

SUMMARY
In Drosophila, the replacement of spent enterocytes (ECs) relies on division of intestinal stem cells (ISCs) and differentiation of their progeny, the enteroblasts (EBs). Recent studies have revealed a role for JAK/STAT signaling in the modulation of the rate of ISC division in response to environmental challenge. Here, we demonstrate the critical role of the Upd3 cytokine in the JAK/STAT-dependent response to enteric infection. We show that upd3 expression is activated in ECs and in EBs that massively differentiate in response to challenge. We show that the Upd3 cytokine, which is secreted basally and accumulates at the basement membrane, is required for stimulation of JAK/STAT signaling in EBs and visceral muscles (VMs). We further show that stimulation of ISC division requires active JAK/STAT signaling in EBs and VMs, but apparently not in ISCs. Our results suggest that EBs and VMs modulate the rate of the EGFR-dependent ISC division through upd3-dependent production of the EGF ligands Spitz and Vein, respectively. This study therefore supports the notion that the production of the Upd3 cytokine in stem cell progeny (ECs and EBs) stimulates intestinal stem cell division through modulation of JAK/STAT signaling in the stem cell microenvironment (EBs and VMs).
Keywords: stem cell, differentiation, JAK, STAT, Upd3, cytokine, infection, challenge, regeneration, intestinal homeostasis, Erwinia
INTRODUCTION
The Drosophila intestine shares striking structural and functional similarities with its mammalian counterpart (Casali and Batlle, 2009). It is composed of an outer layer of longitudinal and circular smooth muscles that execute peristalsis, and an inner layer of specialized epithelial cells that constitutes the intestinal epithelium. The outer and inner layers are connected by a basement membrane. Seminal studies have recently demonstrated that, in addition to specialized epithelial cells, the intestinal epithelium is also populated with intestinal stem cells (ISCs) (Micchelli and Perrimon, 2006; Ohlstein and Spradling, 2006). ISCs are small cells located at the basement membrane, where they contribute to the renewal of the intestinal epithelium (Ohlstein and Spradling, 2007). Division of ISCs gives rise to one cell that retains stem cell properties, and one progenitor, termed enteroblast (EB), which will ultimately differentiate into enterocytes or enteroendocrine cells displaying absorptive and secretory functions, respectively (Ohlstein and Spradling, 2007).
Under normal physiological conditions, homeostasis of the intestinal epithelium is maintained by the balance between the production of new cells by stem cell division and the removal of old cells by apoptosis. In both mammals and Drosophila, the entire intestine is renewed within 5 to 7 days. Recent studies conducted in Drosophila revealed that the rate of stem cell division increases dramatically in response to stressful conditions, including aging (Biteau et al., 2008), chemical (Amcheslavsky et al., 2009) or bacterial challenges (Apidianakis et al., 2009; Buchon et al., 2009b; Cronin et al., 2009; Jiang et al., 2009). Buchon et al. first investigated the role of JAK/STAT signaling in the stimulation of ISC division in response to Erwinia infection (Buchon et al., 2009a). Jiang et al. investigated the role of JAK/STAT signaling in response to several challenges including apoptosis, stress signaling and Pseudomonas entomophila infection (Jiang et al., 2009). These studies and a recent study published by Lemaitre and colleagues (Osman et al., 2012) led to a current model of intestinal homeostasis in which challenged enterocytes produce the Upd2 and Upd3 cytokines, that activate the mitogenic activity of JAK/STAT signaling in ISCs, thereby increasing their rate of division in response to challenge. In this report, we show that Upd3, a ligand of the JAK/STAT signaling pathway in Drosophila, is a major cytokine controlling the rate of ISC division in response to enteric infection. We show that upd3 is expressed in ECs, as previously proposed (Buchon et al., 2009a; Buchon et al., 2009b; Jiang et al., 2009), but also in differentiating EBs, which suggests a previously unappreciated role for the differentiation process in ISC division. Surprisingly, our genetic analyses suggest that ISC division requires activation of JAK/STAT signaling in EBs and in the visceral muscles (VMs), but apparently not in ISCs. Our results support a model of intestinal homeostasis in which the production of the Upd3 cytokine in ECs and in differentiating EBs regulates JAK/STAT signaling in EBs and VMs, which potentially control the rate of EGFR-dependent division of ISCs through EGF ligand production. We discuss the conceptual implications of these findings.
RESULTS
upd3 expression is activated in response to challenge
To determine the potential contribution of the known ligands of the Drosophila JAK/STAT signaling pathway to the stimulation of ISC division observed in response to challenge, we analyzed the expression profile of upd, upd2 and upd3 in various models of enteric infection. It was previously shown that all three cytokines were induced in response to Pseudomonas entomophila infection (Jiang et al., 2009). Similarly, we found that all three cytokines were induced in response to Erwinia carotovora (Ecc15) and Serratia marcescens infection (Fig. S1A, B). However, upd3 consistently displayed the highest levels of induction, suggesting that the Upd3 cytokine may play a major role in the control of ISC division in response to challenge (Fig. S1A, B). We generated transgenic flies displaying upd3-lacZ reporter and we showed that, in response to Ecc15 infection, upd3 was strongly expressed in the posterior midgut (compare Fig, 1A and 1C, PM and Fig. 1B and 1D, upd3-lacZ). This region of the midgut displayed a high rate of ISC division in response to challenge (compare Fig.1B and 1D, pH3). These results showed a correlation between the local level of upd3 expression and the rate of stem cell division in the posterior midgut intestine, as previously proposed (Buchon et al., 2009a; Buchon et al., 2009b; Cronin et al., 2009; Jiang et al., 2009).
Fig. 1. upd3 is expressed in the posterior midgut in response to challenge.

(A–D) Fluorescence images showing upd3-lacZ expression and pH3-positive nuclei in response to mock-treatment (A and B) and 18-hour Ecc challenge (C and D). Whole intestine are shown in (A) and (C). (B) and (D) correspond to magnification of the squared regions displayed in (A) and (C), respectively. AM: anterior midgut; PM: posterior midgut; asterisk: hindgut proliferating zone.
Upd3 is required for ISC division in response to challenge
To demonstrate the involvement of the Upd3 cytokine in ISC division, we generated an upd3 deletion mutant by mobilizing a P element located in the third intron of the upd3 locus (Fig. 2A). The Δupd3#9 mutant animals were homozygous viable and displayed normal longevity as compared to wild-type animals (Fig. S2). However, the increase in the rate of ISC division observed in wild-type animals in response to Ecc15 infection was no longer observed in Δupd3#9 animals (Fig. 2B, Ecc15). The stimulation of ISC division was also compromised in Δupd3#9 animals in response to Pseudomonas and Serratia infections (Figure 2B, Pe and Sm). Expression of a UAS-upd3 transgene under the control of the upd3-GAL4, tub-GAL80ts system (Fig. 2C, Δupd3#9 upd3>upd3) led to a ~200-fold increase in the levels of upd3 expression at 29°C (data not shown). Under these conditions, we observed a significant increase in the number of mitotic cells in unchallenged Δupd3#9 mutant animals, indicating that upd3 over-expression is sufficient to stimulate ISC division (Fig. 2C). In addition, expression of UAS-upd3 restored ISC division in response to Ecc15 infection in the Δupd3#9 mutant animals (Fig. 2C). These data indicate that the production of the Upd3 cytokine is necessary and probably sufficient to stimulate ISC division in response to Ecc15 infection.
Fig. 2. The Upd3 cytokine is required for stimulation of ISC division in response to challenge.

(A) Structural organization of the upd3 locus in the parental strain (d00871) and in the upd3 deletion mutant (Δupd3#9). (B) pH3-positive nuclei were scored in wild-type (w1118), heterozygote (Δupd3#9/w1118), and upd3 mutant (Δupd3#9) animals infected for 18 hours with Erwinia carotovora caratovora 15 (Ecc15) (OD600=50), Pseudomonas entomophila (Pe), or Serratia marcescens (Sm). Averages of three representative experiments (n = 12 guts/experiment) with standard deviation are shown. P values were calculated by two-tailed, unpaired t-tests.
For Δupd3#9 vs. w1118: mock, P = 0.0437 (*); Ecc15 infection, P = 0.0015 (**); Pe infection, P = 0.0132 (*); Sm infection, P = 0.0010 (**).
For infection vs. mock, w1118: Ecc15, P =0.0044 (**); Pe, P = 0.0021 (**); Sm, P = 0.0066 (**).
For infection vs. mock, Δupd3#9: Ecc15, P =0.2698 (ns); Pe, P = 0.0111 (*); Sm, P = 0.7408 (ns).
(C) Genetic rescue of the upd3 mutant. pH3-positive nuclei were scored in wild-type (w1118), upd3 mutant (Δupd3#9) or rescued upd3 mutant animals (Δupd3#9;upd3>upd3: ectopic expression of UAS-Upd3 with upd3-GAL4, tub-Gal80ts). P values were calculated by two-tailed, unpaired t-tests. For mock infection, Δupd3#9 vs. w1118, P =0.2995 (ns) and wild-type vs. rescue, P<0.0001 (***). For Ecc15 infection, Δupd3#9 vs. w1118, P<0.0001 (***) and upd3>upd3 vs. w1118, P =0.38 (ns).
Ecc15 infection stimulates EB-to-EC differentiation
Under normal physiological conditions, the replacement of spent ECs relies on ISC division and EB differentiation. It is now well established that the rate of ISC division increases dramatically in response to challenge. In contrast, the dynamics of EB-to-EC differentiation in response to challenge has been less characterized. To address these aspects further, we used the EC-specific marker Myo1A (Morgan et al., 1994) and the EB-specific marker Su(H)GBE-lacZ (Furriols and Bray, 2001). In unchallenged animals, the posterior midgut epithelium was constituted of ECs, which are large Myo1A-positive/Su(H)GBE-lacZ-negative cells (Fig. 3A,B, Myo1A>GFP). Basally located with respect to ECs, we observed small EBs, which are Myo1A-negative/Su(H)GBE-lacZ-positive cells (Fig. 3A,B, Su(H)GBE-lacZ, asterisk). In challenged animals, the first signs of response to infection was an apparent increase in Su(H)GBE-lacZ immuno-detection along the midgut (Fig. 3C,D), which correlated with the appearance of Su(H)GBE-lacZ-positive cells of intermediate size, located at the basal side of the epithelium (Fig. 3D, asterisks, and Fig. S3). Su(H)GBE-lacZ-positive EBs began to express the EC marker Myo1A and displayed inflated nuclei, a likely reflection of the onset of the endoreplication process leading to polyploidy in mature ECs (Fig. 3D, asterisks, and Fig. S3). Myo1A-positive/Su(H)GBE-lacZ-positive cells with inflated nuclei were detected as early as 4 hours after infection and they represented the majority of Su(H)GBE-lacZ-positive cells (EBs) in the posterior midgut 8 hours after infection (Fig. S3). After 18 hours of infection, we observed Myo1A-positive/Su(H)-positive cells as large as mature Myo1A-positive/Su(H)GBE-lacZ-negative ECs, indicating an advanced stage in the differentiation process (Fig. 3E,F). Thus, in addition to the observed increase in the rate of stem cell division, Ecc15 infection leads to a massive wave of EB-to-EC differentiation.
Fig. 3. upd3 is expressed and required in differentiated ECs and in differentiating EBs.

(A, C and E) Confocal microscopy images of posterior midgut (10× objective, XY section). (B, D, F, G, H and I) Confocal microscopy images of posterior midgut (60× objective). Bottom panels show XY section. Dotted lines indicate Y coordinates for XZ reconstruction (top panels).
(A–F) Expression of the EC-specific marker Myo1A and EB-specific marker Su(H)-lacZ in mock-treated animal (A and B), animals infected for 8 hours with Ecc15 (C and D) and animals infected for 18 hours with Ecc15 (E and F).
(G–I) Expression of the EB-specific marker Su(H)>GFP and the upd3-lacZ reporter in mock-treated animal (G), animals infected for 8 hours with Ecc15 (H) and animals infected for 18 hours with Ecc15 (I). (J and K) pH3-positive nuclei were scored in Myo1A-Gal4ts/+ (w1118) or Myo1A-Gal4ts/UAS-iupd3 (UAS-iupd3) (J) and Su(H)-GAL4ts/+ (w1118) or Su(H)-Gal4ts/UAS-iupd3 (UAS-iupd3) (K) animals infected with Ecc15 for 8 hours. Averages of three representative experiments (n = 12 guts/experiments) with s.d. are shown. (J) UAS-iupd3 vs. w1118, P = 0.0462 (*). (K) UAS-iupd3 vs. w1118, P = 0.0164 (*).
Upd3 is produced in differentiated ECs and in differentiating EBs
We used the Myo1A>GFP and Su(H)GBE>GFP markers in combination with the upd3-lacZ reporter to determine the identity of the Upd3-producing cells. In unchallenged animals, upd3-lacZ expression was hardly detectable above background levels (Fig. 3G, upd3-lacZ). 8 hours after challenge, upd3-lacZ was expressed in large Myo1A-expressing cells, which were presumably ECs prior to challenge (Fig. 3H, arrowhead and Fig. S4A, apical Z section). We also observed upd3-lacZ expression in Su(H)GBE>GFP-positive cells (Fig. 3H, top and bottom panel, asterisks). The intermediate size of these Su(H)GBE-GFP-positive cells, with respect to large ECs, as well as their basal location in the epithelium unambiguously identified them as differentiating EBs (Fig. 3H, top panel, asterisk, and Fig. S4, basal Z section). Accordingly, these differentiating EBs also expressed the Myo1A marker (Fig. S4B, arrowheads). We also investigated upd3 expression 18 hours after infection, a time point when most EBs present prior to challenge underwent advanced EB-to-EC differentiation, as demonstrated in Fig. 3E,F. Large Su(H)GBE>GFP-positive cells expressed the upd3-lacZ reporter, indicating that EBs that underwent EB-to-EC differentiation expressed the Upd3 cytokine (Fig. 3I, asterisk). To further determine the respective contribution of Upd3 production in ECs and EBs, we silenced upd3 expression using the EC-specific driver Myo1A-GAL4 or the EB-specific driver Su(H)GBE-GAL4. Silencing upd3 in ECs or in EBs led to a 50% decrease in the rate of ISC division after 8 hours of infection (Fig. 3J,K). We conclude that both ECs and differentiating EBs contribute to the Upd3-mediated modulation of the rate of ISC division in response to Ecc15 infection. These results therefore uncover an important role for EB differentiation in the production of the Upd3 cytokine and the control of the Upd3-dependent ISC division.
Upd3 is required for activation of JAK/STAT signaling in EBs and visceral muscles in response to challenge
To identify the cells responding to Upd3 signaling, we used the 10X-STAT-GFP reporter, that faithfully reports on the activity of the JAK/STAT signaling pathway (Bach et al., 2007). As previously shown, the 10X-STAT-GFP reporter was expressed in EBs and ISCs throughout the posterior midgut in unchallenged animals (Beebe et al., 2010; Buchon et al., 2009b; Cronin et al., 2009; Jiang et al., 2009; Lin et al., 2010; Liu et al.) (Fig. 4A,B, w1118, asterisk). In unchallenged upd3 mutant animals, the 10X-STAT-GFP reporter was expressed in a similar pattern (Fig. 4C,D, Δupd3#9, asterisk), indicating that the activity of the JAK/STAT reporter in EBs and ISCs may be due to Upd and/or Upd2 signaling, but not Upd3 signaling. The 10X-STAT-GFP reporter was strongly expressed in wild-type animals in response to Ecc15 challenge (Fig. 4E, w1118), clearly showing active JAK/STAT signaling in Su(H)GBE-lacZ-positive cells of intermediate size (Fig. 4F, top panels, w1118, asterisks), which are differentiating EBs, and in visceral muscles (VMs) (Fig. 4F, bottom panels). In agreement with a major role for the Upd3 cytokine in the activation of JAK/STAT signaling in response to challenge, the 10X-STAT-GFP reporter was weakly expressed in challenged upd3 mutant animals (Fig. 4G, Δupd3#9). EBs did not undergo complete differentiation, as determined by the absence of Su(H)GBE-lacZ-positive cells of intermediate size (compare the size of EBs (asterisks) in Fig. 4F, w1118, and Fig. 4H, Δupd3#9). Strikingly, the strong activation of JAK/STAT signaling observed in visceral muscles in wild-type animals (Fig. 4F, bottom panel) was absent in upd3 mutant animals (Fig. 4H, bottom panel). We conclude that, in response to challenge, the Upd3 cytokine produced in ECs and differentiating EBs is required for activation of JAK/STAT signaling in EBs and in VMs.
Fig. 4. Upd3 is required for activation of JAK/STAT signaling in EBs and visceral muscles in response to challenge.

(A–H) Low-magnification (left panels) and high-magnification (right panels) confocal microscopy images of 10XSTAT-GFP and Su(H)GBE-lacZ expression in the posterior midgut. Bottom right panels show XY sections. Dotted lines indicate Y coordinates for XZ reconstruction (top right panels). Images corresponding to mock-treated (A and B) and challenged (E and F) wild-type (w1118) and mock-treated (C and D) and challenged (G and H) upd3 mutant (Δupd3#9) animals.
The secreted Upd3 cytokine locates to the basement membrane and signals to VMs
Our previous results suggested that the Upd3 cytokine produced in ECs and in EBs activates JAK/STAT signaling in VMs. To further demonstrate the paracrine nature of Upd3 signaling, we generated a GFP-tagged version of the Upd3 cytokine. We found that the Upd3-GFP cytokine decorated the lining of the circular visceral muscle fibers, as determined by co-staining of F-actin, which is present at high level in the smooth muscles underlying the midgut epithelium (Fig. 5A). These data suggested that the Upd3 cytokine is secreted basally and locates to the basement membrane of the intestine, where it potentially interacts with the extracellular matrix (ECM). In agreement with this notion, we found that the Upd3 cytokine co-localized with the ECM marker Viking (Fig. 5B,C). To further demonstrate that Upd3 production in EBs (or ECs) would activate JAK/STAT signaling in visceral muscles, we over-expressed the UAS-upd3 transgene using the EB-specific driver Su(H)GBE-Gal4. In control animals (Su(H)ts/+), the 10X-STAT-GFP reporter displayed strong expression in EBs, but only basal level expression in visceral muscles (Fig. 5D, asterisk). However, in animals over-expressing the Upd3 cytokine in EBs (Su(H)ts>UAS-upd3), the 10X-STAT-GFP reporter displayed a striking high expression level in the underlying muscles (Fig. 5E, asterisk). Moreover, we noticed the presence of cells of intermediate size displaying 10X-STAT-GFP expression, which were presumably differentiating EBs (Fig. 5E, arrowhead). These results indicate that the Upd3 cytokine produced in EBs locates to the basement membrane, where it displays the potential to trigger EB differentiation and activate JAK/STAT signaling in visceral muscles. As expected, we found that over-expressing the Upd3 cytokine in ECs using the Myo1A-GAL4ts driver led to similar results (data not shown). We conclude that basal secretion of the Upd3 cytokine in producing cells leads to activation of JAK/STAT signaling in responsive cells that interact with the basement membrane, which includes EBs and visceral muscles.
Fig. 5. The Upd3 cytokine produced in EB locates to the extracellular matrix and activates JAK/STAT signaling in EB and in visceral muscles.
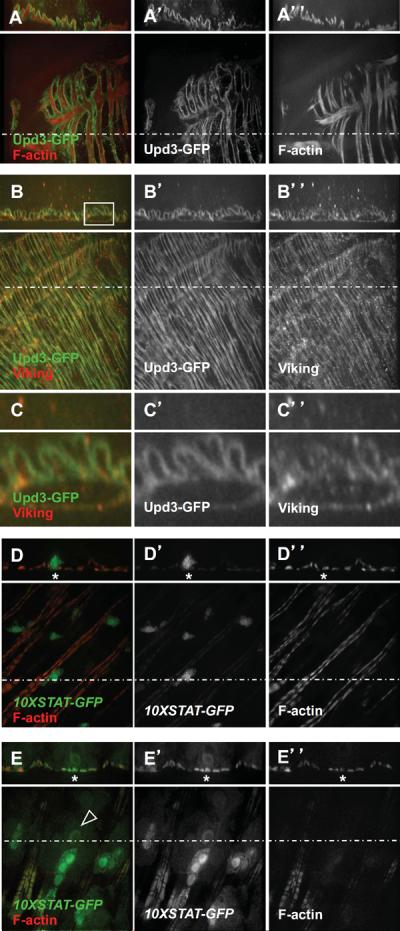
(A) The Upd3-GFP fusion decorates muscles as visualized by F-actin staining.
(B) The Upd3-GFP fusion protein co-localizes with the extracellular matrix marker Viking.
(C) Magnification of the squared area shown in (B).
(D) The 10X-STAT-GFP reporter is expressed in EBs and display basal expression level in visceral muscles.
(E) Over-expression of the UAS-upd3 transgene using the Su(H)-Gal4ts driver leads to 10X-STAT-GFP expression in visceral muscles (asteriks) and differentiation of EBs (arrowhead pointing to the nucleus of a differentiating EBs).
JAK/STAT signaling is required in EBs and in visceral muscles for stimulation of ISC division
To investigate the role of JAK/STAT signaling in various cell types with respect to ISC division, we used the Myo1A-GAL4 (Morgan et al., 1994), Dl-GAL4 (Zeng et al., 2010), Su(H)GBE-GAL4 (Zeng et al., 2010) and How-GAL4 (Brand and Perrimon, 1993) drivers, which confer specific expression in ECs, ISCs, EBs and VMs, respectively. Expression of a dominant negative version of the Drosophila JAK/STAT receptor Domeless indicated that complete stimulation of ISC division in response to challenge requires JAK/STAT signaling in EBs and in VMs, but apparently not in ISCs or ECs (Fig. 6). As opposed to previous conclusions (Buchon et al., 2009a), our results suggest that the activity of JAK/STAT signaling is apparently not required in ISCs (or in ECs) in order to stimulate ISC division in response to Ecc15 infection. Instead, our results indicate that JAK/STAT signaling is required in EBs, the differentiating stem cell progeny, and in VMs, the stem cell niche (Lin et al., 2008), for non-cell autonomous stimulation of ISC division.
Fig. 6. JAK/STAT signaling is required in enteroblasts and visceral muscles for ISC division in response to challenge.

Expression of a dominant negative version of the Domeless receptor (UAS-domeΔCYT) in ISCs and EBs (esg-GAL4), ISCs alone (Dl-GAL4), EBs alone (Su(H)GBE-GAL4), visceral muscles (how-GAL4), or enterocytes (Myo1A-GAL4). Animals were mock-treated (Mock) or infected with Ecc15 for 8 hours (8Ecc). Averages of six independent experiments (n = 12 guts/experiments) with s.e.m. are shown. P values were calculated by two-tailed, unpaired t-tests. Compared to control, esg-GAL4, P = 0.012 (*); Dl-GAL4, P = 0.36 (ns); Su(H)GBE-GAL4, P = 0.017 (*), how-GAL4, P = 0.015 (*); Myo1A-GAL4, P = 0.93 (ns).
Role of Upd3 signaling in stimulation of EGFR-dependent ISC division
Our results suggested an important role for the activity of JAK/STAT signaling in VMs in the modulation of the rate of ISC division in response to challenge. In the Drosophila intestine, VMs act as the stem cell niche (Lin et al., 2008) and produce growth factors, such as the EGF family member Vein, that stimulates EGFR signaling in stem cells and contribute to their maintenance and proliferation (Biteau and Jasper, 2011; Buchon et al., 2010; Jiang et al., 2011; Xu et al., 2011). In agreement with previous findings (Buchon et al., 2010), we found that, in response to Ecc15 challenge, the expression of vein was increased in visceral muscles (Fig. 7A,B). This increase in vein expression relied on upd3 expression (Fig. 7C). However, Vein depletion in VMs did not phenocopy the strong inhibition of ISC division observed in upd3 mutant animals (data not shown), suggesting that EGF family members may display redundant functions in the intestine. In agreement with this assumption, we found that the Upd3 cytokine was also required for activation of the EGF family member gene spitz in response to challenge (Fig. 7F). Analysis of spitz expression pattern revealed that this EGF ligand is expressed in Su(H)GBE-lacZ-positive cells in unchallenged flies (Fig. 7D). After 18 hours of infection, we observed that spitz was expressed in large Su(H)GBE-lacZ-positive cells, which are differentiated EBs (Fig. 7E). Thus, at least two different cell types, EBs and visceral muscle cells, produce Upd3-regulated factors that may regulate EGFR signaling in ISCs. Several groups used the progenitor-specific escargot-GAL4 driver to express a dominant negative version of the EGF receptor and concluded that EGFR signaling is required for stimulation of ISC division (Biteau and Jasper, 2011; Buchon et al., 2010; Jiang et al., 2011; Xu et al., 2011). In agreement with these observations, we showed that blocking EGFR signaling in ISC using the stem-cell specific driver Dl-GAL4 abolished the stimulation of ISC division observed in response to Ecc15 challenge (Fig. 7G). Collectively, these studies support the notion that, in response to challenge, the production of the Upd3 cytokine in differentiating EBs and differentiated ECs leads to activation of JAK/STAT signaling in EBs and VMs, which modulates the JAK/STAT-dependent production of EGF ligands, thereby stimulating the EGFR-dependent ISC division (Fig. 7H).
Fig. 7. Role of Upd3 signaling in EGFR-dependent ISC division.

(A–B) Vein is weakly expressed in visceral muscles as visualized by F-actin staining in mock-treated animals and highly expressed in animals infected for 18 hours with Ecc15.
(C) Quantification of vein expression by RT-PCR in mock-treated animals and in animals infected with Ecc15 for 8 hours (8Ecc). For w1118, 8Ecc vs. Mock, P = 0.0006 (***). For Δupd3#9, 8Ecc vs. Mock, P = 0.5585 (ns).
(D and E) Spitz is expressed in EBs as visualized with the Su(H)GBE-lacZ marker in mock-treated animals and in differentiated EBs in animals infected with Ecc15 for 18 hours.
(F) Quantification of spitz expression by RT-PCR in mock-treated animals and in animals infected with Ecc15 for 8 hours (8Ecc). For w1118, 8Ecc vs. Mock, P = 0.0101 (*). For Δupd3#9, 8Ecc vs. Mock, P = 0.8212 (ns).
(G) EGF signaling was conditionally (using tub-GAL80ts) inhibited in ISCs and EBs (esg-GAL4), or ISCs alone (Dl-GAL4) by expressing a dominant-negative form of the EGF receptor (UAS-EGFRDN). For Ecc15 treatment (8Ecc), escg-GAL4 vs. w1118, P = 0.0414 (*) and Dl-GAL4 vs. w1118, P = 0.0012 (**).
(H) Model of intestinal homeostasis. Erwinia infection activates upd3 expression in mature enterocytes (EC) and in differentiating enteroblasts (EB) (Upd3 production and producing cells are depicted in red). The basal secretion of the Upd3 cytokine activates JAK/STAT signaling in EBs and in visceral muscles (VM) (JAK/STAT signaling depicted in green). Activation of JAK/STAT signaling in EBs and VMs leads to EGF ligand production that stimulates the EGFR-dependent division of ISCs (EGF signaling depicted in blue).
DISCUSSION
In Drosophila, the replacement of spent enterocytes (ECs) relies on division of intestinal stem cells (ISCs) and differentiation of their progeny, the enteroblasts (EBs). The mechanisms coupling environmental challenges and intestinal stem cell division have been recently investigated in Drosophila (Amcheslavsky et al., 2009; Buchon et al., 2009b; Cronin et al., 2009; Jiang et al., 2009). A consensus has emerged in the field supporting a model of intestinal homeostasis in which environmental challenges stimulate cytokine production in challenged ECs, which constitutes a mitogenic signal that stimulates ISC division. The results reported here lead us to propose an alternative model in which in response to environmental challenges, challenged ECs, but also differentiating EBs participate in cytokine production, which modulates the JAK/STAT-dependent activity of the stem cell microenvironment (the niche) and indirectly modulates the rate of EGFR-dependent stem cell division (see model in Fig. 7H). Below, we discussed the implication so this alternative model of intestinal homeostasis.
Is there a role for JAK/STAT signaling in ISCs in the control of ISC division?
Numerous studies on the mammalian intestine have documented the role of JAK/STAT signaling in cell proliferation and tumor formation (Becker et al., 2004; Rigby et al., 2007). Similarly, recent studies reported a major role for JAK/STAT signaling in the control of stem cell division in the Drosophila intestine (Buchon et al., 2009a; Buchon et al., 2009b; Cronin et al., 2009; Jiang et al., 2009). These Drosophila studies relied on genetic manipulations using the escargot-Gal4 driver, which is not only expressed in ISCs, but also in EBs (Micchelli and Perrimon, 2006; Ohlstein and Spradling, 2006). Here, we interfered with JAK/STAT signaling by expressing a dominant negative version of the Domeless receptor with the Dl-GAL4 (Zeng et al., 2010) and Su(H)-GAL4 (Zeng et al., 2010) drivers, which confer specific expression in ISCs and EBs, respectively. These experiments revealed that, in order to stimulate ISC division, JAK/STAT signaling is apparently not required in ISCs, but in EBs. Interestingly and in agreement with the notion that JAK/STAT signaling is not required in ISCs for ISC division, several groups have reported the outcome of genetic experiments clearly indicating that STAT−/− clones displayed normal cell numbers, but accumulated cells with small nuclei. This indicates that STAT−/− clones proliferate normally, but their progeny fails to differentiate into large ECs (examples of STAT −/− clones can be found in Figure 4D, E, I from (Jiang et al., 2009), Figure 3B, D from (Beebe et al., 2010), and Figure 3D, E, F, H from (Lin et al., 2010)). Altogether, these data suggest that JAK/STAT signaling in ISCs plays little role in ISC division under normal physiological conditions, as well as in response to environmental challenge.
Regulation of upd3 expression
Our genetic analysis revealed a crucial role for the Upd3 cytokine in the control of ISC division in response to challenge. Several studies have proposed that the Upd3 cytokine may be expressed by challenged ECs in response to bacterial infection (Buchon et al., 2009b; Jiang et al., 2011). However, the identity of the producing cells had never been unambiguously demonstrated using specific markers. Here, we used Myo1A and Su(H)GBE-lacZ and showed that, as previously proposed, the Upd3 cytokine is expressed in mature ECs early on during infection. In addition, our study reveals that bacterial challenge triggers a massive wave of differentiation that triggers upd3 expression in differentiating EB. The mechanism(s) regulating upd3 expression in ECs and differentiating EBs are unresolved. The possibility that upd3 expression may be regulated by NF-kB signaling in response to bacterial infection has been tested and excluded (Buchon et al., 2009b). It was suggested that the infection process leads to the production of reactive oxygen species (ROS) (Buchon et al., 2009a) that may activate signaling events involved in stress responses, such as JNK signaling. In this context, ROS production may activate upd3 expression in challenged ECs. We also note that ROS have been shown to regulate progenitor differentiation in Drosophila hematopoietic lineage (Owusu-Ansah and Banerjee, 2009). It is therefore possible that ROS production may trigger the massive wave of differentiation observed in response to infection, thereby leading to upd3 expression in EBs. In addition to infection, various treatments leading to tissue damage, including feeding toxic compounds or genetic manipulation of ECs, lead to upd3 activation (Buchon et al., 2009b; Jiang et al., 2009). It is therefore tempting to speculate that upd3 activation results from the damage caused to the epithelium by the infection process. For instance, the loss of EC-EB cell-cell contacts, as a consequence of the damages inflicted by the infection process, may trigger the signaling events leading to upd3 expression in ECs and in EBs. We note that this mode of regulation may not only take place in response to extensive damage caused by bacterial infection and may also occur locally under physiological conditions when spent ECs undergo apoptosis and leave the epithelium (our unpublished observation).
Stem cell microenvironment and the control of stem cell division
Numerous studies have emphasized the crucial role of the stem cell niche in the maintenance of stem cells (Morrison and Spradling, 2008; Ohlstein et al., 2004). In the Drosophila intestine, the visceral muscles act as a niche and provide growth factors that stimulate stem cell division (Jiang and Edgar, 2009; Lin et al., 2008). Our results show that, in order to modulate the rate of stem cell division in response to challenge, JAK/STAT signaling is required in EBs and in visceral muscles, where it controls the expression of growth factors that potentially stimulate stem cell division. Thus, our results indicate that Upd3/JAK/STAT signaling control the rate of stem cell division through modulation of the signaling activity of the stem cell microenvironment in the Drosophila intestine. Interestingly, a similar situation was recently described in studies on the mammalian bladder, where the activity of the Hedgehog/Wnt and BMP signaling pathways in the stem cell microenvironment modulate the rate of stem cell division in response to urinary infection (Mysorekar et al., 2009; Shin et al., 2011). Thus, modulation of stem cell microenvironment signaling activity is a conserved feature of the proliferative response to infection in Drosophila and in mammals.
A role for stem cell progeny in the control of stem behavior
Recently, Ohlstein and colleagues showed that insulin signaling in EBs control the rate of ISC division through modulation of cell-cell contacts (Choi et al.). Here, we demonstrate that differentiating EBs produce signaling molecules, such as Upd3 cytokine, that control the rate of stem cell division. Thus, the importance of stem cell progeny and their differentiation in the control of stem cell division is an emerging theme in stem cell biology. The differentiation process is often regarded as a mere consequence of stem cell division and daughter cell specification. However, our results indicate that the differentiation process and production of signaling molecules instruct stem cell division though modulation of the microenvironment activity in the Drosophila intestine. In the mammalian intestine, stem cell progeny are transit-amplifying cells that ultimately differentiate into one of the mature intestinal cell lineages (Radtke and Clevers, 2005). Given the striking structural and functional similarities displayed by the Drosophila and the mammalian intestine, it is possible that similar to the findings reported here the differentiation process plays a critical role in the modulation of stem cell division. The Drosophila intestine therefore constitutes a powerful system to investigate the role of the differentiation process in orchestrating the maintenance of intestinal homeostasis under physiological conditions and in response to challenge.
MATERIALS AND METHODS
Bacterial stocks
Erwinia carotovora carotovora 15 (Ecc15) (Basset et al., 2000) was grown in LB medium at 30C. Pseudomonas entomophila (Pe) (Vodovar et al., 2005) and Serratia marcescens Db11 (Sm) (Kurz et al., 2003) were grown in LB medium at 37C.
Fly stocks and culture
Δupd3#9, upd3-lacZ, UAS-upd3, UAS-upd3-GFP and upd3-GAL4 lines are described in the Methods section. The following fly strains were used: w1118 (BestGene); 10XSTAT-GFP (Bach et al., 2007); Su(H)GBE-lacZ (Furriols and Bray, 2001);UAS-domeΔCYT (Brown et al., 2001); esg-GAL4,UAS-GFP (DGRC, Kyoto, Japan); Su(H)-GAL4 and dl-GAL4 (Zeng et al., 2010); how-GAL4 (Brand and Perrimon, 1993); Myo1A-GAL4 (Morgan et al., 1994); spitz-GAL4 (Jiang and Edgar, 2009); tub-GAL80ts (Bloomington Drosophila Stock Center); UAS-EGFRDN(stock 5364, Bloomington Drosophila Stock Center); vn-lacZ (stock 11749, Bloomington Drosophila Stock Center). Flies were maintained on standard media at room temperature. For GAL4ts experiments, F1 progeny were raised at 18°C throughout their development. GAL4ts activity was controlled by placing 5 day-old F1 adults at either restrictive (29°C) or permissive (18°C) temperatures.
Generation of the upd3 mutant line
The various deletion alleles of the upd3 locus were generated by excision of P element P(XP)upd3d00871 inserted in the third intron of the upd3 gene. P(XP)upd3d00871 /FM7 females were crossed to P[ry+, Delta 2–3]99B/TM3 males and non-FM7 F1 males were then crossed to attached-X females. F2 males were scored for potential P element excision on the basis of eye color. Excision of the P element was confirmed by PCR amplification. DNA sequencing of the Δupd3#9 mutant line revealed a deletion spanning from position + 3476 to position + 6706 with reference to the start codon (Figure 2A). This results in the production of a truncated polypeptide displaying the first 217 amino-acid residues of Upd3 plus 7 C-terminal amino acid residues (VSVVHMP) resulting from deletion of the acceptor splicing sites of exon 4 and translation termination in (unspliced) intron 3.
Generation of upd3-lacZ
The upd3-lacZ reporter construct was generated by PCR amplification of the 4kb DNA region upstream from the upd3 start codon using oligonucleotides 5' upd3 4kb-PRM-KpnI (5'-GGTACCGTTACCCGGTGATCATCACG -3') and 3' upd3 PRM-BamHI (5′-GGATCCGTTGGCTGGGAATTCCGCAC -3′) as primers, and genomic DNA as template. The resulting PCR product was cloned into pPelican and injected into embryos.
Generation of UAS-upd3
The 5' end of the upd3 cDNA was determined by RACE using total RNA extracted from adult flies and the SMART RACE cDNA amplification kit (Clontech). The entire upd3 cDNA was cloned by PCR amplification using oligonucleotides 5' upd3 RACE end-KpnI (5'-GGTACCGTGCGTGTCAATCAAGTACG -3') and 3' upd3 -XbaI (5'-TCTAGACTAGAGTTTCTTCTGGATC -3') as primers and from adult fly cDNA as template. The resulting amplicon was cloned into pCR-TOPO 2.1 (Invitrogen). DNA sequencing confirmed the electronic annotation (www.flybase.org), except for a large intron in the 5' region (position +3 to +1832, with respect to the start codon). The transcription start site at position −595 was inferred from the sequence of the largest clone identified by RACE. The position of the second, third and fourth intron was confirmed by DNA sequencing. Transgenic flies expressing the UAS-upd3(p) 1M rescue construct were generated by cloning the KpnI-XbaI cDNA fragment into pUASp and injected into embryos.
Generation of upd3-GAL4
Transgenic flies expressing the upd3-GAL42kb-1M reporter construct was generated by PCR amplification of the 2 kb DNA region located upstream from the upd3 start codon using oligonucleotides 5' upd3 PRM-KpnI (5'-GGTACCATGACACCGATCACCA -3') and 3' upd3 PRM-BamHI (5'-GGATCCGTTGGCTGGGAATTCCGCAC -3') as primers and genomic DNA as template. The resulting PCR product was cloned into pGaTB. The DNA fragment harboring the upd3-GAL4 construct was excised from the corresponding plasmid using KpnI and NotI and cloned into pCaSpeR4 and injected into embryos.
Generation of UAS-upd3-GFP
The upd3 cDNA was PCR amplified using oligonucleotides 5' upd3-KpnI (5'-GGTGGTACCGTGCGTGTCAATCAAGTACG -3') and 3' upd3-XmaI (5'-CCCCCCGGGCGAGTTTCTTCTGGATCGCCTTTGGC -3') as primers and cloned into pEGFP-N1 (Clonetech, Inc) generating pUpd3-EGFP-N1. Transgenic flies expressing the UAS-upd3-GFP rescue construct were generated by cloning the EcoRI/NotI upd3-GFP DNA fragment from pUpd3-EGFP-N1 into pUASp and injected into embryos.
Enteric infection
One-week-old females were starved for 2 hours, and then placed in empty vials containing Kimwipe soaked in 5% (w/v) sucrose solution (mock infection), or soaked in bacteria pellet re-suspended with 5% sucrose. Erwinia carotovora carotovora was administered at OD600 = 200, unless noted otherwise. Pseudomonas entomophila: OD600 = 20; Serratia marcescens: OD600 = 50. The flies were then kept at 29°C for the duration of the infection.
Immunofluorescence and microscopy
Guts were dissected in 1X phosphate-buffered saline (PBS) and fixed at room temperature for 1 hour in 1X PBS, 2mM MgCl2, 4% paraformaldehyde. Samples were washed and blocked for 1 hour in 1X PBS, 1% Bovine Serum Albumin (BSA), 0.1% TritonX-100. All subsequent stainings and washes were done in 1X PBS, 1% BSA, 0.1% TritonX-100. The samples were stained overnight with primary antibodies at 4°C, and for 2 hours with secondary antibodies at room temperature. The samples were mounted using a DABCO-based anti-fade media. The following primary antibodies were used: rabbit anti-pH3 (Millipore; 1:1,000); mouse anti-β-Galactosidase (Promega; 1:1,000); chicken anti-GFP (Abcam; 1:10,000). Fluorescent secondary antibodies used were: Alexa488 or Alexa594-coupled anti-mouse or anti-rabbit antibodies (Invitrogen); FITC-coupled anti-chicken antibody (Jackson ImmunoResearch). DNA and F-actin were stained using Hoechst and Phalloidin, respectively (Molecular Probes).
Fluorescent microscopy was conducted on a TE 2000 microscope (Nikon) and images were processed using the MetaMorph software (Molecular Devices). Confocal microscopy was conducted on a spinning-disc confocal microscope (Improvision) and images were processed using the Volocity software (PerkinElmer).
Supplementary Material
Highlights
Upd3 is required for stimulation of ISC division in response to challenge
Upd3 is produced in differentiated ECs and in differentiating EBs
Upd3 is required for activation of JAK/STAT signaling in EBs and in visceral muscles
JAK/STAT signaling is required in EBs and in visceral muscles for ISC division
ACKNOWLEDGMENTS
This work was supported by the ASTAR agency (F.Z.) and the National Institutes of Health Grants F32-AI07138803 (A.W.R.) and R01-AI068814 (H.A.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Amcheslavsky A, Jiang J, Ip YT. Tissue damage-induced intestinal stem cell division in Drosophila. Cell Stem Cell. 2009;4:49–61. doi: 10.1016/j.stem.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apidianakis Y, Pitsouli C, Perrimon N, Rahme L. Synergy between bacterial infection and genetic predisposition in intestinal dysplasia. Proc Natl Acad Sci U S A. 2009;106:20883–20888. doi: 10.1073/pnas.0911797106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach EA, Ekas LA, Ayala-Camargo A, Flaherty MS, Lee H, Perrimon N, Baeg GH. GFP reporters detect the activation of the Drosophila JAK/STAT pathway in vivo. Gene Expr Patterns. 2007;7:323–331. doi: 10.1016/j.modgep.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Basset A, Khush RS, Braun A, Gardan L, Boccard F, Hoffmann JA, Lemaitre B. The phytopathogenic bacteria Erwinia carotovora infects Drosophila and activates an immune response. Proc Natl Acad Sci U S A. 2000;97:3376–3381. doi: 10.1073/pnas.070357597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker C, Fantini MC, Schramm C, Lehr HA, Wirtz S, Nikolaev A, Burg J, Strand S, Kiesslich R, Huber S, Ito H, Nishimoto N, Yoshizaki K, Kishimoto T, Galle PR, Blessing M, Rose-John S, Neurath MF. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity. 2004;21:491–501. doi: 10.1016/j.immuni.2004.07.020. [DOI] [PubMed] [Google Scholar]
- Beebe K, Lee WC, Micchelli CA. JAK/STAT signaling coordinates stem cell proliferation and multilineage differentiation in the Drosophila intestinal stem cell lineage. Dev Biol. 2010;338:28–37. doi: 10.1016/j.ydbio.2009.10.045. [DOI] [PubMed] [Google Scholar]
- Biteau B, Hochmuth CE, Jasper H. JNK activity in somatic stem cells causes loss of tissue homeostasis in the aging Drosophila gut. Cell Stem Cell. 2008;3:442–455. doi: 10.1016/j.stem.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biteau B, Jasper H. EGF signaling regulates the proliferation of intestinal stem cells in Drosophila. Development. 2011;138:1045–1055. doi: 10.1242/dev.056671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Brown S, Hu N, Hombria JC. Identification of the first invertebrate interleukin JAK/STAT receptor, the Drosophila gene domeless. Curr Biol. 2001;11:1700–1705. doi: 10.1016/s0960-9822(01)00524-3. [DOI] [PubMed] [Google Scholar]
- Buchon N, Broderick NA, Chakrabarti S, Lemaitre B. Invasive and indigenous microbiota impact intestinal stem cell activity through multiple pathways in Drosophila. Genes Dev. 2009a;23:2333–2344. doi: 10.1101/gad.1827009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchon N, Broderick NA, Kuraishi T, Lemaitre B. Drosophila EGFR pathway coordinates stem cell proliferation and gut remodeling following infection. BMC Biol. 2010;8:152. doi: 10.1186/1741-7007-8-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchon N, Broderick NA, Poidevin M, Pradervand S, Lemaitre B. Drosophila intestinal response to bacterial infection: activation of host defense and stem cell proliferation. Cell Host Microbe. 2009b;5:200–211. doi: 10.1016/j.chom.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Casali A, Batlle E. Intestinal stem cells in mammals and Drosophila. Cell Stem Cell. 2009;4:124–127. doi: 10.1016/j.stem.2009.01.009. [DOI] [PubMed] [Google Scholar]
- Choi NH, Lucchetta E, Ohlstein B. Nonautonomous regulation of Drosophila midgut stem cell proliferation by the insulin-signaling pathway. Proc Natl Acad Sci U S A. 108:18702–18707. doi: 10.1073/pnas.1109348108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronin SJ, Nehme NT, Limmer S, Liegeois S, Pospisilik JA, Schramek D, Leibbrandt A, Simoes RD, Gruber S, Puc U, Ebersberger I, Zoranovic T, Neely GG, von Haeseler A, Ferrandon D, Penninger JM. Genome-Wide RNAi Screen Identifies Genes Involved in Intestinal Pathogenic Bacterial Infection. Science. 2009;325:340–343. doi: 10.1126/science.1173164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furriols M, Bray S. A model Notch response element detects Suppressor of Hairless-dependent molecular switch. Curr Biol. 2001;11:60–64. doi: 10.1016/s0960-9822(00)00044-0. [DOI] [PubMed] [Google Scholar]
- Jiang H, Edgar BA. EGFR signaling regulates the proliferation of Drosophila adult midgut progenitors. Development. 2009;136:483–493. doi: 10.1242/dev.026955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Grenley MO, Bravo MJ, Blumhagen RZ, Edgar BA. EGFR/Ras/MAPK signaling mediates adult midgut epithelial homeostasis and regeneration in Drosophila. Cell Stem Cell. 2011;8:84–95. doi: 10.1016/j.stem.2010.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Patel PH, Kohlmaier A, Grenley MO, McEwen DG, Edgar BA. Cytokine/Jak/Stat signaling mediates regeneration and homeostasis in the Drosophila midgut. Cell. 2009;137:1343–1355. doi: 10.1016/j.cell.2009.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz CL, Chauvet S, Andres E, Aurouze M, Vallet I, Michel GP, Uh M, Celli J, Filloux A, De Bentzmann S, Steinmetz I, Hoffmann JA, Finlay BB, Gorvel JP, Ferrandon D, Ewbank JJ. Virulence factors of the human opportunistic pathogen Serratia marcescens identified by in vivo screening. EMBO J. 2003;22:1451–1460. doi: 10.1093/emboj/cdg159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin G, Xu N, Xi R. Paracrine Wingless signalling controls self-renewal of Drosophila intestinal stem cells. Nature. 2008;455:1119–1123. doi: 10.1038/nature07329. [DOI] [PubMed] [Google Scholar]
- Lin G, Xu N, Xi R. Paracrine unpaired signaling through the JAK/STAT pathway controls self-renewal and lineage differentiation of drosophila intestinal stem cells. J Mol Cell Biol. 2010;2:37–49. doi: 10.1093/jmcb/mjp028. [DOI] [PubMed] [Google Scholar]
- Liu W, Singh SR, Hou SX. JAK-STAT is restrained by Notch to control cell proliferation of the Drosophila intestinal stem cells. J Cell Biochem. 2010;109:992–999. doi: 10.1002/jcb.22482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micchelli CA, Perrimon N. Evidence that stem cells reside in the adult Drosophila midgut epithelium. Nature. 2006;439:475–479. doi: 10.1038/nature04371. [DOI] [PubMed] [Google Scholar]
- Morgan NS, Skovronsky DM, Artavanis-Tsakonas S, Mooseker MS. The molecular cloning and characterization of Drosophila melanogaster myosin-IA and myosin-IB. J Mol Biol. 1994;239:347–356. doi: 10.1006/jmbi.1994.1376. [DOI] [PubMed] [Google Scholar]
- Morrison SJ, Spradling AC. Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell. 2008;132:598–611. doi: 10.1016/j.cell.2008.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mysorekar IU, Isaacson-Schmid M, Walker JN, Mills JC, Hultgren SJ. Bone morphogenetic protein 4 signaling regulates epithelial renewal in the urinary tract in response to uropathogenic infection. Cell Host Microbe. 2009;5:463–475. doi: 10.1016/j.chom.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlstein B, Kai T, Decotto E, Spradling A. The stem cell niche: theme and variations. Curr Opin Cell Biol. 2004;16:693–699. doi: 10.1016/j.ceb.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Ohlstein B, Spradling A. The adult Drosophila posterior midgut is maintained by pluripotent stem cells. Nature. 2006;439:470–474. doi: 10.1038/nature04333. [DOI] [PubMed] [Google Scholar]
- Ohlstein B, Spradling A. Multipotent Drosophila intestinal stem cells specify daughter cell fates by differential notch signaling. Science. 2007;315:988–992. doi: 10.1126/science.1136606. [DOI] [PubMed] [Google Scholar]
- Osman D, Buchon N, Chakrabarti S, Huang YT, Su WC, Poidevin M, Tsai YC, Lemaitre B. Autocrine and paracrine unpaired signalling regulate intestinal stem cell maintenance and division. J Cell Sci. 2012 doi: 10.1242/jcs.113100. [DOI] [PubMed] [Google Scholar]
- Owusu-Ansah E, Banerjee U. Reactive oxygen species prime Drosophila haematopoietic progenitors for differentiation. Nature. 2009;461:537–541. doi: 10.1038/nature08313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radtke F, Clevers H. Self-renewal and cancer of the gut: two sides of a coin. Science. 2005;307:1904–1909. doi: 10.1126/science.1104815. [DOI] [PubMed] [Google Scholar]
- Rigby RJ, Simmons JG, Greenhalgh CJ, Alexander WS, Lund PK. Suppressor of cytokine signaling 3 (SOCS3) limits damage-induced crypt hyper-proliferation and inflammation-associated tumorigenesis in the colon. Oncogene. 2007;26:4833–4841. doi: 10.1038/sj.onc.1210286. [DOI] [PubMed] [Google Scholar]
- Shin K, Lee J, Guo N, Kim J, Lim A, Qu L, Mysorekar IU, Beachy PA. Hedgehog/Wnt feedback supports regenerative proliferation of epithelial stem cells in bladder. Nature. 2011;472:110–114. doi: 10.1038/nature09851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vodovar N, Vinals M, Liehl P, Basset A, Degrouard J, Spellman P, Boccard F, Lemaitre B. Drosophila host defense after oral infection by an entomopathogenic Pseudomonas species. Proc Natl Acad Sci U S A. 2005;102:11414–11419. doi: 10.1073/pnas.0502240102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu N, Wang SQ, Tan D, Gao Y, Lin G, Xi R. EGFR, Wingless and JAK/STAT signaling cooperatively maintain Drosophila intestinal stem cells. Dev Biol. 2011;354:31–43. doi: 10.1016/j.ydbio.2011.03.018. [DOI] [PubMed] [Google Scholar]
- Zeng X, Chauhan C, Hou SX. Characterization of midgut stem cell- and enteroblast-specific Gal4 lines in drosophila. Genesis. 2010;48:607–611. doi: 10.1002/dvg.20661. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.