from smooth muscle to endothelial cells via gap junctions (Nausch et al, 2012). Others have provided evidence that these particular arteries have myoendothelial gap junction communication to support this conclusion (Dora et al, 2003; Saliez et al, 2008). This increase in endothelial cell calcium at the myoendothelial junction potentially stimulates eNOS, as it has recently been shown that eNOS can reside in the myoendothelial junction in mouse arteries (Straub et al, 2011). It is unknown whether phosphorylation of eNOS also occurs during this process.
In this study, we sought to determine whether phenylephrine stimulation of intact arteries results in eNOS regulation via phosphorylation. We show that in intact, cannulated mouse mesenteric arteries, phenylephrine induced eNOS Ser1179 phosphorylation within 5 minutes, even as the artery reached maximal constriction. As the artery partially dilated in reaction to phenylephrine-induced stimulation of NOS, there was no further increase in Ser1179 phosphorylation, nor was there any further increase when acetylcholine was added to induce maximal dilation, despite its NOS dependence. These data indicate that phosphorylation of eNOS on Ser1179 does not necessarily correspond to vasodilatory responses and full phosphorylation at this site does not result in maximal dilation, but suggests that this phosphorylation may act permissively when other mechanisms of regulation are activated.
2. Methods
2.1. Measurement of In Vitro Artery Function
All experimental procedures were approved by the William & Mary Institutional Animal Care and Use Committee and are in accordance with the Guide for the Care and Use of Laboratory Animals (National Research Council). Male C57BL/6 mice (~12 weeks old) were anesthetized with sodium pentobarbital (Nembutal, 50 mg/kg body weight, intra-peritoneal), the intestines were removed and euthanasia resulted from exsanguination. The intestines along with the mesenteric arcade were placed in cold PSS-MOPS buffer (in mM: 145 NaCl, 4.7 KCl, 2.0 CaCl2, 1.17 MgSO4, 1.2 NaH2PO4, 2.0 MOPS, 0.02 EDTA, 5.0 Glucose, 2.0 Pyruvate, pH 7.4). Two first order mesenteric arteries were removed from each mouse, cannulated in separate vessel chambers (Danish Myo Technology, Aarhus, DK) filled with PSS-MOPS, secured with nylon suture (8-0, S & T, Neuhausen, Switzerland), gently cleared of blood, and pressurized to 75 mmHg with PSS-MOPS + 1% bovine serum albumin perfusion, with no subsequent flow through the lumen. The chambers were mounted on stages of two separate pressure myograph microscope systems (Danish Myo Technology) and equilibrated at 37 °C for 30 min, with constant superfusion (superfusate not re-circulated) before testing functional responses. Luminal diameter was measured using a 10X objective, ccd camera, and VediView Software (Danish Myo Technology) on one system and Ionoptix software (Milton MA) on the other system using on-screen cursors.
After the 30 minute equilibration, mounted vessels were tested in one of 6 different protocols. 1) No treatment (NT), which consisted of 20 minutes of equilibration with the superfusate re-circulated (100 ml total volume). This treatment acted as a time control in which no drugs were added, and diameter was measured before and after equilibration. 2) Phenylephrine treatment (10−5 M in 100 ml superfusate) for 5 minutes (PE5) with diameter measured every minute. 3) Phenylephrine treatment (10−5 M in 100 ml superfusate) for 15 minutes (PE15) with diameter measured every minute. 4) Acetylcholine treatment (10−4 M in 100 ml superfusate) for 5 min (PE+ACh) after pre-constriction with 10−5 M phenylephrine for 15 min. Diameter was measured every minute after phenylephrine addition and subsequent acetylcholine addition. 5) Treatment with L-NAME (10−4 M, in lumen perfusate and in superfusate) beginning during cannulation, followed by treatment with phenylephrine and acetylcholine as in treatment #4 (L-NAME), to determine how much of the constriction and dilation with these drugs was due to nitric oxide synthase/s. 6) Arteries were treated with air through the lumen for 10–15 min to destroy the endothelium, followed by treatment with phenylephrine and acetylcholine as in treatment #4 (Denuded), to determine how much of the constriction and dilation to drugs were endothelial dependent. All arteries were frozen immediately in liquid nitrogen (and stored at −80 °C) after each of these treatments for western blot analysis, with the exception of the L-NAME arteries. Maximal diameter was measured in the L-NAME group after superfusion with Ca++-free PSS-MOPS + 1 mM EGTA for at least 15 min. In the other groups, maximal diameter was estimated by the initial diameter upon chamber mounting when the vessel was bathed in cold PSS-MOPS. Previous results in our laboratory have shown this to be a close estimate given that the vessel is fully relaxed in this state and there is no basal tone.
All groups had similar luminal diameters at the initial measurement in cold PSS (Mean ± SEM: NT: 230 ± 5 μm; PE5: 235 ± 6 μm; PE15: 234 ± 6 μm; PE+ACh: 242 ± 8 μm; L-NAME: 224 ± 12; Denuded: 232 ± 11). In four of the groups the baseline diameter after 30 min of equilibration at 37° C was either not changed (PE5: 229 ± 8 μm; PE15: 235 ± 7 μm), or increased slightly but significantly (p<0,05, paired t-test) (NT: 235 ± 5 μm; PE+ACh: 245 ± 8 μm), indicating an absence of basal tone, which is consistent with our previous results (Looft-Wilson et al, 2008). However, the L-NAME and Denuded groups had significantly decreased (p<0.05, paired t-test) diameters after the 30 min equilibration (L-NAME: 195 ± 11 μm; Denuded: 198 ± 12 μm), indicating basal nitric oxide release from the endothelium in this artery type.
In a separate set of experiments arteries underwent one of three treatments: 1) NT, 2) PE15, or 3) treatment with phenylephrine for 15 min in the presence of the alpha1-adrenergic receptor antagonist, prazosin (500 nM) in the superfusate beginning at the 30 min equilibration (Prazosin). These experiments were performed to confirm that the constriction to phenylephrine was entirely dependent on alpha1-adrenergic receptor stimulation. These arteries had similar initial diameters (NT: 256 ± 24; PE15: 238 ± 15; Prazosin: 241 ± 7) with no change after 30 minutes of equilibration (NT: 260 ± 22; PE15: 241 ± 14; Prazosin: 245 ± 6).
In a preliminary set of experiments, arteries were treated with increasing doses of phenylephrine (10−9 M – 10−4 M) in a cumulative manner, with 5 minutes allowed after each dose for equilibration. These experiments were performed to determine the dose of phenylephrine that produces the maximal constriction, the variability with each dose, and the effective concentration to produce a 50% constriction (EC-50). This curve indicated that a dose of 10−5 M phenylephrine produced a maximal response with low variability (SEM) (Supplementary Figure 1), so this dose of phenylephrine was chosen as the single treatment dose to assess functional responses and eNOS phosphorylation status in the experiments described above. Although this dose produces a maximal drug response, greater constriction is still possible in this artery when endothelium-dependent vasodilation is impaired either through denudation or NOS inhibition.
Vessel diameter responses were calculated by: diameter with drug/baseline diameter. Vessel function experiments were performed in two arteries from each mouse, but different treatments were performed in each of the two arteries.
2.2. Phosphorylation of eNOS in Mesenteric Arteries After Drug Treatment
Western blotting was performed to quantify the relative phosphorylation of eNOS at Ser1179 and Thr495 in pooled mesenteric arteries (4–5 arteries for a given treatment per western blot). Tissues were homogenized in 80–100 μl lysis buffer with phosphatase inhibitors [50 mM Tris-HCl, 100 mM NaF, 15 mM Na4P2O7, 1 mM Na3VO4, 1% Triton X-100, and 1:200 protease inhibitor cocktail solution (#P2714, Sigma, St. Louis, MO); pH=7.6], incubated for 1 hr at 4 °C, and centrifuged (14,000 rpm, 10 min) to remove insoluble material. Proteins were separated by 10% SDS-PAGE (4% stacking gel) using 25 μl of protein. Proteins were transferred to a nitrocellulose membrane and total protein was visualized on the membrane with ponceau-S (in some but not all membranes). Membranes were cut in half between the 75 and 50KDa markers, and the higher molecular weight half was immuno-labeled first for phosphorylated eNOS, either pSer1179-eNOS (1:750; BD Biosciences, San Jose, CA) or pThr495-eNOS (1:1000, BD Biosciences), followed by HRP-conjugated secondary antibody (anti-mouse 1:5000, Pierce Biotechnology, Rockford, IL), and visualized with enhanced chemiluminescence (Pierce Biotechnology) captured on film. The membrane was then stripped for 20 minutes (Restore Western Blot Stripping Buffer, Thermo Scientific, Waltham, MA), and re-probed with eNOS (1:1000, BD Biosciences) using the same procedure as with the phosphorylated forms. We have previously shown that this stripping protocol results in negligible residual labeling of the phosphorylated form even with long film exposure times (Looft-Wilson et al, 2008). The half of the membrane containing the lower molecular weight proteins was labeled for gapdh (1:1000; Millipore, Temucula, CA) or β-actin (1:1000; Cell Signaling Technology, Danvers, MA).
2.3. Data Analysis
Statistics were performed using Prism software (GraphPad Software, Inc., San Diego, CA). Vessel responses were compared between groups by two-way ANOVA with repeated measures (Bonferroni post-hoc analysis). Protein bands were quantified by scanning the film and determining the density of each band using Image J software (NIH). Single bands were detected on the membranes at the appropriate sizes: gapdh (38 KDa), pS1179eNOS (140 KDa), pT495-eNOS (140 KDa), eNOS (140 KDa), beta-actin (45 KDa). Phosphorylated eNOS protein was normalized to total eNOS protein and this ratio was normalized to the no treatment (No T) values in each blot to determine the relative change in eNOS phosphorylation at each site in response to the drug treatments, and to allow combining of different blots. Treatments were compared by one-way ANOVA (Bonferroni post-hoc analysis). Initial diameters between groups were compared by one-way ANOVA (Bonferroni post-hoc analysis), and comparisons of initial diameters to baseline diameters after 30 min equilibration were compared within each group by paired t-test. Significance was at P<0.05. The EC-50 for phenylephrine was calculated for each vessel in the phenylephrine cumulative dose-response trial using GraphPad Prism’s sigmoidal plot function.
3. Results
Addition of phenylephrine to mesenteric arteries caused an initial constriction followed by a slow dilation over the 15 minutes (Fig. 1A and 1B), similar to the responses observed in mouse thoracodorsal arteries (Straub et al, 2011). This reactive slow dilation was dependent upon NOS activation because it was blocked by incubation with L-NAME (10−4 M), and required the presence of the endothelium because it was eliminated by denudation. Subsequent addition of acetylcholine resulted in full dilation that was ~50% dependent upon NOS activation as indicated by the impaired dilation after 5 min of acetylcholine addition, and fully dependent upon an intact endothelium as dilation was nearly eliminated by denudation. The early dilation after acetylcholine addition (1–2 min) is less dependent upon NOS as indicated by the near maximal dilation (relative to baseline diameter) in the presence of L-NAME at these time-points. In addition, we have previously provided evidence that this early response in these arteries is attributable to a mechanism consistent with endothelium-dependent hyperpolarization (EDH), because it is blocked by high K+ and is not due prostacyclin and only partially due to NOS (Looft-Wilson et al, 2008). We also showed in this previous study that the artery will remain ~50% dilated for at least 30 minutes after acetylcholine addition and this dilation is due to NOS, as it is completely blocked by L-NAME. When these responses were examined in terms of absolute diameter changes (Figure 1B), it is clear that L-NAME and denuation resulted in greater basal tone and a deeper constriction, which is consistent with results found by others (Dora et al, 2000).
Figure 1.

Artery diameter responses (mean ± SEM) to phenylephrine (10−5 M) superfusion followed by acetycholine (10−4 M) without (Control) or with L-NAME (10−4 M) (L-NAME) in the superfusate and luminal perfusate, or with denudation (Denuded). (A) Diameter responses are represented as % of baseline diameter measured at 30 min post-equilibration. (B) Absolute diameter values are plotted for the same vessels as in panel A. Maximal diameters (estimated from initial diameter in cold PSS) were similar between the groups, indicating that the L-NAME and Denuded groups had basal tone after equilibration. * significantly (p<0.05) different from both L-NAME and Denuded. # significantly (p<0.05) different from L-NAME. Twenty-two of the Control arteries are the same ones depicted in the PE+ACh group in Figure 2 and analyzed by western blot in Figures 3 and 4, and three of the Control arteries and three of the Denuded arteries were analyzed by western blot in Figure 5. Values for one of the Control arteries were estimated at times 1–5 min based on group average due to a computer crash during these time-points for this artery.
Arteries in the different treatment groups had similar vasomotor responses to agonists up to the time-point at which they were frozen (Figure. 2). The corresponding eNOS phosphorylation events at the various time-points are shown in Figures 3 and 4 (Individual western blot films and quantifications are displayed in Supplementary Figure 4 and the Supplementary Table). Phosphorylation of eNOS at Ser1179 relative to total eNOS expression (Figure 3) was significantly increased by more than 2-fold after 5 min of phenylephrine treatment, and remained similarly increased after 15 min of phenylephrine treatment and after acetylcholine addition. Even though the arteries in PE15 and PE+ACh groups were 63% and 199% more dilated, respectively, than PE5 (% dilation: PE5 = 30.1 ± 9.6%; PE15 = 49.1 ± 12.8%; PE+ACh = 90.1 ± 10.1%), the relative phosphorylation at Ser1179-eNOS was not different. Phosphorylation of eNOS at Thr495 (Figure 4) showed a trend toward decreasing in all groups relative to the control group (NT), but did not reach signficance. Total eNOS expression was not different between the groups when normalized to the housekeeping gene gapdh (Supplementary Figure 3).
Figure 2.
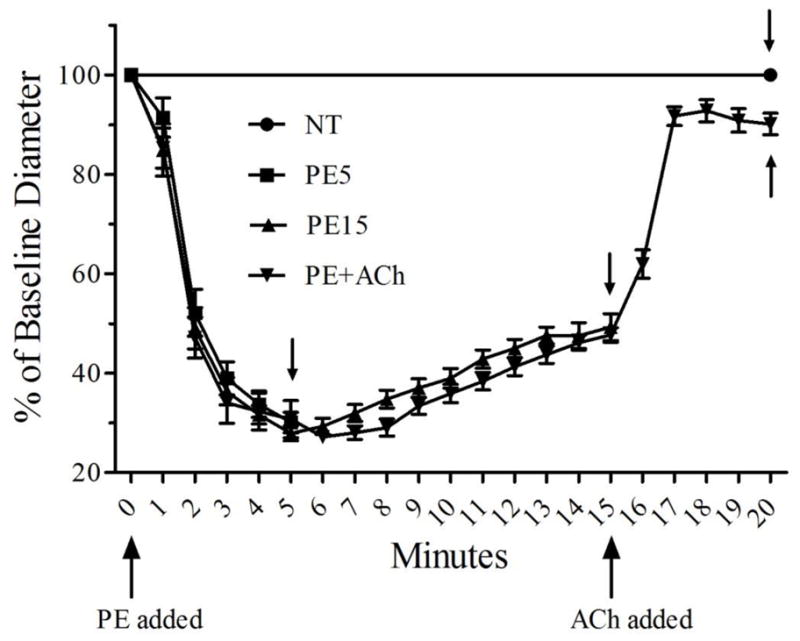
Artery diameter responses for all vessels in each of the four treatments used to detect eNOS phosphorylation. Each treatment group contains 22 total arteries, with 4–5 arteries used in each of the 5 western blot trials presented in Figures 3 and 4. Arteries were frozen immediately after the last diameter measurement in each of the four different treatments, which corresponds to min 20 for the NT and PE+ACh groups, min 5 for the PE5 group and min 15 for the PE15 group (small arrows show the time-point when arteries were frozen in a given group). Values for min 1–5 are missing for one vessel in the PE+ACh group due to a computer crash.
Figure 3.
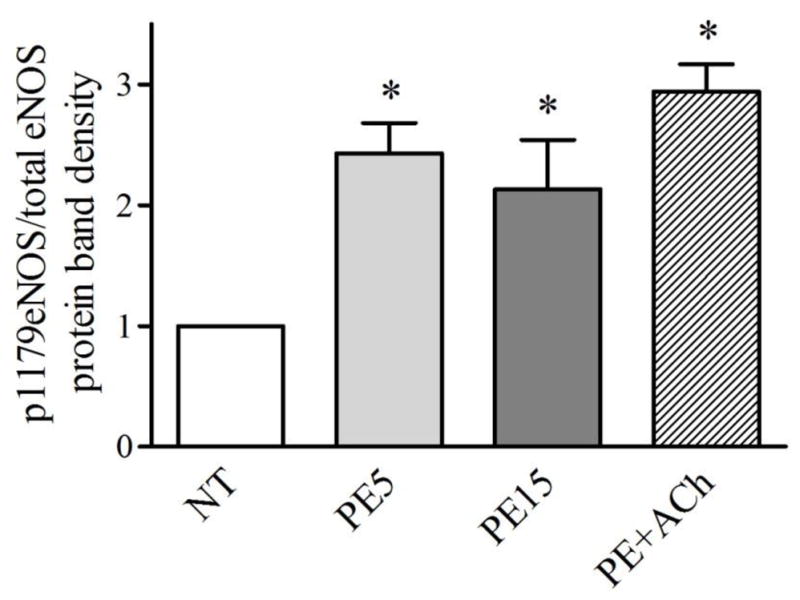
eNOS phosphorylation at Ser1179 increased significantly (p<0.05) relative to control arteries (NT) after treatment with phenylephrine (10−5M) for 5 (PE5) or 15 min (PE15), or treatment with PE for 15 min followed by 5 min with acetylcholine (10−4M) (PE+ACh). There were no differences in phosphorylation between the drug treatment groups. Data are the mean of 5 western blots, with 4–5 arteries pooled for each treatment in a given western blot.
Figure 4.
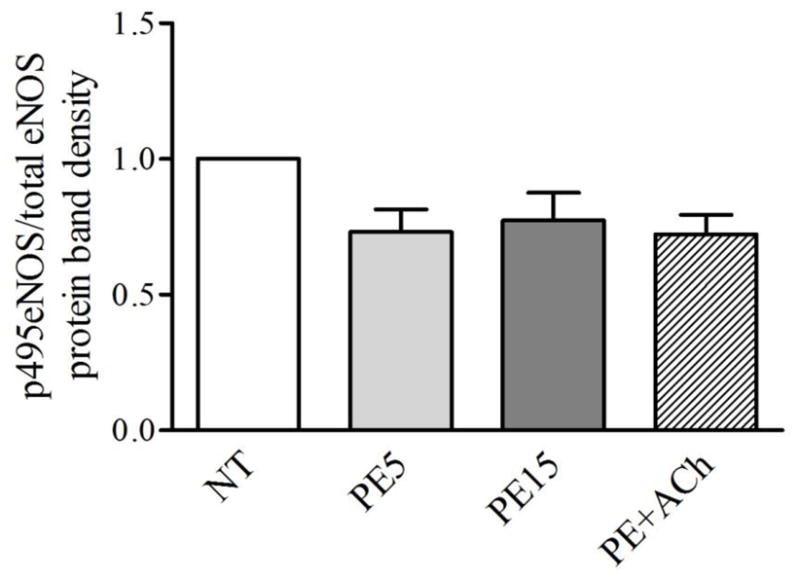
eNOS phosphorylation at Thr495 was decreased, but not significantly, relative to control arteries (NT) after treatment with phenylephrine (10−5M) for 5 (PE5) or 15 min (PE15), or treatment with PE for 15 min followed by 5 min with acetylcholine (10−4M) (PE+ACh). There were no differences in phosphorylation between the drug treatment groups. Data are the mean of 5 western blots, with 4–5 arteries pooled for each treatment in a given western blot.
When eNOS expression was compared between three of the Control arteries in Figure 1 and three of the Denuded arteries in Figure 1, it is clear that the majority of eNOS is expressed in the endothelium because the eNOS signal in the Denuded group was only ~5% as strong as the signal in the Control group (Figure 5). The residual signal in the Denuded group could mean that a minor amount of eNOS is expressed in other cell types in the vessel wall, or that denudation was no complete. Incomplete denudation is a possibility because there are many endothelial cell projections that extend through the basal lamina to the smooth muscle and eNOS has been indentified in these projections (Straub et al, 2011).
Figure 5.

eNOS is highly expressed in Control arteries (n=3 pooled arteries), but relative expression declines by ~95% in Denuded arteries (n=3 pooled arteries), when normalized to the housekeeping gene, beta-actin. This indicates that eNOS expression is largely endothelial. The lower membrane was first probed for gapdh, but the signal was too weak, so it was stripped and re-probed for beta-actin. Integrated density values (measured with Image J) are shown below each band. The ratio of these values for eNOS/beta-actin are calculated below the films, and then normalized to the Control ratio for relative comparison between the two groups.
4. Discussion
This study is the first to show the time-course of eNOS phosphorylation with alpha1-adrenergic stimulation of smooth muscle and it revealed some unexpected results. Because stimulation of intact arteries with phenylephrine has been shown to result in both increased endothelial cell calcium and eNOS activation, it might be assumed that the increased calcium was responsible for the activation of eNOS, particularly because both the increase in calcium and expression of eNOS are found in the endothelial projections at the myoendothelial junction in mouse thoracodorsal arteries (Straub et al, 2011). It is unexpected that phosphorylation of eNOS would occur as a result of phenylephrine treatment and suggests that additional communication occurs from smooth muscle to endothelial cell either through gap junctions or possibly via a smooth muscle autacoid that activates enzymatic processes leading to phosphorylation of eNOS at Ser1179. There are several enzymes known to phosphorylate eNOS at this site, including AKT1, AMPK, PKA, and CaMKII (Chen et al, 1999; Fleming et al, 2001; Fulton et al, 1999; Gallis et al, 1999; Harris et al, 2001; Michell et al, 2001; Michell et al, 1999). It is unclear which of these pathways are involved and how they are activated by alpha1-adrenergic stimulation, but CaMKII is activated by increased intracellular calcium, so this is a possible mechanism (Fleming et al, 2001).
Also, unexpectedly, the vasomotor tone of the intact vessel did not correlate with phosphorylation status of eNOS. Firstly, 5 minutes after phenylephrine stimulation, the artery was at its maximal point of constriction, yet eNOS was also at a level of Ser1179 phosphorylation that appears to be maximal. Blockade of nitric oxide synthases or denudation at this time-point did not increase the relative magnitude of constriction (% constriction) indicating that the influence of eNOS on vasomotor tone at this time-point is minimal. It should be noted, however, that the absolute constriction is greater due to the increase in basal tone with NOS inhibition and denudation, as illustrated in Figure 1B. Secondly, NOS and an intact endothelium was critical for dilation that occurred from minutes 5–15 after phenylephrine stimulation, and at its greatest dilation at 15 minutes (~50% dilation), but there was no additional increase in eNOS phosphorylation at Ser1179. Finally, full dilation induced by acetylcholine, which was ~50% NOS-dependent at 5 min after stimulation, did not result in any further Ser1179 phosphorylation. These data indicate that artery dilation induced by either phenylephrine or acetylcholine was not accompanied by active phosphorylation at Ser1179. Rather, it appears that phenylephrine stimulation induced Ser1179 phosphorylation before the functional contribution of eNOS and thus may play a permissive role in its activation by other mechanisms. For example, it may increase the sensitization to intracellular calcium as shown in cultured endothelial cells (McCabe et al, 2000). This study does not provide direct evidence, however, as to the mechanism by which eNOS is ultimately activated, which may involve one or more mechanisms.
Typically, endothelial cells stimulated by agonists have decreased phosphorylation of eNOS on Thr495 (Fleming et al, 2001; Harris et al, 2001). In this study, there was a trend for reduced Thr495 phosphorylation, but it did not reach significance. It is possible that intact arteries have lower basal levels of Thr495 phosphorylation than cultured endothelial cells, such that agonist-induced stimulation has a negligible effect at this site, or that this site is regulated differently in intact arteries compared to cultured endothelial cells.
This study utilized first order mesenteric arteries as a model for several reasons. They are large enough to examine both functional responses and to provide enough tissue (even though it has to be pooled from several arteries) for western blot analysis. It also has relatively robust reliance on eNOS for both phenylephrine-induced dilation and endothelium-dependent dilation (Figure 1). In smaller mesenteric arteries this NOS-dependence is less important and there is more reliance on endothelium-dependent hyperpolarization (EDH) in mice (Nausch et al, 2012) and rats (Garland & McPherson, 1992; Parsons et al, 1994; Shimokawa et al, 1996). This is likely the reason why a recent study by Nausch et al. (Nausch et al, 2012) found that with phenylephrine stimulation, the resulting vasodilation was not dependent upon NOS, but rather endothelial K+ channel activation consistent with EDH mechanisms. The first order mesenteric arteries do rely on mechanisms consistent with EDH for the intial dilation to acetycholine (Looft-Wilson et al, 2008), but all the slow dilation secondary to phenylephrine stimulation appears to be NOS-dependent as shown in Figure 1.
The final reason for using this model is that examination of phenylephrine-induced responses in this vessel are of physiological importance. The mesenteric vascular bed is highly innervated by sympathetic nerves and contributes significantly to vascular resistance in rodents (Christensen & Mulvany, 1993; Long & Segal, 2009). Thus, it is likely that vasodilatory mechanisms will occur in the presence of some degree of underlying sympathetic tone. Moreover, it has been shown in mouse mesenteric arteries that the norepinephrine released from the sympathetic varicosities is the primary sympathetic neurotransmitter responsible for the slow dilation that occurs in response to sympathetic nerve stimulation, and that the alpha1-adrenergic receptors are the adrenergic receptors involved (Nausch et al, 2012). We confirmed in this study that constriction to phenylephrine is completely blocked by the alpha1-adrenergeric receptor blocker prazosin (Supplementary Figure 2). Therefore, phenylephrine stimulation of the intact artery, as performed in this study, is a legitimate way to mimic the effects of nerve stimulation in vitro in a targeted way.
A possible limitation of using phenylephrine superfusion to mimic the effects of norepinephrine release from sympathetic nerves is that the phenylephrine likely diffuses to the endothelial cells and could interact with receptors on these cells if present. This is unlikely, however, because at least in hamster arterioles, there are no alpha-adrenergic receptors expressed in endothelial cells and these vessels respond to phenylephrine treatment in a similar way as mesenteric arteries, with an increase in endothelial intracellular calcium (Jackson et al, 2008). Moreover, in mouse mesenteric arteries it has been shown that picospritzing phenylephrine onto the endothelium (such that it does not interact with the smooth muscle) does not result in increased endothelial cell calcium (Nausch et al, 2012). Thus, it is very unlikely that the artery responses are due to phenylephrine action on endothelial cells.
5. Conclusions
In summary, this study shows for the first time the correlation between intact artery vasomotor tone and the time-course of eNOS phosphorylation at Ser1179 and Thr495 during alpha1-adrenergic receptor stimulation and endothelium-dependent vasodilation. The significant increase in Ser1179 phosphorylation occurred shortly after phenylephrine stimulation and remained at the same level over many minutes of phenylephrine stimulation and acteylcholine stimulation, despite changes in vasomotor tone. These data suggest that the phosphorylation at this site is not controlling the NOS-dependent effect on vasomotor tone, but may be acting permissively to enhance eNOS stimulation by other mechanisms.
Supplementary Material
Acknowledgments
The authors thank Dr. Brennan Harris for helpful comments in the study design and manuscript preparation. This work was supported by NIH-1R15HL082647 and NIH-1R15HL102742 (R.C. Looft-Wilson) and The American Physiological Society’s Undergraduate Summer Research Fellowship (S.E. Todd, 2009; S.M. Mutchler, 2011).
Footnotes
Disclosures
The authors of this manuscript have no conflicts of interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Atochin DN, Wa0ng A, Liu VW, Critchlow JD, Dantas AP, Looft-Wilson R, Murata T, Salomone S, Shin HK, Ayata C, Moskowitz MA, Michel T, Sessa WC, Huang PL. The phosphorylation state of eNOS modulates vascular reactivity and outcome of cerebral ischemia in vivo. The Journal of clinical investigation. 2007;117:1961–1967. doi: 10.1172/JCI29877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boric MP, Figueroa XF, Donoso MV, Paredes A, Poblete I, Huidobro-Toro JP. Rise in endothelial-derived NO after stimulation of rat perivascular sympathetic mesenteric nerves. Am J Physiol. 1999;277:H1027–H1035. doi: 10.1152/ajpheart.1999.277.3.H1027. [DOI] [PubMed] [Google Scholar]
- Brandes RP, Schmitz-Winnenthal FH, Feletou M, Godecke A, Huang PL, Vanhoutte PM, Fleming I, Busse R. An endothelium-derived hyperpolarizing factor distinct from NO and prostacyclin is a major endothelium-dependent vasodilator in resistance vessels of wild-type and endothelial NO synthase knockout mice. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:9747–9752. doi: 10.1073/pnas.97.17.9747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZP, Mitchelhill KI, Michell BJ, Stapleton D, Rodriquez-Crespo I, Witters LA, Power DA, Ortiz de Montellano PR, Kemp BE. AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett. 1999;443:285–289. doi: 10.1016/s0014-5793(98)01705-0. [DOI] [PubMed] [Google Scholar]
- Christensen KL, Mulvany MJ. Mesenteric arcade arteries contribute substantially to vascular resistance in conscious rats. J Vasc Res. 1993;30:73–79. doi: 10.1159/000158978. [DOI] [PubMed] [Google Scholar]
- Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- Dora KA, Doyle MP, Duling BR. Elevation of intracellular calcium in smooth muscle causes endothelial cell generation of NO in arterioles. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:6529–6534. doi: 10.1073/pnas.94.12.6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dora KA, Hinton JM, Walker SD, Garland CJ. An indirect influence of phenylephrine on the release of endothelium-derived vasodilators in rat small mesenteric artery. British journal of pharmacology. 2000;129:381–387. doi: 10.1038/sj.bjp.0703052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dora KA, Sandow SL, Gallagher NT, Takano H, Rummery NM, Hill CE, Garland CJ. Myoendothelial gap junctions may provide the pathway for EDHF in mouse mesenteric artery. J Vasc Res. 2003;40:480–490. doi: 10.1159/000074549. [DOI] [PubMed] [Google Scholar]
- Fleming I, Fisslthaler B, Dimmeler S, Kemp BE, Busse R. Phosphorylation of Thr(495) regulates Ca(2+)/calmodulin-dependent endothelial nitric oxide synthase activity. Circ Res. 2001;88:E68–75. doi: 10.1161/hh1101.092677. [DOI] [PubMed] [Google Scholar]
- Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399:597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallis B, Corthals GL, Goodlett DR, Ueba H, Kim F, Presnell SR, Figeys D, Harrison DG, Berk BC, Aebersold R, Corson MA. Identification of flow-dependent endothelial nitric oxide synthase phosphorylation sites by mass spectrometry and regulation of phosphorylation and nitric oxide production by the phosphatidylinositol 3-kinase inhibitor LY294002. The Journal of biological chemistry. 1999;274:30101–30108. doi: 10.1074/jbc.274.42.30101. [DOI] [PubMed] [Google Scholar]
- Garland JG, McPherson GA. Evidence that nitric oxide does not mediate the hyperpolarization and relaxation to acetylcholine in the rat small mesenteric artery. British journal of pharmacology. 1992;105:429–435. doi: 10.1111/j.1476-5381.1992.tb14270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris MB, Ju H, Venema VJ, Liang H, Zou R, Michell BJ, Chen ZP, Kemp BE, Venema RC. Reciprocal phosphorylation and regulation of endothelial nitric oxide synthase in response to bradykinin stimulation. The Journal of biological chemistry. 2001;276:16587–16591. doi: 10.1074/jbc.M100229200. [DOI] [PubMed] [Google Scholar]
- Jackson WF, Boerman EM, Lange EJ, Lundback SS, Cohen KD. Smooth muscle alpha1D-adrenoceptors mediate phenylephrine-induced vasoconstriction and increases in endothelial cell Ca2+ in hamster cremaster arterioles. British journal of pharmacology. 2008;155:514–524. doi: 10.1038/bjp.2008.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlencordt PJ, Gyurko R, Han F, Scherrer-Crosbie M, Aretz TH, Hajjar R, Picard MH, Huang PL. Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease in apolipoprotein E/endothelial nitric oxide synthase double-knockout mice. Circulation. 2001;104:448–454. doi: 10.1161/hc2901.091399. [DOI] [PubMed] [Google Scholar]
- Long JB, Segal SS. Quantifying perivascular sympathetic innervation: regional differences in male C57BL/6 mice at 3 and 20 months. J Neurosci Methods. 2009;184:124–128. doi: 10.1016/j.jneumeth.2009.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Looft-Wilson RC, Ashley BS, Billig JE, Wolfert MR, Ambrecht LA, Bearden SE. Chronic diet-induced hyperhomocysteinemia impairs eNOS regulation in mouse mesenteric arteries. Am J Physiol Regul Integr Comp Physiol. 2008;295:R59–66. doi: 10.1152/ajpregu.00833.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe TJ, Fulton D, Roman LJ, Sessa WC. Enhanced electron flux and reduced calmodulin dissociation may explain “calcium-independent” eNOS activation by phosphorylation. The Journal of biological chemistry. 2000;275:6123–6128. doi: 10.1074/jbc.275.9.6123. [DOI] [PubMed] [Google Scholar]
- Michell BJ, Chen ZP, Tiganis T, Stapleton D, Katsis F, Power DA, Sim AT, Kemp BE. Coordinated control of endothelial nitric oxide synthase phosphorylation by protein kinase C and the cAMP-dependent protein kinase. The Journal of biological chemistry. 2001;276:17625–17628. doi: 10.1074/jbc.C100122200. [DOI] [PubMed] [Google Scholar]
- Michell BJ, Griffiths JE, Mitchelhill KI, Rodriquez-Crespo I, Tiganis T, Bozinovski S, de Montellano PR, Kemp BE, Pearson RB. The Akt kinase signals directly to endothelial nitric oxide synthase. Curr Biol. 1999;9:845–848. doi: 10.1016/s0960-9822(99)80371-6. [DOI] [PubMed] [Google Scholar]
- Murohara T, Asahara T, Silver M, Bauters C, Masuda H, Kalka C, Kearney M, Chen D, Symes JF, Fishman MC, Huang PL, Isner JM. Nitric oxide synthase modulates angiogenesis in response to tissue ischemia. The Journal of clinical investigation. 1998;101:2567–2578. doi: 10.1172/JCI1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nausch LW, Bonev AD, Heppner TJ, Tallini Y, Kotlikoff MI, Nelson MT. Sympathetic nerve stimulation induces local endothelial Ca2+ signals to oppose vasoconstriction of mouse mesenteric arteries. American journal of physiology Heart and circulatory physiology. 2012;302:H594–602. doi: 10.1152/ajpheart.00773.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons SJ, Hill A, Waldron GJ, Plane F, Garland CJ. The relative importance of nitric oxide and nitric oxide-independent mechanisms in acetylcholine-evoked dilatation of the rat mesenteric bed. British journal of pharmacology. 1994;113:1275–1280. doi: 10.1111/j.1476-5381.1994.tb17136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafikov R, Fonseca FV, Kumar S, Pardo D, Darragh C, Elms S, Fulton D, Black SM. eNOS activation and NO function: structural motifs responsible for the posttranslational control of endothelial nitric oxide synthase activity. J Endocrinol. 2011;210:271–284. doi: 10.1530/JOE-11-0083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudic RD, Shesely EG, Maeda N, Smithies O, Segal SS, Sessa WC. Direct evidence for the importance of endothelium-derived nitric oxide in vascular remodeling. The Journal of clinical investigation. 1998;101:731–736. doi: 10.1172/JCI1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saliez J, Bouzin C, Rath G, Ghisdal P, Desjardins F, Rezzani R, Rodella LF, Vriens J, Nilius B, Feron O, Balligand JL, Dessy C. Role of caveolar compartmentation in endothelium-derived hyperpolarizing factor-mediated relaxation: Ca2+ signals and gap junction function are regulated by caveolin in endothelial cells. Circulation. 2008;117:1065–1074. doi: 10.1161/CIRCULATIONAHA.107.731679. [DOI] [PubMed] [Google Scholar]
- Scotland RS, Morales-Ruiz M, Chen Y, Yu J, Rudic RD, Fulton D, Gratton JP, Sessa WC. Functional reconstitution of endothelial nitric oxide synthase reveals the importance of serine 1179 in endothelium-dependent vasomotion. Circ Res. 2002;90:904–910. doi: 10.1161/01.res.0000016506.04193.96. [DOI] [PubMed] [Google Scholar]
- Sessa WC. eNOS at a glance. J Cell Sci. 2004;117:2427–2429. doi: 10.1242/jcs.01165. [DOI] [PubMed] [Google Scholar]
- Shesely EG, Maeda N, Kim HS, Desai KM, Krege JH, Laubach VE, Sherman PA, Sessa WC, Smithies O. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:13176–13181. doi: 10.1073/pnas.93.23.13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimokawa H, Yasutake H, Fujii K, Owada MK, Nakaike R, Fukumoto Y, Takayanagi T, Nagao T, Egashira K, Fujishima M, Takeshita A. The importance of the hyperpolarizing mechanism increases as the vessel size decreases in endothelium-dependent relaxations in rat mesenteric circulation. Journal of cardiovascular pharmacology. 1996;28:703–711. doi: 10.1097/00005344-199611000-00014. [DOI] [PubMed] [Google Scholar]
- Straub AC, Billaud M, Johnstone SR, Best AK, Yemen S, Dwyer ST, Looft-Wilson R, Lysiak JJ, Gaston B, Palmer L, Isakson BE. Compartmentalized connexin 43 s-nitrosylation/denitrosylation regulates heterocellular communication in the vessel wall. Arterioscler Thromb Vasc Biol. 2011;31:399–407. doi: 10.1161/ATVBAHA.110.215939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuttle JL, Falcone JC. Nitric oxide release during alpha1-adrenoceptor-mediated constriction of arterioles. American journal of physiology Heart and circulatory physiology. 2001;281:H873–881. doi: 10.1152/ajpheart.2001.281.2.H873. [DOI] [PubMed] [Google Scholar]
- Yu J, deMuinck ED, Zhuang Z, MD, Kauser K, Rubanyi GM, Qian HS, Murata T, Escalante B, Sessa WC. Endothelial nitric oxide synthase is critical for ischemic remodeling. mural cell recruitment, and blood flow reserve. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:10999–11004. doi: 10.1073/pnas.0501444102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.