

Abstract
This Letter describes the continued optimization of an MLPCN probe molecule (ML137) with a focused effort on the replacement/modification of the isatin moiety present in this highly selective M1 PAM. A diverse range of structures were validated as viable replacements for the isatin, many of which engendered sizeable improvements in their ability to enhance the potency and efficacy of acetylcholine when compared to ML137. Muscarinic receptor subtype selectivity for the M1 receptor was also maintained.
Keywords: Muscarinic acetylcholine receptor 1, M1, Allosteric, Positive allosteric modulator (PAM), ML137, VU0448350
Acetylcholine (ACh) is a critical neurotransmitter influencing a broad range of physiological processes both inside and outside of the central nervous system by binding to both nicotinic (nAChR) and muscarinic acetylcholine receptors (mAChR). The challenges associated with developing highly selective mAChR ligands interacting at the orthosteric binding site of ACh are most significantly impacted by the high level of receptor homology displayed across the five mAChR subtypes (M1 through M5).1 Despite this seemingly insurmountable roadblock towards the production of truly selective muscarinic agonists, a great deal of information has been learned from M1–5 KO mice2 and the testing of less-than-selective muscarinic agonists in preclinical and clinical experiments. The clinical promise for dual, or potentially individual, M1/M4 mAChR activators in treating aspects of both Alzheimer’s disease and schizophrenia has been demonstrated with the orthosteric agonist xanomeline;3,4 however, this clinical candidate’s limited selectivity (particularly against M3) ultimately caused its withdrawal from further clinical trials irrespective of the encouraging positive outcomes. A new and exciting chapter in the search for selective/specific mAChR activators commenced with the identification of novel positive allosteric modulators (PAMs) and allosteric agonists through the use of functional high throughput screening (HTS) assays.5 These next-generation ligands have the novel benefit of interacting at sites removed from the orthosteric binding site, where the potential for selective interactions with just one of the five mAChR is greatly enhanced. This revitalized interest in selectively activating the mAChRs has been very successful, providing a number of M1 PAMs (BQCA,6 ML137,7 and ML1698), M4 PAMs (ML1089, ML173,10 ML253,11 and ML29312) and M5 PAMs (ML12913, and ML17214). Here we describe the continued structural exploration around the M1 PAM, MLPCN probe molecule ML137.
Figure 1 shows the development of two M5 PAMs and one M1 PAM from a common HTS lead, VU0119498, all of which share the isatin core in common. Although VU0119498 displayed PAM activity at the M1, M3, and M5 mAChRs,13 specific structural modifications resulted in highly preferring (ML129) or completely selective (ML172) M5 PAMs and a highly selective M1 PAM (ML137). The ability to dial in muscarinic receptor subtype selectivity was facilitated by the wide variety of commercially available substituted isatins and the efficiency of analog preparation, clearly demonstrating that this scaffold can be very advantageous. However, it was also appreciated that the isatin motif has an established history of appearing in a wide range of medicinal chemistry targets across the full spectrum of potential clinical indications.15 Additionally, HTS hits containing the isatin core are so frequent that the isatin scaffold has earned a position of notoriety in some measures of HTS promiscuity.16 As such, it seemed reasonable to explore replacements, or modifications, to the isatin core in the context of ML129, ML137, and ML172.
Figure 1.

HTS lead VU0119498 parlayed into selective M1 and M5 PAMs.
Our first efforts focused on a large and extensive library which thoroughly and rapidly explored small modifications to the isatin core employing commercially available benzyl, biphenyl and biaryl ethers similar to those found in ML129 and ML172. As is often the case with steep allosteric SAR,5 this effort proved unfruitful (data and structures not shown). We then hypothesized that a more focused effort directed solely at replacing the isatin moiety, while retaining the exact biaryl from ML137, could be more productive. These analogs and the requisite phenylpyrazoles were prepared according to the general synthesis route appearing in Scheme 1.
Scheme 1.

Reagents and conditions: (a) PdCl2(dppf)•DCM, Cs2CO3, THF/H2O, 160 °C, 10 min; (b) Ghosez’ reagent (1-chloro-N,N,2-trimethylprop-1-en-1-amine), DCM, rt, 15 h; (c) base (i.e. K2CO3, NaH, etc.), aprotic solvent (i.e. DCM, DMF, MeCN), 0 °C to rt.
Readily available 4-bromobenzyl alcohols 1 undergo Suzuki reactions with pyrazole boronic acids/esters 2 employing standard conditions and provided the biaryl benzyl alcohols 3. Ghosez’ reagent (1-chloro-N,N,2-trimethylprop-1-en-1-amine) in DCM smoothly converted the benzylic alcohol into the corresponding benzyl chloride 4. With ample quantities of 4 in place, isatin or a variety of isatin replacements could be alkylated, employing a range of bases, to give analogs 5 and the majority of structures appearing in Figure 2 and Table 1. Alternative routes towards the preparation of these initial analogs can be seen in Schemes 2 and 3.
Figure 2.

Rapid foray into isatin replacements while holding the benzyl pyrazole constant.
Table 1.
Structure and activities of analogs 23a–r.

| ||||||||
|---|---|---|---|---|---|---|---|---|
| Compd | *a | R1 | R2 | X | R3 | hM1 EC50 (μM)b | AChmax (%)b,c | pEC50b |
| 23a | ± | Me | H | F | Me | >10 | 53 ± 2 | |
| 23b | ± | NH2 | H | F | Me | 1.3 | 50 ± 3 | 5.90 ± 0.04 |
| 23c | ± | NH2 | Me | F | Me | >10 | 28 ± 2 | |
| 23d | ± | NMe2 | Me | H | Me | >10 | 48 ± 2 | |
| 23e | ± | OH | Me | F | Me | >10 | 49 ± 3 | |
| 23f | ± | OH | CH2F | F | Me | >10 | 59 ± 2 | |
| 23g | ± | OH | CF3 | F | Me | 4.8 | 51 ± 3 | 5.32 ± 0.07 |
| 23h | A | OH | CF3 | F | Me | 3.3 | 57 ± 1 | 5.48 ± 0.07 |
| 23i | B | OH | CF3 | F | Me | - | ||
| 23j | ± | OH | CF3 | F | Et | 3.3 | 37 ± 2 | 5.48 ± 0.07 |
| 23k | ± | OH | CF3 | F | n-Pr | - | ||
| 23l | ± | OH | CF3 | F | i-Pr | - | ||
| 23m | ± | OH | CF3 | F | H | - | ||
| 23n | ± | OMe | CF3 | F | Me | - | ||
| 23o | ± | F | CF3 | F | Me | >10 | 25 ± 2 | |
| 23p | ± | F | F | F | Me | - | ||
| 23q | ± | OH | CF3 | Cl | Me | >10 | 29 ± 2 | |
| 23r | ± | OH | CF3 | H | Me | 4.0 | 66 ± 2 | 5.40 ± 0.05 |
Absolute stereochemistry is unknown, but each resolved enantiomer displayed an ee of >95% as determined by analytical chiral HPLC
Values represent the mean ± standard error of the means of at least three independent determinations performed in triplicate.
“-” indicates an inactive compound showing no PAM activity up to the highest concentration tested (30 μM).
When hM1 EC50 is reported as >10, this value represents the percent maximal ACh response when the test compound was present at its highest concentration (30 μM).
Scheme 2.

Reagents and conditions: (a) K2CO3, MeCN, rt, overnight; (b) TMS-CF3, CsF, THF, rt, overnight; (c) 2, PdCl2(dppf)•DCM, Cs2CO3, THF/H2O, 160 °C, 10 min.
Scheme 3.

Reagents and conditions: (a) NEt3, DCM, 30 min., 93% yield; (b) CuI(cat.), K3PO4, DMEDA (N, N′-dimethylethylene-diamine), toluene, degassed with argon (10 minutes), sealed, 110 °C for 3 h, 59% yield.
Scheme 2 depicts the alkylation of isatin with a 4-bromobenzyl bromide 6 using a carbonate base in acetonitrile to give N-benzylated isatins 7. Next, a trifluoromethyl group was introduced at the ketone carbon employing the conditions of Shreeve, et al.,17 to yield the aryl bromides 8. A Suzuki reaction analogous to the first step in Scheme 1 could then provide analogs 9, as well as a diverse range of alternative biaryls, simply by changing the boronate in the final step.
Scheme 3 shows the route used in the preparation of sultam 13 (See Figure 2 for structure), which was necessary due to a lack of reactivity and selectivity (N versus C benzylation) associated with the desired parent sultam. Starting from commercially available benzylic amine 11 and sulfonyl chloride 10, the linear sulfonamide 12 was prepared in 93% yield. The nitrogen-aryl bond was then formed using Cu(I) catalysis in degassed toluene to give the desired sultam 13, in 59% yield.
Figure 2 shows the functional activity for two active analogs along with a host of unsuccessful isatin replacements (inactive at human M1 (hM1) up to a concentration of 30 μM). Although not as potent (EC50) as their parent ML137 (hM1 EC50 = 0.60 μM, AChmax = 45 ± 3 %, pEC50 = 6.22 ± 0.02),18 both 13 and 14 produced comparable efficacy (% AChmax) at the highest concentration tested of 30 μM and, more importantly, no longer contained the isatin core. Indolinone 14 appeared to be more promising than sultam 13 from activity and synthesis standpoints and so was explored further. While reduction of the isatin ketone was gratifyingly tolerated, attachment of the biaryl at the resultant methylene (as in 15) or further reduction of the amide carbonyl to the indoline 16 was not tolerated. The overall planarity of the indolinone core appeared to be important for PAM activity since scission of the 5-membered lactam to give the N-acetyl amide 17 provided an inactive compound. However, annulation of a second aryl ring, as in 18, proved detrimental to activity as well, despite maintaining a planar system. The remaining analogs in Figure 2 (19–22) attempted to mimic various heteroatom interactions potentially occurring with ML137 or 14, but all retained no hM1 PAM activity.
While full reduction of the isatin ketone to arrive at 14 appeared to be the most tolerated modification so far, the decrease in potency suggested that the introduction of various substituents to this methylene might restore some of the lost activity. Numerous small modifications were made to 14 based on the chemistries appearing in Schemes 1 and 2, as well as through straightforward derivatization of ML137 to provide the compounds appearing in Table 1. The introduction of a methyl group onto the indolinone methylene diminished hM1 PAM activity for 23a. However, the introduction of an amino group at the same location, increased the potency of 23b 2-fold over 14. Unfortunately, this primary amino group instilled an undesirable level of chemical instability, presumed to be air oxidation or self-condensation based on the brightly colored byproducts observed. Attempts to block this degradation with methyl groups provided only weakly active PAMs (23c and 23d). Replacing the amino group with a hydroxyl group provided the stable compound 23e, now with improved efficacy relative to 23c. Fluorination of the methyl group in 23e was beneficial, providing racemic 23g which displayed a measurable EC50 of 4.8 μM. Although not routinely performed during these early stages of structure-activity relationship (SAR) development, the enantiomers of 23g were resolved employing HPLC chiral column chromatography to demonstrate that all the hM1 PAM activity resided in a single enantiomer of unknown absolute configuration (compare 23h and 23i).
With 23g showing promise as another viable replacement for the isatin core, we explored alternatives to the N-methyl pyrazole. Although a wide range of heterocycles could be efficiently introduced at the terminal location of the biaryl utilizing the chemistry in Scheme 2, none was found to be superior to the pyrazole (data and structures not shown for about 30 diverse alternative aryl groups). Only the most minor of modifications was tolerated in that the N-ethyl pyrazole 23j was found to be of comparable potency, but with reduced efficacy. Alkyl groups larger than ethyl (23k and 23l), or the non-alklyated pyrazole 23m resulted in a complete loss of activity. Additional modifications to 23g appeared detrimental towards hM1 PAM activity (23n–q) with the exception of deleting the fluorine on the central phenyl ring providing 23r, a compound of similar potency when compared to 23g.
Unfortunately, none of the analogs appearing in Table 1 dramatically surpassed the potency of 14. However, the relative simplicity of 14 inspired us to explore more benzo-fused bicycles which did not contain a 5-membered ring as the point of attachment to the biaryl southern region. In direct analogy to 14, expansion of the 5-membered lactam to a 6- or 7-membered version caused a drop in activity (Table 2, 24a and 24b). Although 24a retained a measurable hM1 EC50 of 5.6 μM, its unsaturated quinolinone analog 24c possessed a hM1 EC50 > 10 μM, but with slightly improved efficacy. Deletion of the phenyl group from 24c to arrive at pyridone 24d resulted in a loss of all PAM activity, indicating the importance of this benzo-fused motif. Placing an oxygen adjacent to the aryl ring (24e) resulted in a compound of similar potency to 24a, while the carbamate and urea analogs 24f and 24g, respectively, maintained reasonable hM1 PAM efficacy but with EC50s greater than 10 μM. The thio version of 24e, compound 24h, appears to have improved potency relative to 24e; however, these two potency values are not statistically different as seen by comparing their experimentally determined pEC50 values. Oxidized versions of 24h and its carbonyl-reduced analog 24k all showed moderately reduced activity (compounds 24i and 24j) or no activity at all (compound 24k). This complete lack of activity for 24k was not a surprise given the results obtained for 16, and served to reiterate the importance of this carbonyl. This survey of expanded-ring analogs in Table 2 barely scratches the surface of possible structures and a more focused optimization of any slightly active structure could prove highly productive.
Table 2.
Structures and activities of analogs 24a–k attached to the preferred biaryl.

| ||||
|---|---|---|---|---|
| Compd | hM1 EC50 (μM)a | AChmax (%)a,b | pEC50a | |
| 24a |

|
5.6 | 44 ± 3 | 5.25 ± 0.07 |
| 24b |
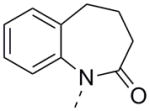
|
- | ||
| 24c |

|
>10 | 51 ± 3 | |
| 24d |

|
- | ||
| 24e |

|
5.2 | 52 ± 3 | 5.29 ± 0.09 |
| 24f |

|
>10 | 62 ± 2 | |
| 24g |
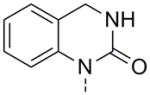
|
>10 | 53 ± 2 | |
| 24h |

|
3.2 | 30 ± 2 | 5.49 ± 0.12 |
| 24i |

|
>10 | 46 ±3 | |
| 24j |

|
>10 | 36± 2 | |
| 24k |

|
- | ||
Values represent the mean ± standard error of the means of at least three independent determinations performed in triplicate.
“-” indicates an inactive compound showing no PAM activity up to the highest concentration tested (30 μM).
When hM1 EC50 is reported as >10, this value represents the percent maximal ACh response when the test compound was present at its highest concentration (30 μM).
The utility of selective M1 PAMs as a therapeutic agent has not been explored to the extent where it is definitively known whether a high efficacy (high fold shift of an ACh concentration-response) PAM or a low efficacy PAM is preferable under the various disease states for which they might provide clinical efficacy. However, ML137, with an ability to shift the ACh concentration-response by just 2.7, rests firmly in the latter category. Therefore, it was desirable to characterize tool compounds spanning a wide range of fold-shift values. Although none of the hM1 PAM potencies presented here rivaled that of the lead compound ML137, improvements with respect to efficacy were being realized and likely contributed to the improved ACh fold-shift values appearing in Table 3. Reduction of the isatin carbonyl (ML137 to 14) caused a 4-fold decrease in hM1 PAM EC50, but at the same time resulted in a 10-fold improvement in ACh fold-shift. As a group, the novel compounds appearing in Table 3 all shared a reduced potency relative to ML137, while at the same time, displayed improved fold-shifts ranging from 6.5 to 27.2. A graphical representation of four hM1 PAMs, and the vehicle control for ACh, as part of a fold-shift experiment is shown in Figure 3. Compound 14 (VU0448350-1), with the highest fold-shift, shifted the ACh concentration-response curve (CRC) furthest to the left, indicating its superior ability to enhance the activity of ACh among this set of compounds. By way of comparison, ML137 (VU0366369-1), which displays an order of magnitude lower fold-shift value relative to 14, produces the first concentration-response curve just to the left (open triangles) of the vehicle control (ACh alone, open squares).
Table 3.
ACh fold-shifts for selected hM1 PAMs.
| Compd | hM1 ACh fold-shifta |
|---|---|
| ML137 | 2.7 ± 0.2 |
| 14 | 27.2 ± 0.6 |
| 23g | 7.4 ± 0.6 |
| 23h | 12 ± 2 |
| 23e | 6.5 ± 0.3 |
| 23f | 8.0 ± 0.3 |
| 23r | 15.2 ± 0.5 |
Leftward shift of an ACh CRC in the presence of 30 μM compound relative to the ACh CRC control. Values represent the mean ± standard error of the means of at least three independent determinations.
Figure 3.

Fold-shift CRCs for the hM1 PAMs: ML137 (VU0366369-1), 14 (VU0448350-1), 23g (VU0422337-1), and 23r (VU0449055-1).
Seemingly minor modifications to allosteric ligands can result in large changes to both potency and receptor subtype selectivity (off-target activity in general) and this observation has been embodied within the concept of the ‘molecular switch’.5, 19 With this idea in mind, we chose to profile the highest efficacy compound (14) across all five of the human muscarinic receptor subtypes; encouragingly, we found little-to-no erosion of muscarinic subtype selectivity (VU0448350-1, Figure 4). Although it would appear from the graph that 14 displays very weak hM4 PAM activity, examination of the raw calcium traces from the functional assay indicated this was most likely an artifact resulting from an unexplained baseline increase at the two highest concentrations tested (data not shown). Fundamentally, the identification of a selective M1/M4 PAM would be a significant achievement in light of the positive clinical outcomes observed with xanomeline;3,4 unfortunately, compound 14 does not represent a step along that path.
Figure 4.

Muscarinic selectivity for 14 (VU0448350-1).
In summary, a number of bicyclic benzo-fused isatin replacements have been identified which retain reasonable hM1 PAM activity and may represent novel starting points for continued optimization. Compound 14, which showed the largest improvement in efficacy compared to ML137, was also demonstrated to be very selective for the hM1 receptor over the other muscarinic receptor subtypes. The continued optimization of ML137 along alternative lines will be reported shortly.
Acknowledgments
The authors thank the NIH (U54MH084659) and NIMH (1RO1MH082867) for support of our M1 program. Vanderbilt University is a Specialized Chemistry Center within the MLPCN, and all ML# probes are freely available upon request.20
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Dencker D, Thomsen M, Wörtwein G, Weikop P, Cui Y, Jeon J, Wess J, Fink-Jensen A. ACS Chem Neurosci. 2012;3:180–89. doi: 10.1021/cn200110q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wess J, Eglen RM, Gautam D. Nat Rev Drug Discov. 2007;6:721–733. doi: 10.1038/nrd2379. [DOI] [PubMed] [Google Scholar]
- 3.Bymaster FP, Whitesitt CA, Shannon HE, DeLapp N, Ward JS, Calligaro DO, Shipley LA, Buelke-Sam JL, Bodick NC, Farde L, Sheardown MJ, Olesen PH, Hansen KT, Suzdak PD, Swedberg MDB, Sauerberg P, Mitch CH. Drug Dev Res. 1997;40:158–170. [Google Scholar]
- 4.Shekhar A, Potter WZ, Lightfoot J, Lienemann J, Dube S, Mallinckrodt C, Bymaster FP, McKinzie DL, Lelder CC. Am J Psychiatry. 2008;165:1033–1039. doi: 10.1176/appi.ajp.2008.06091591. [DOI] [PubMed] [Google Scholar]
- 5.Melancon BJ, Hopkins CR, Wood MR, Emmitte KA, Niswender CM, Christopoulos A, Conn PJ, Lindsley CW. J Med Chem. 2012;55:1445–1464. doi: 10.1021/jm201139r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ma L, Seager MA, Wittmann M, Jacobson M, Bickel D, Burno M, Jones K, Graufelds VK, Xu G, Pearson M, McCampbell A, Gaspar R, Shughrue P, Danziger A, Regan C, Flick R, Pascarella D, Garson S, Doran S, Kreatsoulas C, Veng L, Lindsley CW, Shipe W, Kuduk S, Sur C, Kinney G, Seabrook GR, Ray WJ. Proc Natl Acad Sci USA. 2009;106:15950–15955. doi: 10.1073/pnas.0900903106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.ML137 (akaVU0366369) Bridges TM, Kennedy JP, Noetzel MJ, Breininger ML, Gentry PR, Conn PJ, Lindsley CW. Bioorg Med Chem Lett. 2010;20:1972–1975. doi: 10.1016/j.bmcl.2010.01.109.
- 8.ML169 (aka VU0405652) Reid PR, Bridges TM, Sheffler DJ, Cho HP, Lewis LM, Days E, Daniels JS, Jones CK, Niswender CM, Weaver CD, Conn PJ, Lindsley CW, Wood MR. Bioorg Med Chem Lett. 2011;21:2697–2701. doi: 10.1016/j.bmcl.2010.12.015.
- 9.ML108 (akaVU0152100) Brady AE, Jones CK, Bridges TM, Kennedy JP, Thompson AD, Heiman JU, Breininger ML, Gentry PR, Yin H, Jadhav SB, Shirey JK, Conn PJ, Lindsley CW. J Pharmacol Exp Ther. 2008;327:941–953. doi: 10.1124/jpet.108.140350.
- 10.ML173 (aka VU0359508 aka compound 21n) Kennedy JP, Bridges TM, Gentry PR, Brogan JT, Kane AS, Jones CK, Brady AE, Shirey JK, Conn PJ, Lindsley CW. ChemMedChem. 2009;4:1600–1607. doi: 10.1002/cmdc.200900231.
- 11.ML253 (aka VU0448088) Le U, Melancon BJ, Bridges TM, Vinson PN, Utley TJ, Lamsal A, Rodriguez AL, Venable D, Sheffler DJ, Jones CK, Blobaum AL, Wood MR, Daniels JS, Conn PJ, Niswender CM, Lindsley CW, Hopkins CR. Bioorg Med Chem Lett. 2012 doi: 10.1016/j.bmcl.2012.10.073. manuscript accepted.
- 12.ML293 (aka VU0409524) Salovich JM, Vinson PN, Sheffler DJ, Lamsal A, Utley TJ, Blobaum AL, Bridges TM, Le U, Jones CK, Wood MR, Daniels JS, Conn PJ, Niswender CM, Lindsley CW, Hopkins CR. Bioorg Med Chem Lett. 2012;22:5084–5088. doi: 10.1016/j.bmcl.2012.05.109.
- 13.ML129 (aka VU0238429 akacompound 42) Bridges TM, Marlo JE, Niswender CM, Jones CK, Jadhav SB, Gentry PR, Plumley HC, Weaver CD, Conn PJ, Lindsley CW. J Med Chem. 2009;52:3445–3448. doi: 10.1021/jm900286j.
- 14.ML172 (akaVU0400265 aka compound 10) Bridges TM, Kennedy JP, Cho HP, Breininger ML, Gentry PR, Hopkins CR, Conn PJ, Lindsley CW. Bioorg Med Chem Lett. 2010;20:558–562. doi: 10.1016/j.bmcl.2009.11.089.
- 15.MacDonald JP, Badillo JJ, Arevalo GE, Silva-García A, Franz AK. ACS Comb Sci. 2012;14:285–293. doi: 10.1021/co300003c. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baell JB, Holloway GA. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 17.Singh RP, Cao G, Kirchmeier RL, Shreeve JM. J Org Chem. 1999;64:2873–2876. doi: 10.1021/jo982494c. [DOI] [PubMed] [Google Scholar]
- 18.Although the M1 PAM EC50 has been previously reported as 0.83 μM with a %AChmax = 65% (Ref. 7), the inherent variability associated with functional assays supports that the most recent EC50 = 0.60 μM for ML137 is equally valid and can serve as a baseline comparator between the two papers.
- 19.Wood MR, Hopkins CR, Brogan JT, Conn PJ, Lindsley CW. Biochemistry. 2011;50:2403–2410. doi: 10.1021/bi200129s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.For information on the MLPCN and information on how to request any of the ML# probe compounds, such as ML137, see: http://mli.nih.gov/mli/mlpcn/