

Abstract
Components of cytoplasmic Processing bodies (P-bodies) and stress granules can be subverted during viral infections to modulate viral gene expression. Because hepatitis C virus (HCV) RNA abundance is regulated by P-body components such as microRNA miR-122, Argonaute 2 and RNA helicase RCK/p54, we examined whether HCV infection modulates P-bodies and stress granules during viral infection. It was discovered that HCV infection decreased the number of P-bodies, but induced the formation of stress granules. Immunofluorescence studies revealed that a number of P-body and stress granule proteins co-localized with viral core protein at lipid droplets, the sites for viral RNA packaging. Depletion of selected P-body proteins decreased overall HCV RNA and virion abundance. Depletion of stress granule proteins also decreased overall HCV RNA abundance, but surprisingly enhanced the accumulation of infectious, extracellular virus. These data argue that HCV subverts P-body and stress granule components to aid in viral gene expression at particular sites in the cytoplasm.
Keywords: hepatitis C virus, Processing bodies, stress granules, lipid droplets, viral RNA assembly
Introduction
Individuals chronically infected with Hepatitis C virus (HCV) are at risk to develop liver cirrhosis and hepatocellular carcinoma (Seeff, 2009). Unfortunately, treatments directed against HCV are limited and a protective vaccine is not available. Thus, an understanding of the HCV lifecycle and virus-host interactions is crucial for the development of new strategies that target HCV. HCV is an enveloped RNA virus of the Flaviviridae family (Lindenbach and Rice, 2001). The 9.6 kb single-stranded positive-sense genome contains conserved 5′ and 3′ untranslated regions (UTRs), and a single open reading frame that is translated by an internal ribosome entry site mechanism to yield ten viral proteins: Core, envelope glycoprotein 1 (E1), E2, p7, nonstructural protein 2 (NS2), NS3, NS4A, NS4B, NS5A and NS5B. The viral NS proteins direct replication of the HCV genome and also participate in the assembly of viral particles on lipid droplets (reviewed in (Bartenschlager et al., 2011). The assembled viral particles contain the structural proteins Core, E1 and E2. To maintain viral RNA abundance in the infected liver, HCV RNA forms a specific interaction with the liver-specific microRNA miR-122 at two sites within the 5′ UTR (Jopling et al., 2008; Jopling et al., 2005; Machlin et al., 2011). This oligomeric RNA complex enhances HCV RNA stability (Norman and Sarnow, 2010), and its anatomy and intracellular localization is under intense scrutiny.
The stability of both cellular and viral RNAs can be modulated in specific cytoplasmic granular structures, known as Processing bodies (P-bodies) and stress granules. P-bodies are discrete cytoplasmic foci where nontranslating mRNAs are either decapped and degraded (Cougot et al., 2004; Sheth and Parker, 2003). Additionally, some mRNAs may be stored in P-bodies and later returned to the cytoplasm as translation-competent mRNAs (Brengues et al., 2005). However, this model has recently been challenged (Arribere et al., 2011). P-bodies are constitutively present in cells, although their number and size depend on the abundance of RNAs sequestered for storage and turnover (Cougot et al., 2004). Components of P-bodies include mRNAs, the decapping enzyme complex Dcp1-Dcp2 (van Dijk et al., 2002), the 5′-3′ exoribonuclease Xrn1 (Ingelfinger et al., 2002), mRNA deadenylase CCR4, activators of decapping such as Lsm1-7 proteins, DEAD-box helicase RCK/p54 (also known as DDX6), RNA-associated protein 55 (RAP55), hEDC3, and Ge-1 (reviewed in Eulalio, 2007 #137}. The finding that Argonaute proteins, microRNAs and microRNA-targeted mRNAs reside in P-bodies as well, argues for a role for P-bodies in microRNA-directed gene regulation (Bhattacharyya et al., 2006; Liu et al., 2005; Pauley et al., 2006; Pillai et al., 2005).
Interestingly, P-bodies often reside next to stress granules, which store a subset of cellular mRNAs that are translationally arrested in response to environmental stimuli, such as oxidative stress, nutrient deprivation or viral infections (reviewed in (Anderson and Kedersha, 2009b; Buchan and Parker, 2009). In addition to the translationally stalled mRNAs, stress granules also harbor translation initiation factors eIF2, eIF3, eIF4A, eIF4E and eIF4G, 40S ribosomal subunits, and poly(A) binding protein (Kedersha and Anderson, 2007). Furthermore, proteins such as T-cell restricted intracellular antigen-1 (TIA-1), TIA-1 related protein (TIAR), the RasGAP-associated endonuclease G3BP1 (Tourriere et al., 2003), ubiquitin-specific processing protease 10 (USP10) (Soncini et al., 2001), Caprin1 (Solomon et al., 2007), OGFOD1 (Wehner et al., 2010) and Argonaute proteins (Leung et al., 2006) are also recruited to stress granules.
To investigate the role of P-body and stress granule constituents in the HCV infectious cycle, we examined the fates of P-bodies and stress granules during HCV infection. Formation of stress granules and dramatic changes in P-body number were observed upon viral infection; and depletion of selected P-body and stress granule proteins greatly affected the abundance of HCV RNA, proteins and virus particles. These findings reveal important roles for these cytoplasmic structures in the HCV life cycle and suggest that they can be targeted for antiviral intervention.
Results
Dispersion of P-bodies following HCV infections
To examine the fate and morphology of P-bodies during HCV infection, Huh7 cells were infected with the genotype 2 JFH-1 strain of HCV, and the distribution of P-bodies in uninfected and infected cells was examined by confocal microscopy using antibodies directed against the P-body protein RCK/p54. RCK/p54 localized to distinct P-bodies in uninfected cells (Fig. 1A). However, the number of these structures greatly diminished in JFH-1-infected cells, as identified by NS5A-positive staining. To determine if the reduced number of P-bodies in JFH-1-infected cells was significant, we counted the number of P-bodies per cell in uninfected and infected cells. The number of P-bodies decreased by 50% in infected cells (Fig. 1B). Interestingly, JFH-1 infection did not completely abrogate P-bodies and similar numbers of P-bodies, albeit decreased, remained 1, 2 and 3 days post-infection (p.i.). The dispersal of P-bodies was not specific to the fast replicating genotype 2 JFH-1 virus, because electroporation of infectious genotype 1 H77c RNA dispersed P-bodies as well (Fig. S1A). P-bodies were also dispersed following electroporation of a genotype 1 replicon RNA (H77ΔE1/p7) that lacks the viral envelope and p7 genes, and does not give rise to infectious particles but undergoes RNA replication (Fig. S1A). These findings suggest that dispersion of P-bodies does not require virus-receptor interactions, viral packaging or the formation of infectious particles. Furthermore, proteins required for viral mRNA translation and RNA replication are sufficient to accomplish the dispersal of P-bodies.
Figure 1. Dispersion of P-bodies during JFH-1 infection.

(A) Distribution of P-bodies. Huh7 cells were infected with JFH-1 virus and 3 days p.i., the distribution of P-bodies in uninfected and infected cells was examined by confocal microscopy. P-bodies and HCV-positive cells were identified by staining with specific antibodies detecting RCK/p54 (red) or HCV NS5A (green) proteins. Representative images are shown. (B) Number of P-bodies in uninfected and JFH-1-infected cells 1, 2, and 3 days p.i. The number of P-bodies was quantified using ImageJ software. Mean values and standard deviations of four independent experiments are shown, *** P<0.001 and ** P<0.002. (C) Abundances of P-body proteins (RCK/p54, Lsm1, Dcp1a, Dcp2, Ge-1, Xrn1, Ago2), and viral NS5A and core proteins in JFH-1 infected and uninfected Huh7 cells at 1, 2, and 3 days p.i. were examined by western blot analysis. The abundance of actin serves as a loading control.
Effects of HCV infection on the abundance of P-body proteins
To determine whether P-body dispersion after JFH-1 infection affected the abundance or integrity of specific P-body proteins, numerous P-body proteins were examined by western blot in uninfected or JFH-1-infected cells. Kinetic analysis showed that neither the abundance nor molecular weights of P-body proteins RCK/p54, Lsm1, Dcp1a, Dcp2, Ge-1, Xrn1 or Ago2 changed during infection with JFH-1 (Fig. 1C). These data argue that the dispersal of P-bodies during viral infection does not result from a change in the abundance of the examined P-body proteins. Given that P-bodies are known to be involved in many aspects of microRNA function in the cytoplasm, we also examined the abundance of miR-122 at early (1 or 2 days) or late (3 days) times after viral infections (Fig. S1B). No differences in miR-122 abundances were observed, arguing that P-body dispersion does not affect miR-122 turnover or synthesis during HCV infection.
Localization of viral core protein with the P-body protein RCK/p54 and lipid droplets
To examine whether HCV interacts with some or all dispersed P-body proteins, the localization of viral core with several P-body proteins was determined by confocal microscopy three days after virus infection. Co-localization of proteins was quantitated by determination of the Pearson’s correlation coefficient (Adler and Parmryd, 2010) (see Materials and Methods). This analysis showed that HCV core partially localized with P-body proteins RCK/p54 (Fig. 2B), Lsm1, Dcp2 and Xrn1 (Fig. S2) in infected cells. It is known that HCV core protein localizes to lipid droplets that accumulate in infected cells (Fig. 2C) (Barba et al., 1997; Miyanari et al., 2007). Therefore, we examined whether core-associated RCK/p54 protein localized to lipid droplets during viral infection. Indeed, RCK/p54 partially localized to lipid droplets in infected cells (Fig. 2D and Fig. S2), but not in uninfected cells (Fig. 2A). The co-localization of RCK/p54 with core and lipid droplets was similar to diglyceride acyltransferase-1 (DGAT-1; Fig. S2), a protein known to facilitate core-mediated virion assembly at lipid droplets (Herker et al., 2010). These findings show that JFH-1 infection directs the redistribution of the P-body proteins such as RCK/p54 and Lsm1 to a complex that harbors viral core protein, cellular lipid droplets and DGAT-1. In contrast, none of the P-body proteins examined co-localized with viral dsRNA or the replication protein NS5A (data not shown).
Figure 2. Localization of RCK/p54, core and lipid droplets during JFH-1 infection.

(A) Localization of RCK/p54 and lipid droplets in uninfected Huh7 cells. (B–D) Localization of RCK/p54, HCV core and lipid droplets in JFH-1 infected Huh7 cells 3 days p.i. Bodipy 493/503-was used to stain lipid droplets, and images were acquired with a Zeiss LSM5 confocal microscope. RCK/p54 (B, D) and HCV core (B) in the representative image have been re-colored image to show co-localization. The Pearson’s correlation coefficient (r) quantifying the degree of co-localization is presented in the merged images.
Decrease of HCV RNA and protein abundances after siRNA-mediated depletion of certain P-body proteins
To examine the effect of P-body protein abundances on JFH-1 gene expression, the abundances of nine different P-body proteins were reduced using siRNAs. Western blot analyses showed that each P-body protein could be effectively depleted (Fig. 3A). While siRNA-mediated depletion of many P-body proteins slightly decreased HCV protein (Fig. 3A) and RNA abundances (Fig. 3B), depletion of Lsm1, Ge-1, Ago2, and in particular RCK/p54, strongly decreased the abundance viral RNA and proteins (Fig. 3A, 3B, 3C). In contrast, depletion of P-body proteins did not change the abundance of cellular actin, miR-122 or U6 RNAs (Fig. 3B, Fig. S3A). These findings argue that HCV RNA abundance is enhanced by the presence of P-body proteins RCK/p54, Lsm1, Ge-1 and Ago2.
Figure 3. Effects of depletion of P-body proteins on JFH-1 protein and RNA abundances.
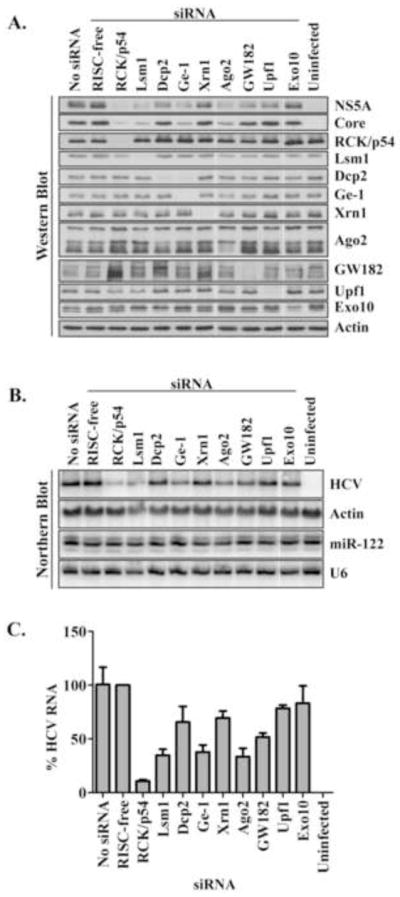
Huh7 cells were depleted of P-body proteins and infected with JFH-1 virus. (A) Abundances of HCV NS5A and core proteins, and P-body proteins RCK/p54, Lsm1, Dcp2, Ge-1, Xrn1, Ago2, GW182, Upf1 and Exo10 during JFH-1 infection were examined by western blot analysis. Transfection reagent alone (no siRNA) and the transfection of a RISC-free siRNA were included as controls. (B) Abundances of HCV RNA and miR-122. Representative northern blots are shown. Actin mRNA and U6 snRNA were used as RNA loading controls. (C) Quantitation of HCV RNA. HCV RNA abundances were normalized to actin mRNA and to control RISC-free siRNA. Mean values and standard deviations from three independent experiments are shown.
Formation of stress granules during HCV infection
It is known that stress granules accumulate adjacent to P-bodies during oxidative stress (Kedersha et al., 2005). To examine whether uninfected Huh7 cells form stress granules in response to oxidative stress, cells were incubated without or with 1 mM arsenite. As shown in Figure 4A, addition of arsenite induces the formation of punctate, eIF3η-containing stress granules in the cytoplasm. Similarly, infection of Huh7 cells with JFH-1 virus resulted in the formation of eIF3- and TIA-1-positive stress granules (Fig. 4A, and data not shown). Quantitation showed that, in contrast to uninfected cells, the formation of stress granules in infected cells increased during infection (Fig. 4B). Oxidative cell stress and stress granule formation is usually accompanied by the phosphorylation of eIF2α. This event blocks translational initiation and shuttles untranslated mRNAs and 40S ribososmal subunits to newly formed stress granules (Anderson and Kedersha, 2009a; Kedersha and Anderson, 2002; Kedersha et al., 2005). To determine whether JFH-1 infection affected the phosphorylation status of eIF2α, the phosphorylation status of eIF2α was examined at different times after infection. Surprisingly, by western blot analysis eIF2α was not phosphorylated, and immunofluorescence studies showed that the localization of phosphorylated eIF2α did not change in JFH-1-infected Huh7 cells (Fig. S4 and data not shown). However, JFH-1 did not inhibit the phosphorylation of eIF2α or the formation of new stress granules following arsenite treatment (data not shown). Thus, JFH-1-induced stress granule formation in Huh7 cells can occur by a mechanism requires little phosphorylation of eIF2α. The abundances of six stress granule proteins remained unchanged during JFH-1 infection (Fig. 4C). Therefore, the formation of stress granules during HCV infection was not a result of enhanced intracellular abundance of the examined stress granule proteins. Instead, stress granule formation during HCV infection most likely reflects a localized concentration of stress granule proteins.
Figure 4. Formation of stress granules during JFH-1 infection.

(A) Induction of stress granules by arsenite and JFH-1 infection. Uninfected and JFH-1 infected Huh7 cells were treated without or with 1 mM arsenite for 30 minutes at 37ºC. Cells were fixed and stained with eIF3η (red) and HCV NS5A (green) specific antibodies. Representative confocal microscopy images are presented. White arrows point to examples of stress granules. (B) Induction of stress granules during JFH-1 infection 1, 2 and 3 days p.i. The appearance of stress granules in uninfected and JFH-1-infected Huh7 cells was quantified using ImageJ software. Mean values and standard deviations of three independent experiments are shown. (C) Abundances of NS5A, core, and stress granule proteins (TIA-1, TIAR, G3BP1, HuR, Ataxin2, and YB-1) from JFH-1 infected and uninfected Huh7 cells at 1, 2, and 3 days p.i. were examined by western blot analysis. Abundance of GAPDH serves as a control.
A number of RNA binding proteins, as well as components of the translation initiation complex localize in stress granules. Components of stress granules in JFH-1 infected cells were examined by confocal microscopy. G3BP1, TIA-1, TIAR, USP10, OGFOD1, and RpS6 co-localized in virus-induced stress granules (Fig. S5 and data not shown). Furthermore, during arsenite-induced stress P-bodies often localize adjacent to stress granules (Buchan et al., 2008). Indeed, during JFH-1 infection RCK/p54-containing P-bodies were found adjacent to and co-localized with G3BP1, TIA-1 and TIAR-containing stress granules (data not shown), indicating that the cytoplasmic granules formed in JFH-1 infected cells are bona fide stress granules.
Localization of stress granule proteins, HCV core and lipid droplets in JFH-1-infected cells
To examine whether HCV core protein co-localized with stress granule proteins, uninfected and HCV-infected cells were fixed and the localization of core, lipid droplets and G3BP1, TIA-1, TIAR, USP10 and OGFOD1 was examined by confocal microscopy. In uninfected cells G3BP1 and USP10 were distributed throughout the cytoplasm, while TIA-1, TIAR and OGFOD1 were predominantly nuclear (data not shown). During JFH-1 infection G3BP1, TIA-1, TIAR, USP10, OGFOD1 and S6 localized with core-containing foci (Fig. 5, Fig. S2 and data not shown). However co-localization analysis show that only G3BP1 and USP10 partially colocalized with core and lipid droplets (Fig. 5 and Fig. S2). In contrast, these stress granule proteins rarely localized with NS5A and viral dsRNA (Fig. S5 and data not shown). Thus, in JFH-1 infected cells, both P-body and stress granule proteins redistribute to granular foci that specifically co-localize with core and lipid droplets.
Figure 5. Localization of core, lipid droplets and stress granule proteins in JFH-1 infected cells.

Localization of HCV core, lipid droplets and stress granule proteins (G3BP1, TIA-1 and USP10) in JFH-1 infected Huh7 cells was examined. Bodipy 493/503-was used to stain lipid droplets, and images were acquired with a Zeiss LSM5 confocal microscope. Representative images show the staining of HCV core (black and white), and co-staining of lipid droplets (green) and individual stress granule proteins (red). The Pearson’s correlation coefficient (r) quantifying the degree of co-localization between lipid droplets (Bodipy 493/503) and stress granules proteins is presented in the merged images.
Diminished HCV protein and RNA abundances following siRNA-directed depletion of stress granule proteins
To examine the role of several proteins that are unique to stress granules in HCV gene expression, TIA-1, G3BP1, HuR, Ataxin2, USP10 and OGFOD1 were depleted by siRNA-mediated mRNA degradation. Western blot analysis showed that each examined stress granule protein was effectively depleted (Fig. 6A). While depletion of each stress granule protein decreased the abundance of viral core and NS5A proteins, concomitant with a decrease in viral RNA (Fig. 6B, 6C), depletion of HuR and USP10 in particular decreased the abundance of viral RNA and protein. Similar to what was observed after depletion of P-body proteins, depletion of the examined stress granule proteins did not affect miR-122 abundance (Fig. S3B). Overall, these co-localization studies suggest that stress granule and P-body proteins modulate HCV RNA abundance by a mechanism that may involve viral core protein and cellular lipid droplets.
Figure 6. Effects of depletion of stress granule proteins on JFH-1 protein and RNA abundances.

(A) Abundances of stress granule proteins TIA-1, G3BP1, HuR, Ataxin2, USP10 and OGFOD1, and viral NS5A and core after siRNA-mediated depletion of stress granule proteins and JFH-1 infection. Western blots are shown. Transfection reagent alone (no siRNA) and the transfection of a RISC-free siRNA were included as controls. The abundance of GAPDH serves as a loading control. (B) Abundances of HCV RNA and miR-122 during depletion of stress granule proteins. Representative northern blots are shown. Actin mRNA and U6 snRNA were used as RNA loading controls. (C) Quantitation of HCV RNA. HCV RNA abundances were normalized to actin mRNA and to RISC-free siRNA. Mean values and standard deviations from three independent experiments are shown.
Localization of ectopically expressed HCV core protein
To examine whether core localizes to P-body and stress granule proteins in the absence of viral infection, Huh7 cells were transfected with control plasmid or with a plasmid that encoded the core gene. Figure 7 shows that ectopically expressed core localized mainly to lipid droplets. Interestingly, RCK/p54-containing P-body foci were not significantly dispersed and did not co-localize with core at lipid droplets in core-expressing cells (Fig. 7). Similarly, G3BP1 did not show co-localization with ectopically expressed core protein (Fig. 7). These data show that expression of core protein in the absence of a viral infection is not sufficient to disperse P-bodies or alter the localization of RCK/p54 and G3BP1, arguing that other viral factors might contribute to the re-localization of P-body and stress granule proteins.
Figure 7. Localization of RCK/p54, G3BP1 and over-expressed HCV core protein.

Distribution of RCK/p54, G3BP1 and over-expressed core protein in Huh7 cells. (A) Localization of lipid droplets (red) and core (green). (B) Localization of RCK/p54 (red) and core (green). (C) Localization of G3BP1 (red) and core (green). Lipid droplets were stained with the neutral lipid dye Bodipy 493/503. Individual and merged images are shown. Images have been re-colored for presentation.
Enhancement of extracellular virion accumulation following depletion of G3BP1
That lipid droplets and core protein co-localize with stress granule and P-body proteins suggest that these proteins might facilitate HCV virus particle production. To test this hypothesis, the effect of depleting selected P-body and stress granule proteins on HCV virion production was examined. Depletion of RCK/p54, Ge-1, HuR, Ataxin2 and USP10 decreased the titer of both cell-associated (Fig. 8A) and extracellular virions (Fig. 8B), as well as the abundance of intracellular core protein (Fig. 8C). Curiously, depletion of USP10 increased the abundance of extracellular core protein (Fig. 8D). Depletion of TIA-1 and OGFOD1 decreased both viral protein and RNA abundances (Fig. 5) yet virus titer was not affected (Fig. 8A, 8B). Interestingly depletion of G3BP1 decreased cell-associated virus titer (Fig. 8A) but increased extracellular titer at least three-fold (Fig. 8B), without any increase in the extracellular abundance of core protein (Fig. 8D), suggesting that the normal function of G3BP1 is to suppress the production of infectious extracellular virus, and that stress granule proteins function at a late step in the HCV infectious cycle.
Figure 8. Cell-associated and extracellular infectious JFH-1 yield following depletion of RCK/p54, Ge-1, TIA-1, G3BP1, HuR, Ataxin2, USP10 and OGFOD1.

Virus yield in cells depleted of RCK/p54, Ge-1, TIA-1, G3BP1, HuR, Ataxin2, USP10 and OGFOD1 proteins. Virus yield was measured in (A) cells (cell-associated) or (B) extracellular medium (extracellular) using the limiting dilution infectivity assay as previously described (Lindenbach et al., 2005). The transfection of a RISC-free siRNA was included as a control. (C) Abundances of cell-associated core protein was determined by western blot analysis and ECF chemifluorescence. (D) Quantitation of extracellular core proteins was analyzed by ELISA. Mean values and standard deviations from three independent experiments are shown. HCV titer and abundance of core protein was normalized to JFH-1 infected cells transfected with the RISC-free siRNA.
Discussion
In this study we show that HCV dispersed P-body foci not by degrading P-body proteins, but likely by modulating the intracellular distribution of these proteins to HCV core protein associated with lipid droplets. Additionally, HCV infection induced the formation of stress granules independent of eIF2α phosphorylation. Stress granule proteins similarly localized with core protein at lipid droplets during HCV infection. Overexpression and localization of core alone did not induce the re-localization of P-body and stress granule proteins to lipid droplets. Additionally RNAi-mediated depletion of specific P-body and stress granule proteins decreased the expression of HCV proteins and RNA, and modulated virion assembly and release.
During HCV infection we observed that RCK/p54, Lsm1, Xrn1 and GW182-containing P-bodies were dispersed (Fig. 1A, Fig. S1 and data not shown). Similarly, early during poliovirus infection P-body foci are dispersed and the exoribonuclease Xrn1, decapping factor Dcp1a and deadenylase protein Pan3 are rapidly degraded (Dougherty et al., 2011). Unlike poliovirus, however, HCV did not induce degradation or modification of P-body proteins (Fig. 1C). Interestingly, West Nile and Dengue viruses also disperse P-body foci late during infection; however, the mechanism by which this occurs is unknown (Emara and Brinton, 2007). We speculated that HCV might disperse P-bodies by modulating the intracellular distribution of P-body components. For example, re-localization of potentially deleterious components such as the exoribonuclease Xrn1 and the decapping enzyme Dcp2 might limit the degradation of viral genomes. Similar to Scheller et al. (2009), however, we find that depletion of Xrn1 and Dcp2 only modestly decreased HCV gene expression, and thus they seem unlikely to affect HCV RNA stability. Alternatively, HCV might sequester key components that are essential to modulate viral gene expression. MiR-122 resides in P-bodies (Bhattacharyya et al., 2006) and is critical to maintaining HCV RNA abundance (Jopling et al., 2008; Jopling et al., 2005). While we cannot exclude the possibility that HCV re-localizes miR-122-containing RISC complexes from P-bodies, sequestration of miR-122 with antisense locked nucleic acid oligonucleotides did not disperse P-bodies (data not shown). However, depleting components of the decapping and RISC complexes, specifically RCK/p54, Lsm1, Ge-1 and Ago2, significantly decreased HCV protein and RNA abundances (Fig. 3). Scheller et al. (2009) demonstrated that HCV 5′ and 3′ UTRs interact with in vitro reconstituted Lsm1-7 rings, and that depletion of Lsm1 decreased viral translation without affecting RNA stability (Scheller et al., 2009). A role for RCK/p54 in HCV translation and replication has previously been proposed (Jangra et al., 2010; Scheller et al., 2009). RCK/p54 was shown to co-immunoprecipitate HCV RNA and cellular mRNA, but did not co-localize with HCV dsRNA (Jangra et al., 2010). Our immunofluorescence localization studies showed RCK/p54, Lsm1, Ge-1, Dcp2, Xrn1 and GW182 were juxtaposed to replication sites stained for dsRNA and NS5A (data not shown). However RCK/p54 and Lsm1 localized with core protein at lipid droplets (Fig. 2 and Fig. S2), a finding that was also reported by Ariumi et al. (Ariumi et al., 2011). In addition Dcp2 and Xrn1, and to a lesser extent Ge-1 and GW182, showed similar localization with core (Fig. S2 and data not shown). That Dcp2 localizes with core at putative assembly sites contrasts that reported by Ariumi et al (Fig. S2 and data not shown; Ariumi, 2011 #230}. Furthermore we find that Dcp2 localization (visualized with three different antibodies) did not localize to large foci, but rather in numerous tiny foci (data not shown). Although the differences in Dcp2 might be attributed to different experimental conditions, the localization of Dcp2 with core is unclear.
Depletion of Ge-1 decreased JFH-1 RNA and protein abundances (Fig. 3), and cell-associated and extracellular virus titers (Fig. 7), suggesting Ge-1 may function at an earlier step in HCV gene expression to ultimately affect virion production. Perez-Vilaro et al (2012) showed that components localized in P-bodies differ during HCV infection (Perez-Vilaro et al., 2012). Indeed, in JFH-1 infected cells we observed small Ge-1-containing foci (data not shown). This finding raises the possibility that Ge-1-complexes localizing with core at lipid droplets may contain different components and functions to those localized in the cytoplasm. Interestingly, the abundance of Dcp2 also decreased following depletion of Ge-1 (Fig. 3A). Since the siRNAs specifically targeted Ge-1, it is unlikely that loss of Dcp2 is the result of an off-target effect. Ge-1 is a central component of P-bodies (Yu et al., 2005); interacts with RCK/p54 (unpublished data), bridges the interaction between Dcp1 and Dcp2 and stimulates Dcp2 activity in vitro (Fenger-Gron et al., 2005). As such in the absence of Ge-1, the decapping complex may disassemble and stimulate Dcp2 turnover. Depletion of RCK/p54 decreased cell-associated and extracellular virus titers, although the amount of core protein released was similar in cells treated with the RISC-free siRNA (Fig. 8). Dhh1, the yeast ortholog of RCK/p54, facilitates the re-localization of brome mosaic virus RNA to P-bodies. Similarly in HCV infected cells, RCK/p54 might facilitate the localization of viral genomes from replication to assembly sites. Recently packaging of the foamy virus genome was shown to require RCK/p54 (Yu et al., 2011). Similarly, depletion of RCK/p54 might limit HCV genome packaging and thus decrease viral titers (Fig. 8). Although the exact role of P-body proteins localizing with core and lipid droplets during HCV infection is elusive, HCV may therefore disperse P-bodies by re-localizing components to virion assembly sites.
Phosphorylation of eIF2α at serine 51 inhibits cellular protein translation and induces the formation of stress granules (Kedersha et al., 1999). Recently HCV was shown to induce phosphorylation of eIF2α via activation of the RNA-dependent protein kinase PKR, resulting in the inhibition of translation of interferon stimulated mRNAs (Garaigorta and Chisari, 2009). We observed that JFH-1 induces the formation of stress granules (Fig. 4). However eIF2α serine 51 was not phosphorylated (Fig. S4). At 3 days p.i., the immunofluorescent distribution of phospho-eIF2α was similar in uninfected and JFH-1 infected Huh7 cells (data not shown). While Garaigorta and Chisari (2009) infected Huh7 cells with JFH1-d183 at an moi of 5 (Garaigorta and Chisari, 2009), we infected Huh7 cells with wild-type JFH-1 at an moi of 0.01 suggesting that the observed differences in eIF2α phosphorylation may be attributed to different experimental approaches. Arnaud et al. (2010) described interferon induction through the RIG-I/MAVS pathway early during infection, and showed immunoprecipitation of phospho-PKR and eIF2α at 15 hours p.i. However, 18 hours p.i. NS3/4A expression stimulated MAVS-cleavage which coincided with a decrease in phosphorylated PKR and eIF2α (Arnaud et al., 2010). That eIF2α remained unmodified in our studies might therefore be attributed to the sensitivity of detection (by western blot analysis) and timing (24 hours p.i.).
Poliovirus infection significantly induced G3BP1-containing stress granules, however as infection progressed the number of G3BP1 granules decreased (White et al., 2007). JFH-1 infection also induced the formation of stress granules, albeit in a low percentage of cells (Fig. 4B). Therefore, does HCV induce or limit stress granule formation, and what are the consequences? West Nile virus re-localized stress granule proteins TIA-1 and TIAR from the nucleus to the cytoplasm but restricted the formation of stress granules (Emara and Brinton, 2007). Interestingly West Nile virus also hindered arsenite-induced stress granule formation by attenuating eIF2α phosphorylation. JFH-1 similarly re-localized stress granule components. In contrast, we found that arsenite treatment induces the phosphorylation of eIF2α in JFH-1 infected cells (data not shown), suggesting that JFH-1 does not inhibit stress granule formation by preventing eIF2α phosphorylation. Although the formation of TIA-1-containing stress granules was not perturbed (Piotrowska et al., 2010), poliovirus limited the formation of G3BP1-containing stress granules by 3C proteinase cleavage of G3BP1 (White et al., 2007). In contrast, we observed that the abundance and integrity of stress granule proteins did not change during JFH-1 infection (Fig. 4C). Furthermore JFH-1 infection does not interfere with the composition of stress granules or localization with P-bodies (Fig. S4 and data not shown). Similar to Ariumi et al (2011), we find that P-body and stress granule proteins only localize with core protein at lipid droplets (Fig. 2, Fig 5 and Fig. S2), and not with endoplasmic reticulum-associated core (data not shown; (Ariumi et al., 2011). Ariumi et al (2011) (Ariumi et al., 2011) suggested that the re-localization of components limits the formation of stress granules in response to heat shock and oxidative stress. Although re-localization of stress granule components to lipid droplets might limit the availability of components for stress granule formation, we found that not all stress granule components re-localize to assembly sites during JFH-1 infection (Fig. 5 and data not shown). Indeed, the number of NS5A-positive cells with arsenite-induced eIF3η-containing stress granules did not differ significantly between uninfected and JFH-1-infected cells.
Depletion of stress granule proteins decreased the abundance of HCV proteins and RNAs (Fig. 4), and G3BP1 and USP10 localized with HCV core protein at lipid droplets. Interestingly HuR, TIA-1 and G3BP1 bind HCV RNA (Harris et al., 2006; Spangberg et al., 2000; Tingting et al., 2006; Yi et al., 2006). Depletion of HuR and USP10 dramatically decreased virus titer. Sindbis virus and other alphaviruses genomes are stabilized by the binding of HuR to a U-rich region in their 3′ UTRs (Sokoloski et al., 2010). As such, the interaction of HuR with the 3′ UTR of both RNA polarities may similarly stabilize HCV RNA (Harris et al., 2006; Spangberg et al., 2000). Depletion of USP10 greatly decreased HCV protein and RNA abundances and virus titers (Fig. 6, Fig. 7). USP10 modulates anterograde and retrograde vesicular trafficking (Cohen et al., 2003a; Cohen et al., 2003b), the localization and stability of p53 (Yuan et al., 2010) and binds Dengue virus RNA (Ward et al., 2011). Thus depletion of USP10 may modulate proteosomal degradation of proteins involved in HCV gene expression, or HCV egress. Interestingly G3BP1 modulates USP10 deubiquitinase activity (Cohen et al., 2003a), as well as interacts and co-localizes with HCV NS5B protein and RNA to modulate HCV replication (Yi et al., 2006; Yi et al., 2011). Similar to Yi et al. we observed that depletion of G3BP1 decreased JFH-1 gene expression (Yi et al., 2011). In contrast however we did not observe significant co-localization of G3BP1 with HCV dsRNA and NS5A-containing replication sites. Our studies and those from Ariumi et al. show G3BP1 localizing with core protein at lipid droplets (Fig. 6 and Fig. S2) (Ariumi et al., 2011). Curiously depletion of G3BP1 increased extracellular titers suggesting that in addition to facilitating genome replication, G3BP1 may restrict the assembly of virions. Overexpressed core protein and G3BP1 do not co-immunoprecipitate (Yi et al., 2011) nor does expression of core protein alone relocalize G3BP1 to lipid droplets (Fig. 7). The mechanism by which G3BP1 regulates virus assembly, although unclear, likely requires an interaction with viral RNA and/or other proteins. Reduced HCV RNA and protein abundances following depletion of TIA-1, G3BP1 and OGFOD1 (Fig. 4) did not significantly affect cell-associated or extracellular virus titers (Fig. 8A and 8B). Keum et al (2012) recently demonstrated that virus particles secreted early compared to late in infection differed in infectivity and composition. Thus two possibilities may explain our data. First, although similar amounts of extracellular core antigen were released (Fig. 8D), the proportion of particles secreted may contain more viral RNA in the absence of TIA-1, G3BP1 and OGFOD1. Second, the composition of the virus particles released in the absence of TIA-1, G3BP1 and OGFOD1 may more resemble virus particles released early during infection namely highly infectious particles, compared to the poorly infectious particles secreted at later times.
Core protein associated with lipid droplets recruits the HCV nonstructural proteins and the replication complex to lipid droplets to facilitate assembly of infectious virions (Bartenschlager et al., 2011). Recently a number of host proteins such as DGAT-1, Annexin A2 and YB-1 have also been shown to facilitate HCV assembly (Backes et al., 2010; Chatel-Chaix et al., 2011; Herker et al., 2010). Proteomic studies of lipid droplets isolated from Huh7 cells and HepG2 cells overexpressing core identified a number of associated proteins including Annexin A2 and DDX3, respectively (Fujimoto et al., 2004; Sato et al., 2006). Consistent with these studies we found that overexpression of core protein alone was insufficient re-localize RCK/p54 or G3BP1 to lipid droplets (Fig. S6). Arsenite-treatment also did not re-localize stress granule proteins to lipid droplets (data not shown). Therefore the re-localization of P-body and stress granule proteins to lipid droplets is not a cellular response to stress and likely requires the expression of other HCV proteins with or without core, and HCV RNA.
In conclusion, we find that depletion of select P-bodies and stress granule proteins decreases HCV gene expression. That HCV re-localizes P-body and stress granule proteins to core protein at lipid droplets highlights an unexpected role for these proteins in the HCV life cycle, possibly to segregate viral RNA for HCV gene expression or assembly.
Materials and Methods
Cells
Huh7 cells were maintained in DMEM and supplemented with 10% FBS, 1% nonessential amino acids and 200 μM L-glutamine.
Small Interfering RNA (siRNA) Transfections
All siRNA oligonucleotides were synthesized by the Stanford Protein and Nucleic Acids facility. The siRNA sequences are as follows: siAgo2, 5′-CAGCCAGCAUCGAACAUGAUU-3′; siAtaxin2, 5′-AAGUGUGAUUUGGUACUUGAU-3′; siDcp2, 5′-CACGGAAACUUCAGGAUAAUU-3′; siExo10, 5′-GUUUCGAGAGAAGAUUGACAAdTdT-3′; siG3BP1, 5′-CAGGAAGACUUGAGGACAUUU-3′; siGe-1, 5′-CCGAGAGUUAAAGAUGUGGUU-3′; siGW182, 5′-GAAAUGCUCUGGUCCGCUAUU-3′; siHuR, 5′-AAGAGGCAAUUACCAGUUUCAUU-3′; siLsm1, 5′-GUGACAUCCUGGCCACCUCACUU-3′; siRCK/p54, 5′-GCAGAAACCCUAUGAGAUUUU-3′; siTIA-1, 5′-CUGGGCUAACAGAACAACUAAUU-3′; siUpf1, 5′-GAUGCAGUUCCGCUCCAUU-3′; siXrn1; 5′-GAGGUGUUGUUUCGCAUUAUU-3′. Oligonucleotides were resuspended in RNase-free water to a 50 μM final concentration. Sense and antisense strands were combined in annealing buffer (150 mM HEPES (pH 7.4), 500 mM potassium acetate, and 10 mM magnesium acetate) to a final concentration of 20 μM, denatured for 1 minute at 95ºC, and annealed for 1 hour at 37ºC. Control RISC-free siRNA was purchased from Dharmacon (Catalog no. D-001220-01-05).
Huh7 cells were seeded in 60 mm tissue culture plates. The following day, Huh7 cells were transfected with 100 nM of siRNA duplexes and Dharmafect I reagent (Dharmacon) according to the manufacturer’s protocol. Twenty-four hours post-transfection, cells were infected with JFH-1 virus as described, and harvested 3 days p.i. The efficiency of siRNA depletion was examined by western blot analysis.
Plasmid transfections
pcDNA3.1 was purchased from Invitrogen. pcDNA-Core 191 was a generous gift from Melanie Ott (Gladstone Institute, UCSF). Huh7 cells seeded in 8-chambered coverglass slides (LabTek II chamber slides, Thermo Scientific) were transfected with 0.8 μg pcDNA3.1 and Core 191 plasmids and Lipofectamine2000 reagent (Invitrogen) according to the manufacturer’s protocol. Twenty-four hours post-transfection, cells were fixed with 4% paraformaldehyde (Electron Microscopy Sciences) in PBS, and processed for immunofluorescence analysis.
JFH-1 infections and lysate preparations
Huh7 cells were infected at 37 ºC with the JFH-1 infectious HCV virus (Wakita et al., 2005) at a multiplicity of infection (moi) of 0.01. Five hours after infection, cells were trypsinized, and replated in duplicate tissue culture plates. Three days p.i. cells were harvested. For western blot analysis, cells were washed with phosphate-buffered saline (PBS) and pelleted briefly at room temperature. Pelleted cells were lysed in RIPA buffer (100 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% sodium deoxycholic acid, 1% Triton X-100, and 0.1% SDS) containing Complete EDTA-free protease inhibitors (Roche) for 20 minutes on ice and pelleted for 20 minutes at 16,000×g at 4ºC. Cell pellets prepared for examination of eIF2α phosphorylation were lysed in RIPA buffer containing Complete EDTA-free protease inhibitors and PhosphoSTOP (Roche). Bradford reagent (BioRad) was used to quantitate the protein concentration of the clarified supernatants. For RNA analysis, cells were lysed in TRIzol reagent (Invitrogen), and RNA was isolated according to the manufacturer’s protocol.
Electroporation of HCV RNA
pH77c and pH77ΔE1/p7 plasmids have been previously described (Jopling et al., 2005). HCV RNA was transcribed in vitro from either pH77c or pH77ΔE1/p7 plasmids by the T7 RNA polymerase, using the Megascript kit (Ambion) according to the manufacturer’s protocol. For electroporation, Huh7 cells were trypsinized, sedimented at 1,000 rpm for 5 minutes, and then washed first with PBS and then Cytomix buffer (120 mM KCl, 150 mM CaCl2, 10 mM K2HPO4, 25 mM HEPES, 2 mM EDTA, and 2 mM MgCl2; pH 7.6). Huh7 cells in Cytomix buffer were electroporated with 10 μg of the respective HCV RNAs in a 0.4 cm Biorad cuvette using the BioRad Genepulser Xcell. Following electroporation, cells were allowed to recover at room temperature for 10 minutes, and then seeded in 8-chambered coverglass slides (LabTek II chamber slides, Thermo Scientific). Five days after electroporation, cells were fixed with 4% paraformaldehyde (Electron Microscopy Sciences) in PBS, and processed for immunofluorescence.
Western blot analysis
For western blot analysis, 20 μg cell lysate was mixed with 5x protein sample buffer (200 mM Tris-HCl (pH 6.8), 50% glycerol, 25% SDS and β-mercaptoethanol), denatured for 5 minutes at 95ºC, and loaded onto a 8%, 10% or 12.5% polyacrylamide-SDS gel. After electrophoresis, proteins were transferred to a PVDF membrane (Millipore) for 1 hour or 1.5 hours at 4 ºC at 100V. The membranes were blocked at room temperature for 1 hour with 5% nonfat dry milk (or bovine serum albumin) in TBS (50 mM Tris-HCl (pH 7.4), and 150 mM NaCl) with 0.1% Tween 20 (TBS/T), and then incubated overnight at 4ºC with primary antibodies. Membranes were washed 3 × 10 minutes with TBS/T prior incubating with the horseradish peroxidase-conjugated secondary antibodies diluted in 5% milk/TBS/T for 1 hour at room temperature. After washing 3 × 10 minutes with TBS/T, bound antibodies were labeled with Pierce ECL Western Blotting Substrate (Thermo Scientific) and detected after exposure to Biomax Light Film (Kodak).
The following primary antibodies were used for western blot analysis: anti-Actin (1:5,000, Sigma A2066), anti-Ago2 (1:10, a gift from Dr. Gunter Meister), anti-Ataxin2 (1:250, BD Transduction Laboratories 611378), anti-Dcp1a (1:1,000, Abcam ab57654), anti-Dcp2 (1:10,000, Bethyl Laboratories A302-597A), anti-DDX6 (RCK/p54) (1:5,000, Bethyl Laboratories A300-460A), anti-eIF2α (K-17) (1:1,000, Santa Cruz sc-30882), anti-PM/Scl-100 (Exo10) (1:5,000, Sigma P41124), anti-G3BP1 (1:20,000, Bethyl Laboratories A302-034A), anti-GAPDH (1:100,000, EMD Chemicals CB1001), anti-GW182 (1:10,000, Bethyl Laboratories A302-329A), anti-HCV core (C7-50) (1:1,000; Abcam ab2740), anti-HCV NS5A (1:10,000; a gift from Dr. Charles Rice), anti-HuR (ELAV1) (19F12) (1;1,000, Abcam ab14371), anti-Lsm1 (1:1,000,Sigma Prestige Antibodies HPA11799), anti-Phospho eIF2α (Ser51) (1:1,000, Cell Signaling 3597), anti-RCD8 (Ge-1) (1:10,000, Bethyl Laboratories A300-745A), anti-Rent1 (Upf1) (1:10,000, Bethyl Laboratories A301-902A), anti-TIA-1 (C-20)(1:2,000, Santa Cruz sc-1751), anti-TIAR (C-18) (1:2,000, Santa Cruz sc-1749), and anti-Xrn1 (1:10,000, Bethyl Laboratories A300-443A). Donkey anti-goat-HRP (sc-2020), donkey anti-mouse-HRP (sc-2314), donkey anti-rabbit-HRP (sc-2313) and goat anti-rat-HRP (sc-2032) were purchased from Santa Cruz Biotechnologies and used at 1:10,000.
To quantitiate cell-associated HCV core abundances, following incubation with primary antibody, membranes were incubated with goat anti-mouse and goat anti-rabbit alkaline phospatase-conjugated antibodies (Invitrogen, 81–6522 and 81–6122) at 1:20,000. Bound antibodies were labeled with ECF substrate (GE Life Sciences) and detected with a STORM phosphorimager.
Northern blot analysis
To visualize specific mRNAs, 10 μg TRIzol-extracted RNA was resuspended in loading buffer (1% MOPS-EDTA-sodium acetate (MESA), 67% formaldehyde, and 50% formamide), denatured for 10 minutes at 65ºC and separated in 1.2% agarose/6.7% formaldehyde gels. Samples were separated in gels in MESA buffer, containing 18% formaldehyde, at 100V. RNA was transferred and UV cross-linked to a Zeta probe membrane (BioRad). Detection of HCV and actin RNA was performed using the ExpressHyb hybridization buffer (Clontech) and [α-32P] dATP-RadPrime DNA labeled (Invitrogen) probes.
To examine small RNAs, 10 μg TRIzol-extracted RNA was resuspended in loading buffer (95% formamide, 1% EDTA), denatured for 5 minutes at 95ºC and separated in 15% polyacrylamide-6.75 M urea gels (Sequagel, National Diagnostics). Gels were electrophoresed in 1x Tris-borate buffer at 1,400 V, 250 mA and 30 W for 1 hour 20 minutes. Subsequently, RNA was transferred to a membrane (Hybond) for 1 hour at 1,400 V, 250 mA and 30 W using Owl Semi Dry Electroblotter (Thermo Scientific). MiR-122 and U6 RNA were detected using the UltraHyb Oligo buffer (Ambion) and oligonucleotide probes. MiR-122 and U6 oligonucleotide probes were 5′-end labeled with PNK kinase and [γ-32P] dATP. Oligonucleotide sequences of miR-122 and U6 probes are 5′-CAAACACCATTGTCACACTCCA-3′, and 5′-CACGAATTTGCGTGTCATCCTTGC-3′ respectively.
Immunofluorescence staining
Uninfected and HCV-infected Huh7 cells were grown in 8-chambered coverglass slides (LabTek II chamber slides, Thermo Scientific) for 3 days. The cells were washed twice with PBS, and then fixed with 4% paraformaldehyde (Electron Microscopy Sciences) in PBS at room temperature for 20 minutes. The cells were again washed twice with PBS, and then permeabilized at room temperature for 15 minutes with 0.5% Triton X-100 in 1% fish gelatin (Sigma) in PBS (1% FG/PBS). Cells prepared for lipid droplet staining were permeabilized at room temperature for 5 minutes with 0.1% Triton X-100 in 1% FG/PBS. After 3 × 10 minute incubations with 1% FG/PBS, the cells were incubated overnight at 4 ºC with primary antibodies diluted in 1% FG/PBS. Cells were washed 2 × 10 minutes with 1% FG/PBS prior incubating with secondary antibodies conjugated to fluorophores for 2 hours at room temperature. Cells were washed 2 × 10 minutes with 1% FG/PBS and then incubated with Hoechst 33258 dye (Sigma) in 1% FG/PBS for 5 minutes at room temperature. After two final 5 minute washes the coverglass slides were mounted with Fluoromount-G (SouthernBiotech), and examined under a laser scanning confocal microscope (Zeiss LSM 510).
The following primary antibodies were used for immunofluorescence: anti-Dcp2 (1:800, Bethyl Laboratories A302-597A), anti-DDX6 (RCK/p54) (1:1,000, Bethyl Laboratories A300-461A), anti-DGAT-1 (H-255) (1:50, Santa Cruz sc-32861), anti-dsRNA (mAb J2) (1:250, Engscicons 10010200), anti-eIF3η N20 (1:200, Santa Cruz sc-16377), anti-PM/Scl-100 (Exo10) (1:1,000, Sigma P41124), anti-G3BP1 (1:1,000, Bethyl Laboratories A302-034A), anti-GW182 (1:1,000, a gift from Dr. Marvin Friztler), anti-HCV Core (C7-50) (1:1,000, Abcam ab2740), anti-HCV NS5A (1:10,000, a gift from Dr. Charles Rice), anti-Lsm1 (1:800, Sigma Prestige Antibodies HPA11799), anti-RCD8 (Ge-1) (1:1,000, Bethyl Laboratories A300-745A), anti-Rent1 (Upf1) (1:1,000, Bethyl Laboratories A301-902A), anti-TIA-1 (C-20) (1:50, Santa Cruz sc-1751), anti-TIAR (C-18) (1:50, Santa Cruz sc-1749) and anti-Xrn1 (1:800, Bethyl Laboratories A300-443A). Secondary antibodies were used at 1:200. Bodipy 493/503 (1 mg/ml, D3922) and donkey anti-Goat AlexaFluor647 (A2144), donkey anti-Mouse AlexaFlour488 (A21202), donkey anti-Mouse AlexaFluor594 (A21203), donkey anti-Rabbit AlexaFluor594 (A21207) and donkey anti-Rabbit AlexaFluor647 (A31573) were purchased from Invitrogen. Sheep anti-Human-TRITC (ab6867) was purchased from Abcam.
Infectivity assays
A previously described limiting dilution assay was used to determine the infectivity of extracellular and cell-associated JFH-1 virus (Lindenbach et al., 2005). Briefly, P-body and stress granule proteins were depleted and cells infected with JFH-1. Infected cells were seeded into 60 mm tissue culture plates, and extracellular virus was collected at 3 days pi. Cell-associated virus was harvested by washing the cell monolayer twice with PBS, and then adding 1 ml media to each plate. Plates were scraped and cells collected. Extracellular and cell-associated virus was freeze-thawed once, and cell debris removed by a brief high-speed spin. Infectivity was determined by a TCID50 assay. Infected cells were detected using the mouse monoclonal HCV Core-specific antibody C7-50 (Abcam) incubated at 1:2,000 at 4ºC overnight, and AlexaFluor488-conjugated goat anti-mouse antibody (Invitrogen) incubated at 1:500 at room temperature for 2 hours, and the TCID50 calculated. The infectivity of extracellular and cell-associated virus was assayed in parallel.
HCV Core antigen ELISA
Quantitiation of extracellular core was assayed with the Quicktiter Core Antigen ELISA kit (Cell Biolabs, Inc) according to the manufacturer’s instructions.
Quantitation of P-body foci and stress granules, and co-localization
Ten images were captured with a laser scanning Zeiss LSM5 confocal microscope. P-body foci and stress granules were visualized after staining of RCK/p54 and eIF3 proteins, repsectively. The number of P-bodies and stress granules in uninfected and JFH-1 infected cells 1, 2 and 3 days p.i. was quantified with ImageJ as described previously (Buchan et al., 2010). The quantitation of co-localization between core and P-body and stress granule proteins, and lipid droplets stained with Bodipy 493/503 was determined using the Pearson’s correlation coffiecient (r) (Adler and Parmryd, 2010). The Pearson’s coefficient ranges from +1 (strong correlation) to 0 (weak to no correlation) to −1 (no correlation). Immunofluorescence images were opened in ImageJ using the LSM toolbox and the degree of co-localization examined with the Just Another Colocalization Plugin (JACoP) (Bolte and Cordelieres, 2006).
Supplementary Material
Infection of cells with hepatitis C virus decreases the number of Processing bodies
Virus infection enhances the number of stress granules
Depletion of P-body residents decreases viral RNA and protein abundance
Depletion of stress granule residents enhances extracellular virus abundance
Acknowledgments
We are grateful to Karla Kirkegaard for valuable discussions, and comments on the manuscript; and Dr. Christopher Potter (Johns Hopkins University) for generous advice and assistance with co-localization analyses. We also thank Charles Rice (Rockefeller University) for the anti-NS5A monoclonal antibody, Gunter Meister (University of Regensburg) for the anti-Ago2 antibody, Marvin Fritzler (University of Calgary) for the anti-GW182 antibody, and Melanie Ott (Gladstone Institute, UCSF) for the pcDNA Core 191 plasmid. This work was supported by grants from the NIH (AI069000, AI47365), by the Damon Runyon Cancer Research Foundation (CTP), the Deutsche Forschungsgemeinschaft (SS) and the Ruth L. Kirschstein National Service Award (TMA).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adler J, Parmryd I. Quantifying colocalization by correlation: the Pearson correlation coefficient is superior to the Mander’s overlap coefficient. Cytometry A. 2010;77:733–742. doi: 10.1002/cyto.a.20896. [DOI] [PubMed] [Google Scholar]
- Anderson P, Kedersha N. RNA granules: post-transcriptional and epigenetic modulators of gene expression. Nat Rev Mol Cell Biol. 2009a;10:430–436. doi: 10.1038/nrm2694. [DOI] [PubMed] [Google Scholar]
- Anderson P, Kedersha N. Stress granules. Curr Biol. 2009b;19:R397–398. doi: 10.1016/j.cub.2009.03.013. [DOI] [PubMed] [Google Scholar]
- Ariumi Y, Kuroki M, Kushima Y, Osugi K, Hijikata M, Maki M, Ikeda M, Kato N. Hepatitis C virus hijacks P-body and stress granule components around lipid droplets. Journal of Virology. 2011;85:6882–6892. doi: 10.1128/JVI.02418-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaud N, Dabo S, Maillard P, Budkowska A, Kalliampakou KI, Mavromara P, Garcin D, Hugon J, Gatignol A, Akazawa D, Wakita T, Meurs EF. Hepatitis C virus controls interferon production through PKR activation. PLoS One. 2010;5:e10575. doi: 10.1371/journal.pone.0010575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arribere JA, Doudna JA, Gilbert WV. Reconsidering movement of eukaryotic mRNAs between polysomes and P bodies. Mol Cell. 2011;44:745–758. doi: 10.1016/j.molcel.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backes P, Quinkert D, Reiss S, Binder M, Zayas M, Rescher U, Gerke V, Bartenschlager R, Lohmann V. Role of annexin A2 in the production of infectious hepatitis C virus particles. Journal of Virology. 2010;84:5775–5789. doi: 10.1128/JVI.02343-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barba G, Harper F, Harada T, Kohara M, Goulinet S, Matsuura Y, Eder G, Schaff Z, Chapman MJ, Miyamura T, Brechot C. Hepatitis C virus core protein shows a cytoplasmic localization and associates to cellular lipid storage droplets. Proc Natl Acad Sci U S A. 1997;94:1200–1205. doi: 10.1073/pnas.94.4.1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartenschlager R, Penin F, Lohmann V, Andre P. Assembly of infectious hepatitis C virus particles. Trends Microbiol. 2011;19:95–103. doi: 10.1016/j.tim.2010.11.005. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya SN, Habermacher R, Martine U, Closs EI, Filipowicz W. Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell. 2006;125:1111–1124. doi: 10.1016/j.cell.2006.04.031. [DOI] [PubMed] [Google Scholar]
- Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- Brengues M, Teixeira D, Parker R. Movement of eukaryotic mRNAs between polysomes and cytoplasmic processing bodies. Science. 2005;310:486–489. doi: 10.1126/science.1115791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchan JR, Muhlrad D, Parker R. P bodies promote stress granule assembly in Saccharomyces cerevisiae. J Cell Biol. 2008;183:441–455. doi: 10.1083/jcb.200807043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchan JR, Nissan T, Parker R. Analyzing P-bodies and stress granules in Saccharomyces cerevisiae. Methods Enzymol. 2010;470:619–640. doi: 10.1016/S0076-6879(10)70025-2. [DOI] [PubMed] [Google Scholar]
- Buchan JR, Parker R. Eukaryotic stress granules: the ins and outs of translation. Mol Cell. 2009;36:932–941. doi: 10.1016/j.molcel.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatel-Chaix L, Melancon P, Racine ME, Baril M, Lamarre D. Y-Box-Binding Protein 1 Interacts with Hepatitis C Virus NS3/4A and Influences the Equilibrium between Viral RNA Replication and Infectious Particle Production. J Virol. 2011;85:11022–11037. doi: 10.1128/JVI.00719-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen M, Stutz F, Belgareh N, Haguenauer-Tsapis R, Dargemont C. Ubp3 requires a cofactor, Bre5, to specifically de-ubiquitinate the COPII protein, Sec23. Nat Cell Biol. 2003a;5:661–667. doi: 10.1038/ncb1003. [DOI] [PubMed] [Google Scholar]
- Cohen M, Stutz F, Dargemont C. Deubiquitination, a new player in Golgi to endoplasmic reticulum retrograde transport. J Biol Chem. 2003b;278:51989–51992. doi: 10.1074/jbc.C300451200. [DOI] [PubMed] [Google Scholar]
- Cougot N, Babajko S, Seraphin B. Cytoplasmic foci are sites of mRNA decay in human cells. Journal of Cell Biology. 2004;165:31–40. doi: 10.1083/jcb.200309008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty JD, White JP, Lloyd RE. Poliovirus-mediated disruption of cytoplasmic processing bodies. Journal of Virology. 2011;85:64–75. doi: 10.1128/JVI.01657-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emara MM, Brinton MA. Interaction of TIA-1/TIAR with West Nile and dengue virus products in infected cells interferes with stress granule formation and processing body assembly. Proc Natl Acad Sci U S A. 2007;104:9041–9046. doi: 10.1073/pnas.0703348104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenger-Gron M, Fillman C, Norrild B, Lykke-Andersen J. Multiple processing body factors and the ARE binding protein TTP activate mRNA decapping. Mol Cell. 2005;20:905–915. doi: 10.1016/j.molcel.2005.10.031. [DOI] [PubMed] [Google Scholar]
- Fujimoto Y, Itabe H, Sakai J, Makita M, Noda J, Mori M, Higashi Y, Kojima S, Takano T. Identification of major proteins in the lipid droplet-enriched fraction isolated from the human hepatocyte cell line HuH7. Biochim Biophys Acta. 2004;1644:47–59. doi: 10.1016/j.bbamcr.2003.10.018. [DOI] [PubMed] [Google Scholar]
- Garaigorta U, Chisari FV. Hepatitis C virus blocks interferon effector function by inducing protein kinase R phosphorylation. Cell Host Microbe. 2009;6:513–522. doi: 10.1016/j.chom.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris D, Zhang Z, Chaubey B, Pandey VN. Identification of cellular factors associated with the 3′-nontranslated region of the hepatitis C virus genome. Mol Cell Proteomics. 2006;5:1006–1018. doi: 10.1074/mcp.M500429-MCP200. [DOI] [PubMed] [Google Scholar]
- Herker E, Harris C, Hernandez C, Carpentier A, Kaehlcke K, Rosenberg AR, Farese RV, Jr, Ott M. Efficient hepatitis C virus particle formation requires diacylglycerol acyltransferase-1. Nature Medicine. 2010;16:1295–1298. doi: 10.1038/nm.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingelfinger D, Arndt-Jovin DJ, Luhrmann R, Achsel T. The human LSm1-7 proteins colocalize with the mRNA-degrading enzymes Dcp1/2 and Xrnl in distinct cytoplasmic foci. RNA. 2002;8:1489–1501. [PMC free article] [PubMed] [Google Scholar]
- Jangra RK, Yi M, Lemon SM. DDX6 (Rck/p54) is required for efficient hepatitis C virus replication but not for internal ribosome entry site-directed translation. Journal of Virology. 2010;84:6810–6824. doi: 10.1128/JVI.00397-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jopling CL, Schutz S, Sarnow P. Position-dependent function for a tandem microRNA miR-122-binding site located in the hepatitis C virus RNA genome. Cell Host Microbe. 2008;4:77–85. doi: 10.1016/j.chom.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science. 2005;309:1577–1581. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- Kedersha N, Anderson P. Stress granules: sites of mRNA triage that regulate mRNA stability and translatability. Biochem Soc Trans. 2002;30:963–969. doi: 10.1042/bst0300963. [DOI] [PubMed] [Google Scholar]
- Kedersha N, Anderson P. Mammalian stress granules and processing bodies. Methods Enzymol. 2007;431:61–81. doi: 10.1016/S0076-6879(07)31005-7. [DOI] [PubMed] [Google Scholar]
- Kedersha N, Stoecklin G, Ayodele M, Yacono P, Lykke-Andersen J, Fritzler MJ, Scheuner D, Kaufman RJ, Golan DE, Anderson P. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J Cell Biol. 2005;169:871–884. doi: 10.1083/jcb.200502088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha NL, Gupta M, Li W, Miller I, Anderson P. RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2 alpha to the assembly of mammalian stress granules. J Cell Biol. 1999;147:1431–1442. doi: 10.1083/jcb.147.7.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung AK, Calabrese JM, Sharp PA. Quantitative analysis of Argonaute protein reveals microRNA-dependent localization to stress granules. Proc Natl Acad Sci U S A. 2006;103:18125–18130. doi: 10.1073/pnas.0608845103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. Complete replication of hepatitis C virus in cell culture. Science. 2005;309:623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- Lindenbach BD, Rice CM. Flaviviridae: The Viruses and Their Replication. 3. Lippincott-Raven Publishers; Philadelphia, Pennsylvania: 2001. [Google Scholar]
- Liu J, Valencia-Sanchez MA, Hannon GJ, Parker R. MicroRNA-dependent localization of targeted mRNAs to mammalian P-bodies. Nature Cell Biology. 2005;7:719–723. doi: 10.1038/ncb1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machlin ES, Sarnow P, Sagan SM. Masking the 5′ terminal nucleotides of the hepatitis C virus genome by an unconventional microRNA-target RNA complex. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:3193–3198. doi: 10.1073/pnas.1012464108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, Bartenschlager R, Wakita T, Hijikata M, Shimotohno K. The lipid droplet is an important organelle for hepatitis C virus production. Nat Cell Biol. 2007;9:1089–1097. doi: 10.1038/ncb1631. [DOI] [PubMed] [Google Scholar]
- Norman KL, Sarnow P. Modulation of hepatitis C virus RNA abundance and the isoprenoid biosynthesis pathway by microRNA miR-122 involves distinct mechanisms. Journal of Virology. 2010;84:666–670. doi: 10.1128/JVI.01156-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauley KM, Eystathioy T, Jakymiw A, Hamel JC, Fritzler MJ, Chan EK. Formation of GW bodies is a consequence of microRNA genesis. EMBO Rep. 2006;7:904–910. doi: 10.1038/sj.embor.7400783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Vilaro G, Scheller N, Saludes V, Diez J. Hepatitis C virus infection alters p-body composition but is independent of p-body granules. J Virol. 2012;86:8740–8749. doi: 10.1128/JVI.07167-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai RS, Bhattacharyya SN, Artus CG, Zoller T, Cougot N, Basyuk E, Bertrand E, Filipowicz W. Inhibition of translational initiation by Let-7 MicroRNA in human cells. Science. 2005;309:1573–1576. doi: 10.1126/science.1115079. [DOI] [PubMed] [Google Scholar]
- Piotrowska J, Hansen SJ, Park N, Jamka K, Sarnow P, Gustin KE. Stable formation of compositionally unique stress granules in virus-infected cells. J Virol. 2010;84:3654–3665. doi: 10.1128/JVI.01320-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S, Fukasawa M, Yamakawa Y, Natsume T, Suzuki T, Shoji I, Aizaki H, Miyamura T, Nishijima M. Proteomic profiling of lipid droplet proteins in hepatoma cell lines expressing hepatitis C virus core protein. J Biochem. 2006;139:921–930. doi: 10.1093/jb/mvj104. [DOI] [PubMed] [Google Scholar]
- Scheller N, Mina LB, Galao RP, Chari A, Gimenez-Barcons M, Noueiry A, Fischer U, Meyerhans A, Diez J. Translation and replication of hepatitis C virus genomic RNA depends on ancient cellular proteins that control mRNA fates. Proc Natl Acad Sci U S A. 2009;106:13517–13522. doi: 10.1073/pnas.0906413106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeff LB. The history of the “natural history” of hepatitis C (1968–2009) Liver Int. 2009;29(Suppl 1):89–99. doi: 10.1111/j.1478-3231.2008.01927.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheth U, Parker R. Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science. 2003;300:805–808. doi: 10.1126/science.1082320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokoloski KJ, Dickson AM, Chaskey EL, Garneau NL, Wilusz CJ, Wilusz J. Sindbis virus usurps the cellular HuR protein to stabilize its transcripts and promote productive infections in mammalian and mosquito cells. Cell Host Microbe. 2010;8:196–207. doi: 10.1016/j.chom.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon S, Xu Y, Wang B, David MD, Schubert P, Kennedy D, Schrader JW. Distinct structural features of caprin-1 mediate its interaction with G3BP-1 and its induction of phosphorylation of eukaryotic translation initiation factor 2alpha, entry to cytoplasmic stress granules, and selective interaction with a subset of mRNAs. Mol Cell Biol. 2007;27:2324–2342. doi: 10.1128/MCB.02300-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soncini C, Berdo I, Draetta G. Ras-GAP SH3 domain binding protein (G3BP) is a modulator of USP10, a novel human ubiquitin specific protease. Oncogene. 2001;20:3869–3879. doi: 10.1038/sj.onc.1204553. [DOI] [PubMed] [Google Scholar]
- Spangberg K, Wiklund L, Schwartz S. HuR, a protein implicated in oncogene and growth factor mRNA decay, binds to the 3′ ends of hepatitis C virus RNA of both polarities. Virology. 2000;274:378–390. doi: 10.1006/viro.2000.0461. [DOI] [PubMed] [Google Scholar]
- Tingting P, Caiyun F, Zhigang Y, Pengyuan Y, Zhenghong Y. Subproteomic analysis of the cellular proteins associated with the 3′ untranslated region of the hepatitis C virus genome in human liver cells. Biochem Biophys Res Commun. 2006;347:683–691. doi: 10.1016/j.bbrc.2006.06.144. [DOI] [PubMed] [Google Scholar]
- Tourriere H, Chebli K, Zekri L, Courselaud B, Blanchard JM, Bertrand E, Tazi J. The RasGAP-associated endoribonuclease G3BP assembles stress granules. J Cell Biol. 2003;160:823–831. doi: 10.1083/jcb.200212128. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- van Dijk E, Cougot N, Meyer S, Babajko S, Wahle E, Seraphin B. Human Dcp2: a catalytically active mRNA decapping enzyme located in specific cytoplasmic structures. EMBO J. 2002;21:6915–6924. doi: 10.1093/emboj/cdf678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward AM, Bidet K, Yinglin A, Ler SG, Hogue K, Blackstock W, Gunaratne J, Garcia-Blanco MA. Quantitative mass spectrometry of DENV-2 RNA-interacting proteins reveals that the DEAD-box RNA helicase DDX6 binds the DB1 and DB2 3′ UTR structures. RNA Biol. 2011;8 doi: 10.4161/rna.8.6.17836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehner KA, Schutz S, Sarnow P. OGFOD1, a novel modulator of eukaryotic translation initiation factor 2alpha phosphorylation and the cellular response to stress. Molecular and Cellular Biology. 2010;30:2006–2016. doi: 10.1128/MCB.01350-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JP, Cardenas AM, Marissen WE, Lloyd RE. Inhibition of cytoplasmic mRNA stress granule formation by a viral proteinase. Cell Host Microbe. 2007;2:295–305. doi: 10.1016/j.chom.2007.08.006. [DOI] [PubMed] [Google Scholar]
- Yi Z, Fang C, Pan T, Wang J, Yang P, Yuan Z. Subproteomic study of hepatitis C virus replicon reveals Ras-GTPase-activating protein binding protein 1 as potential HCV RC component. Biochem Biophys Res Commun. 2006;350:174–178. doi: 10.1016/j.bbrc.2006.09.027. [DOI] [PubMed] [Google Scholar]
- Yi Z, Pan T, Wu X, Song W, Wang S, Xu Y, Rice CM, Macdonald MR, Yuan Z. Hepatitis C virus co-opts Ras-GTPase-activating protein-binding protein 1 for its genome replication. Journal of Virology. 2011;85:6996–7004. doi: 10.1128/JVI.00013-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JH, Yang WH, Gulick T, Bloch KD, Bloch DB. Ge-1 is a central component of the mammalian cytoplasmic mRNA processing body. RNA. 2005;11:1795–1802. doi: 10.1261/rna.2142405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu SF, Lujan P, Jackson DL, Emerman M, Linial ML. The DEAD-box RNA Helicase DDX6 is Required for Efficient Encapsidation of a Retroviral Genome. PLoS Pathog. 2011;7:e1002303. doi: 10.1371/journal.ppat.1002303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Luo K, Zhang L, Cheville JC, Lou Z. USP10 regulates p53 localization and stability by deubiquitinating p53. Cell. 2010;140:384–396. doi: 10.1016/j.cell.2009.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.