

Abstract
The structure-activity relationship study of a diphenylpropanamide series of RORγ selective modulators is reported. Compounds were screened using chimeric receptor Gal4 DNA-binding domain (DBD)-NR ligand binding domain cotransfection assay in a two-step format. Three different regions of the scaffold were modified to assess the effects on repression of RORγ transcriptional activity and potency. The lead compound 1 exhibits modest mouse pharmacokinetics and an acceptable in vitro profile which makes it a suitable in vivo probe to interrogate the functions of RORγ in animal models of disease.

Nuclear hormone receptors (NR) are a highly conserved group of transcription factors that regulate a range of metabolic, endocrine and immunologic disorders including cancer, inflammation, diabetes and atherosclerosis. Members of the nuclear receptor (NR) superfamily are characterized by a highly conserved DNA binding domain and a ligand binding domain.1 48 NRs have been identified in humans and are comprised of classic steroid receptors, RXR heterodimer receptors and xenobiotic receptors. Approximately half of the human NRs are characterized as ligand activated transcription factors regulating the expression of target genes, whereas a large number of these receptors are still classified as orphan due to lack of a characterized natural ligand.2 Retinoic acid receptor-like orphan receptors α and γ (RORs) are examples of such orphan nuclear receptors that play critical roles in immunity, cellular metabolism and circadian rhythms.3
The retinoic receptor-related orphan receptor γ (RORγ) and its isoform RORγt play a critical role in differentiation of Th17 cells and secretion of inflammatory cytokines such as Interleukin 17 (IL-17).4 Th17 cells have been identified as key mediators for immune responses of a wide variety of autoimmune diseases such as multiple sclerosis (MS), Crohn’s disease, and rheumatoid arthritis (RA). While cholesterol and cholesterol sulfate have been put forward as the natural ligands for RORα, the endogenous ligands for RORγ still remain debatable.5 We have shown that various oxysterols that bind to RORα also bind to RORγ and regulate their activity.6 We have also demonstrated that synthetic LXR agonist T0901317 also binds and modulates RORα and γ.7 Recently we were able to identify SR-33358a (synthetic RORα-selective inverse agonist), SR-10018b (dual RORα, γ synthetic inverse agonist), SR-10788c (dual RORα, γ synthetic agonist) and SR-22118d (RORγ selective modulator). Natural products such as digoxin and ursolic acid have also been shown to inhibit Th17 cell differentiation, but their potential as candidates for further development is rather limited due to narrow therapeutic index and selectivity issues over other receptors such as glucocorticoid receptors.9 While corticosteroids are highly efficacious in managing autoimmune disorders, they also induce some serious side effects such as osteoporosis, retinopathy and diabetes. In light of these facts, development of new RORγ selective modulators that specifically control IL-17 expression and Th17 cell differentiation will provide drug-like molecules with improved therapeutic profiles. Herein, we present our SAR efforts to characterize SR- 9805/1 (Fig 1) and its analogs as potent RORγ selective modulators.10
Figure 1.

SR-9805 (1) and analogs as ROR-γ modulators
Based on the lead compound 1 four regions of this scaffold were modified in a step-wise fashion in order to investigate the effects of substituents X, Y, Z, and R on the activity of these compounds. Compounds represented by general structure 6 were synthesized via a two-step protocol starting from commercially available starting materials (Scheme 1). 3,5-dimethoxyphenol 3 was treated with various electron rich cinnamic acids 4 in the presence of trifluoroacetic acid (TFA) as a solvent to furnish dihydrocoumarins 5. Compound 5 was subjected to ring opening using various amines to yield the final compounds 6.11
Scheme 1.

Reagents and conditions: a. TFA, µW, 30 min, 120 °C; b. R1R2NH, THF, 60 °C, 2h.
Compounds were screened using chimeric receptor Gal4 DNA-binding domain (DBD)-NR ligand binding domain cotransfection assay in a two-step format. To determine the effect on the RORγ transcriptional activity, HEK293T cells were cotransfected with Gal4-RORγ along with a UAS-luciferase plasmid. The cells were treated for 20 hr with the compound and relative change was determined by normalizing to cells treated with vehicle. Compounds were first screened at two concentrations (1 µM and 10 µM) to determine the effect on repression of RORγ transcriptional activity and the maximum repression at 10 µM is reported (Table 1–3). A high % repression indicates that the compound is more efficacious at repressing transcription. Compounds that showed more than 50% repression at 1 µM were then fully titrated in a ten-point dose response format to generate IC50 values. As shown in Fig 2A, SR-9805 shows an IC50 of 76 nM on RORγ transcriptional activity. SR-9805 also did not show any activity on related nuclear hormone receptors RORα, LXRα, and FXR (data not shown). These compounds were further evaluated using a competition assay to determine if they can directly bind to RORγ.12 As shown in Fig 2B increasing concentrations of SR-9805 were incubated with 5nM of [3H]-T0903017 and 1 µg of GST-RORγ along with Glutathione-YSi beads to determine IC50 as detailed in the methods.12
Table 1.
Effect of Substituent R1 and R2 on RORγ modulation
 | ||||
|---|---|---|---|---|
| Compound | R1 | R2 | Binding Assay IC50(nM)a |
%Repression @10 µM (GAL4 Assay Ic50)a |
| 1 | 57 | 95(76nM) | ||
| 6a | NTb | 17 | ||
| 6b | NTb | 11 | ||
| 6C | NTb | 4 | ||
| 6d | 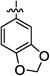 |
NTb | 0 | |
| 6e | NTb | 1 | ||
| 6f | NTb | 68(>1 µM) | ||
| 6g | NTb | 57 | ||
| 6h | 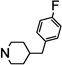 |
NTb | 23 | |
| 6i | NTb | 8 | ||
| 6j | NTb | 83(>1 µM) | ||
| 6k | NA | NTc | ||
| 6l | NA | NTc | ||
| 6m | NA | NTc | ||
| 6n | NA | NTc | ||
| 6o | NA | NTc | ||
| 6p | 96 | 95 (114 nM) | ||
Results are average of at least three replicates. Value = fold change relative to DMSO control at 10 µM compound
NT = not tested; These compounds were tested at 10 µM and 1 µM in the GAL4 Assay and only the compounds which showed more than 50% repression of ROR-γ transcriptional activity at l µM were run in the binding assay.
For these compounds the binding assay was run prior to the GAL4 assay and if the compounds did not show any binding, no GAL4 cell based assay was performed for these compounds (NA= No Activity). All standard deviations ≤ 20%.
Table 3.
Effect of Substituent R4, on RORγ modulation
 | |||
|---|---|---|---|
| Compound | R4 | Binding Assay IC50(nM) |
%Repression @10 uM (GAL4 Assay IC50)a |
| 1 | 3,4-methylenedioxy | 57 | 95(76nM) |
| 6p | 4-OMe | 96 | 95(114nM) |
| 8 | 2-OMe | 106 | 96 (265nM) |
| 9 | H | NTb | 27 |
| 10 | See Scheme 2 | NTb | 0 |
| 11 | see Scheme 2 | NTb | 0 |
| 19 | 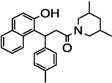 |
NTb | 96 (> 1 µM) |
Results are average of at least three replicates. Value = fold change relative to DMSO control at 10 µM compound;
NT = not tested; These compounds were tested at 10 µM and l µM in the GAL4 Assay and only the compounds which showed more than 50% repression of RORγ transcriptional activity at 1 µM were run in the binding assay. All standard deviations ≤ 20%.
Figure 2.

Demonstration of transcriptional repression and binding of compound 1 to RORγ.
We started the SAR studies by modifying the amide functionality in the lead compound 1 (Substituent R2, Table 1). Compound 1 shows 95% repression of RORγ transcriptional activity at 10 µM with an IC50 of 76 nM in a GAL4 assay and an IC50 of 57 nM in a binding assay. Several five and six membered ring amides were synthesized, however none of them exhibited any improvement with regards to efficacy (Table 1). For example, analogs with a hydroxy substituent on the six member ring (6c, d, i) showed poor repression of RORγ transcriptional activity at 10 µM.
Compounds 6 f and 6 j exhibited modest repression but with micromolar potency. The high repression of RORγ transcriptional activity and nanomolar potency (114 nM) of compound 6p (Table 1) further validates the importance of the 3,5-dimethyl piperidine functionality in this scaffold. As far as the substituent R1 is concerned, modifying 3,4-(methylenedioxy)phenyl to 4-methoxy phenyl (1 vs 6p) did not have a dramatic effect on the efficacy or potency of these compounds. Modifying substituent R2 from 3,5-dimethylpiperidine(6p) to benzylamine (6k), morpholine (6l), piperidine (6m), cyclopentylamine (6n) or pyrrolidine (6o), resulted in loss of activity. In fact as long as the substituent R2 was 3,5-dimethyl piperidine the compounds maintained nanomolar potency (1 vs 6p, Table 1 & 8, Table 3).
Efforts were then focused on modifications to the phenol ring bearing the substituent R3 (Table 2). The compounds shown are only a subset of those actually synthesized, however they are representative of the group. Disubstituted analogs such as 4-Et (7b), 4-OEt (7c), 3-Me (7e), 4-Me (7f) had modest effect on the % repression as well as the potency. Bulky substituents such as 2-naphthyl (7h), 4-OPr (7d) or 3-OPh (7g) were also well tolerated. While these substitutions provided compounds which showed maximum transcriptional repression at 10 µM, IC50’s were significantly right-shifted compared to 1. The nature of this effect is unclear, but is also fairly robust. Hence, we conclude that modifications to this ring are not well tolerated.
Table 2.
Effect of Substituent R3 on RORγ modulation
 | |||
|---|---|---|---|
| Compound | R3 | Binding Assay IC50(nM) |
%Repression @10 µM (GAL4 Assay IC50)a |
| 1 | 3,5-diOMe | 57 | 95 (76 nM) |
| 7a | 3,4-diOMe | NTb | 22 |
| 7b | 4-Et | NTb | 95(>1 µM) |
| 7c | 4-OEt | NTb | 95(>1 µM) |
| 7d | 4-OPr | NTb | 96(>1 µM) |
| 7e | 3-Me | NTb | 96(>1 µM) |
| 7f | 4-Me | NTb | 95(>1 µM) |
| 7g | 3-OPh | NTb | 96(>1 µM) |
| 7h | 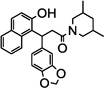 |
NTb | 93(>1 µM) |
Results are average of at least three replicates. Value = fold change relative to DMSO control at 10 µM compound;
NT = not tested; These compounds were tested at 10 µM and l µM in the GAL4 Assay and only the compounds which showed more than 50% repression of RORγ transcriptional activity at 1 µM were run in the binding assay. All standard deviations ≤ 20%.
Modification of substituent R4 also presented a very flat SAR (Table 3). Modifying the substituent from 3,4-(methylenedioxy)phenyl to 4-OMe (6p) or 2-OMe (8) reduced the potency by 1.5 and 3.5 fold respectively in a GAL4 assay while still maintaining a high percentage repression of RORγ transcriptional activity. RORγ binding was also very good. The R4-substituent also has to be something other than hydrogen (9) which leads to loss of activity.
In order to study the effect of amide functionality and the location of 3,5-dimethoxy phenyl ring system with respect to the amide carbonyl, we synthesized analogs 10, 11, and 19 (Scheme 2).
Scheme 2.

Reagents and Conditions: a. DCC, DMF, µW, 30 min, 120 °C; b. (1) H2, Pd-C, 48h, (2) 3,5-Dimethylpiperidine, THF, 40 °C. (c) NaI, TMSCl, DMF, 140 °C; (d) 3,5-Dimethylpiperidine, THF, 40 °C
Compound 10 was synthesized via a two-step protocol which involved coupling of 2-hydroxy-6-methoxybenzaldehyde 12 and p-tolylacetic acid 13 in the presence of DCC to afford coumarin 14.13 Compound 14 was hydrogenated followed by treatment with 3,5-dimethyl piperidine to afford compound 10. Synthesis of compound 11 also involved a two-step sequence starting with a three component reaction.14 Treatment of a mixture of 2-naphthol, p-tolualdehyde and urea along with TMSCl/NaI as a promoter resulted in the naphthoxazinone 18. Compound 18 was treated with 3,5-dimethyl piperidine to afford compound 11. Compound 19 (synthesis not shown) was made as described in Scheme 1.
It was observed that modifying the amide in compound 1 to an urea functionality (11, Table 3) resulted in complete loss of activity. As shown in table 3, the high percentage (96%) repression of RORγ transcriptional activity shown by compound 19 was completely lost upon its modification to the urea 11. Changing the location of the 3,5-dimethoxy phenyl ring system from the β position to the α position of the amide carbonyl (10, Table 3) also resulted in complete loss of activity.
Overall, the scaffold represented by compound 1 provides a very limited area for modification without having any detrimental effect on potency and % repression of RORγ activity.
The in vivo properties of few RORγ selective modulators were examined (Table 4)15. Compound 1 shows low to modest solubility at pH 5 and 7.4. Brain penetration of these compounds was also measured since RORγ is highly expressed in the central nervous system (CNS). Mice were given a 10 mg/kg IP dose of drug, and plasma and brain levels of drug were determined 6h later. The lead compound 1 exhibited limited exposure in plasma, although CNS penetration was good. Compound 1 also exhibited high % inhibition of cytochrome 2C9 and 3A4.
Table 4.
In vitro and In vivo properties of selected compounds
| Entry | Solublilitya |
[Plasma] b µM |
[Brain] b µM |
b.p. (%)c |
p450 % inhibitiond |
||||
|---|---|---|---|---|---|---|---|---|---|
| pH 5.0 ( µM) |
pH 7.4 ( µM) |
1A2 | 2C9 | 2D6 | 3A4 | ||||
| 1c | 15.2 | 15.6 | 0.30 | 0.78 | 260 | 3 | 92 | 15 | 97 |
| 8 | 6.0 | 12.7 | NT | NT | NT | −22 | 53 | −11 | 70 |
| 6p | 19.5 | 18.0 | NT | NT | NT | 4 | 65 | −7 | 75 |
100 µM solutions of compounds were shaken at pH 7.4 (PBS) and pH 5.0 (Sodium Citrate Buffer) at room temperature for 20hr
Mice sacrificed at t = 6h. Brain and plasma levels of drug determined. Mice dosed 10 mg/kg IP in 10:10:80 DMSO:Tween:water
b.p. = brain penetration
% inhibition at 10 µM drug. NT= Not tested.
The SAR on the RORγ selective scaffold (Fig 1) presented herein is very tight. Small modifications to its structure result in total loss of potency. Having the 3,5-dimethyl piperidine as the amine fragment in the amide moiety is pertinent to the efficacy and potency of these compounds. A wide variety of electron-donating substituents are tolerated on ring A (Fig 1) without affecting the % repression of RORγ transcriptional activity. Ring B is likely most tolerant to substitution. We also observed that modifying the amide in compound 1 to an urea functionality (11, Table 3) or changing the location of the 3,5-dimethoxy phenyl ring system from the β position to the α position of the amide carbonyl (10, Table 3) resulted in complete loss of activity. In summary, this RORγ selective scaffold provides a starting point for development of new probes to interrogate the functions of RORγ in animal models of disease. Further work focused on the improvement of efficacy, potency and in vivo profile of these compounds is underway and will be reported in due course.
Acknowledgment
This work was supported, in whole or in part, Grant MH084512 (PI:H Rosen).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schütz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM. Cell. 1995;83:835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kliewer SA, Lehmann JM, Willson TM. Science. 1999;284:757–760. doi: 10.1126/science.284.5415.757. [DOI] [PubMed] [Google Scholar]
- 3.Jetten AM. Nucl. Recept. Signaling. 2009;7:e003. doi: 10.1621/nrs.07003. [DOI] [PMC free article] [PubMed] [Google Scholar]; Solt LA, Griffin PR, Burris TP. Curr. Opin. Lipidol. 2010;21:204–211. doi: 10.1097/MOL.0b013e328338ca18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang XXO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, Ma L, Shah B, Panopoulos AD, Schluns KS, Watowich SS, Tian Q, Jetten AM, Dong C. Immunity. 2008;28:29–39. doi: 10.1016/j.immuni.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]; Ivanov II, Zhou L, Littman DR. Semin. Immunol. 2007;19:409–417. doi: 10.1016/j.smim.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]; Manel N, Unutmaz D, Littman DR. Nature Immunol. 2008;9:641–649. doi: 10.1038/ni.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kallen J, Schlaeppi JM, Bitsch F, Delhon I, Fournier B. J. Biol. Chem. 2004;279:14033–14038. doi: 10.1074/jbc.M400302200. [DOI] [PubMed] [Google Scholar]; Kallen JA, Schlaeppi JM, Bitsch F, Geisse S, Geiser M, Delhon I, Fournier B. Structure. 2002;10:1697–1707. doi: 10.1016/s0969-2126(02)00912-7. [DOI] [PubMed] [Google Scholar]
- 6.Wang Y, Kumar N, Solt LA, Richardson TI, Helvering LM, Crumbley C, Garcia-Ordonez RA, Stayrook KR, Zhang X, Novick S, Chalmers MJ, Griffin PR, Burris TP. J. Biol. Chem. 2010;285:5013–5025. doi: 10.1074/jbc.M109.080614. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wang Y, Kumar N, Crumbley C, Griffin PR, Burris TP. Biochim. Biophys. Acta. 2010;1801:917–923. doi: 10.1016/j.bbalip.2010.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumar N, Solt LA, Conkright JJ, Wang Y, Istrate MA, Busby SA, Garcia-Ordonez RD, Burris TB, Griffin PR. Mol. Pharmacol. 2010;77:228–236. doi: 10.1124/mol.109.060905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Kumar N, Kojetin DJ, Solt LA, Kumar G, Nuhant P, Duckett DR, Cameron MD, Butler AA, Roush WR, Griffin PR, Burris TP. ACS Chem. Biol. 2011;6:218–222. doi: 10.1021/cb1002762. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Solt LA, Kumar N, Nuhant P, Wang Y, Lauer JL, Liu J, Istrate MA, Kamenecka TM, Roush WR, Vidović D, Schürer SC, Xu J, Wagoner G, Drew PD, Griffin PR. Nature. 2011;472:491–494. doi: 10.1038/nature10075. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wang Y, Kumar N, Nuhant P, Cameron MD, Istrate MA, Roush WR, Griffin PR, Burris TP. ACS Chem. Biol. 2010;5:1029–1034. doi: 10.1021/cb100223d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kumar N, Lyda B, Chang MR, Lauer JL, Solt LA, Burris TP, Kamenecka T, Griffin PR. ACS Chem. Biol. 2012;7:672–677. doi: 10.1021/cb200496y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huh JR, Leung MWL, Huang P, Ryan DA, Krout MR, Malapaka RRV, Chow J, Manel N, Ciofani M, Kim SV, et al. Nature. 2011;472:486–490. doi: 10.1038/nature09978. [DOI] [PMC free article] [PubMed] [Google Scholar]; Xu T, Wang X, Zhong B, Nurieva RI, Ding S, Dong C. J. Biol. Chem. 2011;286:22707–22710. doi: 10.1074/jbc.C111.250407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Littman D, Huh JR, Huang R, Huang W, Englund EE. 2011 WO 2011112263. [Google Scholar]
- 11.Li K, Foresee LN, Tunge JA. J. Org. Chem. 2005;70:2881–2883. doi: 10.1021/jo0477650. [DOI] [PubMed] [Google Scholar]; Li K, Tunge JA. J. Comb. Chem. 2008;10:170–174. doi: 10.1021/cc700150q. [DOI] [PubMed] [Google Scholar]
- 12.Cell Culture and Cotransfections: HEK293 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum at 37°C under 5% CO2. Reverse transfections were performed in bulk using 1x106 cells in 6 cm plates, 3 µg of total DNA in a 1:1 ratio of receptor and reporter and FuGene6 (Roche) in a 1:3 DNA: lipid ratio. Following day, cells re plated in 384 well plates at a density of 10,000 cells/well. After 4 hr, the cells were treated with the compound or DMSO as control. The luciferase levels were assayed following additional 20 hour incubation by one-step addition of BriteLite Plus(Perkin Elmer) and read using an Envision (Perkin Elmer). Data was normalized as fold change over DMSO treated cells.Radioligand Binding Assay: The assay contains 0.25 mg of beads (Glutathione YSI; PE # RPNQ0033), 1 μg of GSTRORγ-LBD, 5 nM of [3H] T0901317 as radioligand and varying concentration of SR2211 in the assay buffer (50 mM HEPES, pH 7.4, 0.01% bovine serum albumin, 150 mM NaCl and 5 mM MgCl2, 10% glycerol, 1mM DTT, Complete protease inhibitor from Roche). All the components were gently mixed and incubated for 20 hr and were read in TopCount. The radioligand binding results were analyzed using GraphPad Prism software.
- 13.Matos MJ, Delogu G, Podda G, Santana L, Uriarte E. Synthesis. 2010;16:2763–2766. [Google Scholar]
- 14.Sabitha G, Arundhathi K, Sudhakar K, Sastry BS, Yadav JS. J. Heterocyclic Chem. 2010;47:272–275. [Google Scholar]
- 15.CNS exposure was evaluated in C57Bl6 mice (n = 3). Compounds were dosed at 10 mg/kg intraperitoneally and after 6 h blood and brain were collected. Plasma was generated and the samples were frozen at −80°C. The plasma and brain were mixed with acetonitrile (1:5 v:v or 1:5 w:v, respectively). The brain sample was sonicated with a probe tip sonicator to break up the tissue, and samples were analyzed for drug levels by LCMS/MS. Plasma drug levels were determined against standards made in plasma and brain levels against standards made in blank brain matrix. All procedures were approved by the Scripps Florida IACUC.