

Abstract
Exposure to cigarette smoke has been associated with in increased risk of neurological diseases such as stroke, Alzheimer’s disease and multiple sclerosis. In these studies, serum and brain sections from Lewis rats or those exposed to cigarette smoke and control rats were examined for evidence of increased inflammation and oxidative stress. Immunocytochemical staining of brain sections from CS-exposed rats showed increased expression of class II MHC and, in ELISA, levels of IFN-gamma and TNF-α were higher than for non-exposed rats. In polymerase chain reaction assays there was increased interferon-gamma, TNF-α, IL-1α, IL-1β, IL-23, IL-6, IL-23, IL-17, IL-10, TGF-β, T-bet and FoxP3 gene expression with CS exposure. There was also markedly elevated MIP-1α/CCL3, less prominent MCP-1/CCL2 and no elevation of SDF-1α gene expression. Analysis of samples from CS-exposed and control rats for anti-oxidant expression showed no significant difference in serum levels of glutathione and, in brain, similar levels of superoxide dismutase and decreased thioredoxin gene expression. In contrast, there was increased brain gene expression for the pro-oxidants iNOS and the NADPH components NOX4, dual oxidase 1 and p22phox. Nrf2 expression, which is typically triggered as a secondary response to oxidative stress, was also increased in brains from CS-exposed rats with nuclear translocation of this protein from cytoplasm demonstrated in astrocytes in association with increased expression of the aryl hydrocarbon receptor gene, a Nrf2 target. These studies, therefore, demonstrate that CS exposure in these animals can trigger multiple immune and oxidative responses that may be have important roles in the pathogenesis of CNS inflammatory neurological diseases.
INTRODUCTION
Links between cigarette smoking and neurological complications have been demonstrated in disease states such as stroke and Alzheimer’s disease, and more recently in MS (Cataldo et al., 2010; Shah and Cole, 2010; Sundstrom et al., 2008). Underlying these associations are animal studies which show that smoking can increase levels of circulating proinflammatory markers and markers of oxidative stress, including levels of reactive oxygen species (ROS), and decrease levels of antioxidants. (Churg et al., 2002; D’Hulst A et al., 2005; Khanna et al., 2009; Moerloose et al., 2005). Among studies performed in humans are those which have shown that urinary levels of nucleic acid and lipid oxidative products were increased with smoking and that serum and urine levels of superoxide dismutase (SOD) and glutathione peroxidase (GPx) correlated inversely with levels of the nicotine metabolite cotinine in smokers (Harman et al., 2003; Sobczak et al., 2004). In addition to activating proinflammatory responses and cellular stress, CS and nicotine have effects that can result in immunosuppression (Chen et al., 2007; Geng et al., 1995; Sopori and Kozak, 1998; Sopori et al., 1998). Therefore, using a Lewis rat model we examined these possible effects of cigarette smoke on the expression of markers of proinflammatory and anti-inflammatory responses. We also examined effects on the expression of NADPH oxidase, pro-oxidant and antioxidant genes and on the activation of Nrf2. These studies showed that a number of these responses to be increased in brains of rats with significant CS exposure. This study, which represents the first demonstration of these effects from CS in the brain, provides a basis for future investigations of the specific mechanisms by which cigarette smoking may trigger the development of inflammatory and degenerative nervous system diseases.
MATERIALS AND METHODS
Exposure of rats to cigarette smoke
This study was conducted according to the Guidelines for Animal Experiments of the NIH using CS-exposed and non-exposed Lewis rats (Harlan) (n=8 rats/group). Rats were exposed to cigarette smoke in a specially constructed smoke chamber where the animals were restrained and ventilated by a smoking machine (Khanna et al., 2009). In the machine, smoke is sucked from lit cigarettes by syringes and pumped around a chamber, where the rats inhale the cigarette smoke. The smoking machine was set to inhale and exhale at intervals mimicking human smoking with each cigarette lasting 8–10 minutes. 3R4F research grade cigarettes with a regular amount of nicotine were used in this study (KTRDC Tobacco Biotechnology Group, University of Kentucky USA). Animals were exposed to cigarette smoke 5 days/week for a total of six weeks with four cigarettes given intermittently throughout the day. Control rats were placed in smoking chambers four times a day for a total period of time that was the same as for exposed rats, but control rats were not exposed to cigarette smoke. The rats were under constant supervision to ensure that the animals did develop signs of distress. The efficacy of cigarette smoke exposure was assessed by quantification of levels of cotinine, the metabolite of nicotine, in serum samples from the rats in an ELISA (Calbiotech). All procedures were approved by the University of Maryland, Baltimore Animal Care and Use Committee.
Detection of mRNA by real time PCR
RNA samples were isolated from brain tissues using a kit from Promega (Madison, USA) and reverse-transcribed into cDNAs by using a cDNA synthesis kit from invitrogen (Carlsbad CA). Primers sequences utilized for the PCR reactions are listed in table 1; RORC primers were purchased from SABiosciences. Real-time quantitative RT-PCR was performed using a Bio-Rad iCycler system (Bio-Rad). For each gene the mRNA expression measured in each sample was normalized to that for β-actin mRNA, and relative mRNA gene expression versus the internal control was calculated as previously described (Khanna et al., 2009).
Table 1.
List of PCR Primers
| Gene Name | Sense (5′ → 3′) | Anti-sense (5′ → 3′) |
|---|---|---|
| IL-4 | TGATGTACCTCCGTGCTTGA | GTGAGTTCAGACCGCTGACA |
| IL-6 | TCAAGGGAAAAGAACCAGACA | GGTTTCAAATCACTCACCCATAC |
| IL-10 | AGTGGAGCAGGTGAAGAATGA | TCATGGCCTTGTAGACACCTT |
| IL-1α | AGTTTCAATCAGCCCTTTACTGA | CTGGGTTGGGATGGTCTCTTC |
| IL-1β | TGTGATGAAAGACGGCACAC | CTTCTTCTTTGGGTATTGTTTGG |
| TNF-α | GGTCTGAGTACATCAACCTGGA | GTCTGTGCCCACATGTTCC |
| TGF-β | CCTGCCCCTACATTTGGA | TGGTTGTAGAGGGCAAGGAC |
| IFN-γ | CCACTATGGATCAGGGAAGG | TCACAATGATTCCACCCACA |
| IL-17 | CTTCACCCTGGACTCTGAGC | CCTCAGCGTTGACACAGC |
| IL-18 | GCCTGATATCGACCGAACA | CCTTCCATCCTTCACAGATAGG |
| SDF-1α | GCGCTCTGCATCAGTGAC | GTTGAGGATTTTCAGATGTTTGAC |
| Tbet | GCGCCAGGAAGTTTCATTT | CATTCTGGTAGGCAGTCACG |
| FoxP3 | AAGTGACGTGCCCCGTATC | TCCGAGTCCAGTAGGTGCTT |
| GATA-3 | AAGGCATCCAGACCAGAAAC | GTTAAACGAGCTGTTCTTGGG |
| MCP-1 | AGCATCCACGTGCTGTCTC | GATCATCTTGCCAGTGAATGAG |
| MIP-1α | GCGCTCTGGAACGAAGTCT | GAATTTGCCGTCCATAGGAG |
| p22phox | GCCATTGCCAGTGTGATCTA | AATGGGAGTCCACTGCTCAC |
| iNos | ACCATGGAGCATCCCAAGTA | CAGCGCATACCACTTCAGC |
| NADPH oxidase 4 (NOX4) | GCTTACCTCCGAGGATCACA | TCTGCTTTTATCCAACATCTCC |
| Dual oxidase 1 (DUOX1) | CACCTCCTGGAGACCTTTTC | CCCTTTGTAGCTTGGGGTTC |
| Superoxide dismutase | CTGGACAAACCTCAGCCCTA | CTGGACAAACCTCAGCCCTA |
| Thioredoxin | ATGGCCACACTTTTCTGGAC | ATGGCCACACTTTTCTGGAC |
| Arylhydrocarbon receptor | CTGCTTCATTTGTCGTGTCC | TTTCCTTGGAACTGCATAGTCA |
| β-actin | CCCAGCACAATGAAGATCAA | CGATCCACACGGAGTACTTG |
Quantitation of glutathione levels
Glutathione levels were measured in serum from the exposed and non-exposed rats using the GSH-Glo™ Assay (Promega) according to the directions provided by the manufacturer.
Enzyme-Linked Immunosorbent Assays
Brain tissue lysates (100 μl volumes) from each TG and WT rat were prepared and analyzed in duplicate using commercial ELISA kits for levels of IFN-γ (BD Biosciences), TNF-α (BD Biosciences) and IL-1β (R&D Systems) as previously described. (Royal et al., 2012)
Tissue Staining
Immunoperoxidase staining of 10μ sections of frozen tissue fixed for 10 min in 4% paraformaldehyde was performed using Vectastain kits (Vector Labs) with a 1:200 dilution of anti-class II MHC antibodies (Pierce) according to the kit directions. For immunofluorescence staining the sections were fixed in 100% methanol (Fisher) for 10 minutes and then the slides were washed and then blocked with blocking solution, then incubated with the following primary antibodies: rat anti-GFAP (1:50 dilution; BD Biosciences), mouse anti-TNF-α (1:50 dilution; BD Biosciences) and mouse anti-Nfr2 (1:200 dilution; kind gift of Dr. Anil Jaiswal, University of Maryland). After incubations at 4°C over night with appropriate primary antibody, the sections were washed and then incubated with, as appropriate, either fluorescein isothiocyanate (FITC)-conjugated (Santa Cruz Biotechnology) of Texas Red-conjugated (Invitrogen) secondary antibodies. For all staining, the specificity of the procedures was verified by negative control sections incubated without primary antibody. Imaging was performed using a Zeiss Axiovert 40 microscope equipped with a Axiocam mrc fluorescence camera (Zeiss) and Axiovision LE software (Zeiss) with subsequent image processing performed using SPOT Imaging software (Spot Imaging Solutions).
Statistical Analyses
Relative gene expression levels were compared using the Mann-Whitney test and glutathione levels were compared using the Student’s t-test. A p-value < 0.05 was considered statistically significant.
RESULTS
Cigarette smoke induced neuroinflammation detected in immunostained brain sections from exposed rats
Sections were stained for MHC class II, which is expressed by T cells, B cells, monocytes, macrophages, and, in the brain, also by microglia and astrocytes. There studies showed there to be increased MCH class II staining in parenchymal and in perivascular regions of brain from the CS-exposed rats (figure 1). In contrast, no staining was noted in brain tissue from the control rats. Lysates of brain tissue from the CS-exposed and non-exposed rats were also examined by ELISA for levels of the proinflammatory cytokines IFN-γ and TNF-α. These studies showed that levels of these cytokine were increased for the rats exposed to CS as opposed to non-exposed rats (figure 2).
Figure 1.
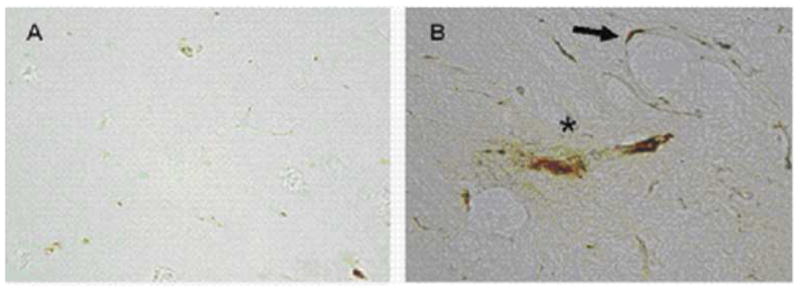
Immunostaining of subcortical white matter brain from a rat not exposed to CS (A) and from a CS-exposed rat (B) for class II MHC. Exposure to CS resulted in expression of MHCII in perivascular regions (arrow) and in the brain parenchyma (asterisk).
Figure 2.

Lysates of brain frontal cortex and subcortical white matter were examined be ELISA for levels of IFN-γ and TNF-α. Levels of these proinflammatory cytokines were higher in brains from CS-exposed rats than from non-exposed rats.
Cigarette smoke exposure induces the expression of inflammatory marker genes
PCR assays were performed to measure brain expression of proinflammatory and anti-inflammatory cytokines and chemokines and of transcription factors that induce the differentiation of proinflammatory and anti-inflammatory T cell subtypes. These studies showed there to be upregulation of the IFN-γ and TNF-α (Th1) cytokine gene expression. In addition, there was increased expression of IL-1-α, IL1-β, and of Th17 (IL-23, IL-6, IL-17), Th2 (IL-10) and Treg cell (TGF-β and IL-10) associated cytokine gene expression (figure 3). Notably, however, IL-4 gene expression was not significantly elevation for CS-exposed versus control rats. Expression of the GATA-3 gene, which activates expression of IL-4, and RORC, which induces Th17 cell differentiation showed, respectively, borderline and no significant increase in the CS-exposed rats. In contrast, there were significant increases in expression of T-bet, which activates expression of the IFN-γ gene, and FoxP3, which induces Treg cell differentiation. Analysis of selected chemokine receptor expression showed there to be significant increases in levels of MIP-1α/CCL3 gene expression but less prominent expression of MCP-1/CCL2 mRNA, and no significant difference in expression for SFD-1β for CS-exposed rats as compared to control rats.
Figure 3.

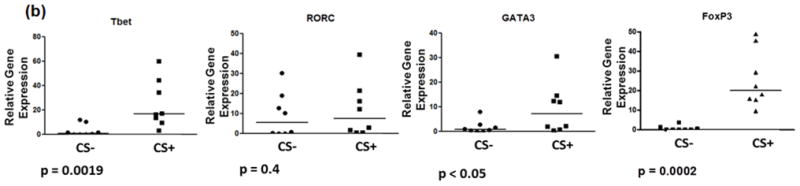
PCR analysis of gene expression for selected (a) cytokines and chemokine receptors and for (b) transcription factors that activate the expression of IFN-γ (T-bet) or IL-4 (GAGA-3) and that induce naïve cell differentiation into Th17 (RORC) or regulatory T cells (FoxP3).
Effect of cigarette smoke exposure on the expression of mediators of reactive oxygen and reactive nitrogen species in rat brain
Samples from CS-exposed and control rats were analyzed for levels of anti-oxidant and pro- and anti-oxidant gene expression. CS exposure resulted in mild elevations in serum levels of the anti-oxidant glutathione as compared to non-exposed rats (11.0 ± 0.37 versus 8.9 ± 0.7, respectively) which (p=0.04). In brain, gene expression levels for iNOS, dual oxidase 1, NOX4 and p22phox, which are components of NADPH, was elevated for rats exposed to CS (figure 4). However, there was decreased expression of thioredoxin and no change in expression of superoxide dismutase mRNA, both which are antioxidants. Also, there was a higher level of expression of Nrf2, a transcription factor that promotes antioxidant protein gene expression.
Figure 4.
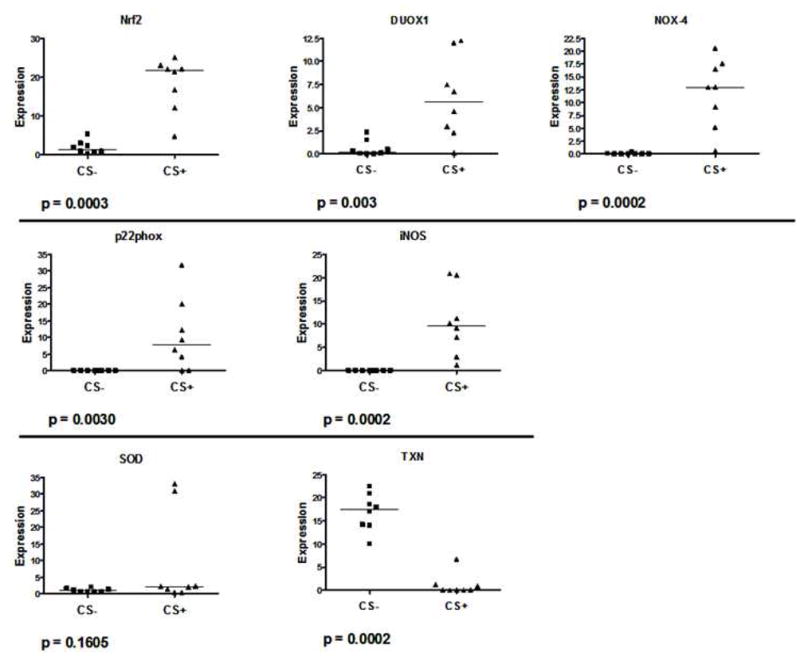
PCR analysis of brain tissue from CS-exposed and control rats for expression of markers of oxidative stress. Levels of Nrf2, DUOX1, NOX4, p22phox, and iNOS gene expression were higher in brains from rats exposed to CS than for control rats. In contrast there was no change in gene expression for superoxide dismutase (SOD) and expression was deceased for thioredoxin (TXN) in brains from rats exposed to CS as compared to brains from control rats.
Cigarette smoke effects on expression of proinflammatory cytokine and Nrf2 in astrocytes and on aryl hydrocarbon receptor gene expression
Frozen sections of brain tissue from the CS-exposed and non-exposed rats were examined by immunofluorescence staining for expression and localization of TNF-α and Nrf2 by astrocytes. These studies showed there to be no expression of TNF-α by these cells. However, there was strong expression of Nrf2 by astrocytes in the sections. Astrocytes in brain from non-exposed rats expressed Nrf2 throughout the cytoplasm and in the nucleus. In contrast, for CS-exposed rats Nrf2 was localized to the perinuclear region and in the nuclei of the stained cells (figure 4). PCR analysis of brian for expression of the aryl hydrocarbon receptor gene, which is an Nrf2 target, showed expression to be increased in brains from the CS-exposed rats (figure 5).
Figure 5.
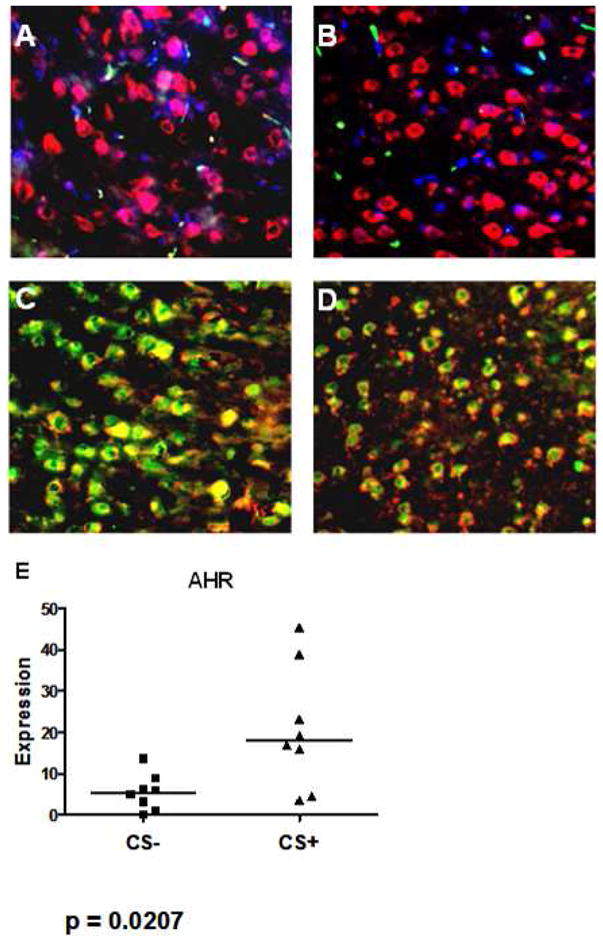
Immunofluorescence staining of brain from a control rat (A and C) and a rat exposed to CS (B and d) stained either with anti-GFAP-Texas Red (A – D) and either with anti-TNF-α-GFAP (A and B) or anti-Nrf2-FITC (C and D). GFAP+ cells in the sections from the CS-exposed and from the control rats showed no staining for TNF-α (A, B). Control rat brain showed large numbers of GFAP+ cells with extensive cytoplasmic and nuclear staining for Nrf2 (C). In sections from the CS-exposed rat Nrf2 staining was also observed, but was primarily perinuclear and nuclear with other regions of cytoplasm staining only for GFAP (D). In brain from CS-exposed rats, expression of an Nrf2 target gene, aryl hydrocarbon receptor (AHR) was increased as compared to control (CS-) rats (E).
DISCUSSION
Our studies demonstrate the presence of inflammatory involvement and increased expression of pro-oxidant markers in the brains of Lewis rats exposed to CS. It has been well documented that tobacco smoke exposure can lead to the development of diseases of a variety of organ systems. We found there to be increased proinflammatory and anti-inflammatory cytokine gene responses in the exposed rat brains. In particular, CS exposure elevated Th1 as well as Th17 and Treg cell-associated cytokine responses. Also, whereas FoxP3 gene expression was increased, there was no change in expression of RORC, which suggests that at the time of euthanization the animals were producing increased numbers of Treg cells relative to Th17 cells. In contrast, expression of the Tbet and GATA-3 genes mirrored those for IFN-γ and IL-4, which suggests that balance of Th1 versus Th2 immune responses may have a more significant role in mediating the immunopathological consequences of CS exposure. This finding also raises the possibility that the increased IFN-γ may underlie the elevated FoxP3 gene expression that was observed in the CS-exposed rats. This possibility is supported by studies in patients with long term renal transplants where IFN-γ was shown to stimulate the production of CD4+CD25+FoxP3+IFN-γ+ from FoxP3+ regulatory T cells, which in such patients can suppress allogeneic responses (Daniel et al., 2011; Feng et al., 2008). Also, in a murine model of infectious colitis IL-12 was shown to induce the conversion of FoxP3+ regulatory T cells to FoxP3+IFN-γ+ regulatory T cells in association with decreased proinflammatory responses against a microbial antigen (Feng et al., 2008). There was also a slightly increase in expression of the gene for the macrophage marker IL-1β; however, decreased expression of this cytokine has been shown to result from the presence of high levels of IFN-γ (De Boer et al., 2001). Also observed was elevation of genes for chemokine and chemokine receptors that are associated with increased T cell and mononuclear phagocyte trafficking into the nervous system. In our studies, there was also positive staining of brain tissue from the CS-exposed rats for class II MHC, which is expressed by activated astrocytes as well as by inflammatory cells (Wong et al., 1984). When activated, astrocytes can also express the cytokine TNF-α (Robbins et al., 1987), which we did not observed in our studies.
In cells the NADPH oxidase (NOX) enzymes, which include NOX4 and the dual oxidase DUOX1, are major contributors to the production of ROS (De et al., 2000; Geiszt et al., 2000). NOX4 is widely distributed throughout the body whereas DUOX1, originally identified in the thyroid, is produced both in the lungs and in the brain (Lambeth, 2004). In response to growth factors, cytokines and calcium signals the NADPH oxidases produce reactive oxygen in a regulated manner (Lambeth, 2004). ROS are enzymatically eliminated from cells by superoxide dismutase (SOD)1, catalase and peroxidases. Also, the cellular stress that results from ROS production triggers the cytoplasmic accumulation of the transcription factor Nrf2 (nuclear factor (erythroid-derived 2)-like 2), which is then translocated to the nucleus where it activates the transcription of a number of antioxidant genes.
In our studies we observed decreased cytoplasmic and increased perinuclear and nuclear staining for Nrf2 in astrocytes as well as increased expression of AHR, a Nrf2 target gene, in the brains of the animals. This suggests that the exposure to cigarettes triggered antioxidant responses that occur as a result of cellular stress. The activation of Nrf2 by CS has been shown to occur in other studies, and in mice exposed to CS expression can be demonstrated in respiratory tract tissue (Gebel et al., 2004). Astrocytes have been known for some time to be a rich source of Nrf2, and there is evidence which suggests that Nrf2 expression by astrocytes may confer protection in individuals at risk for Parkinson’s disease and might underlie the observed potent anti-Parkinson’s disease effect of smoking (Baumann et al., 1980; Grandinetti et al., 1994; Jakel et al., 2007; Johnson et al., 2008; Tanaka et al., 2010; Thacker et al., 2007). The apparent CS-induced nuclear translocation of Nrf2, as well as the demonstrated increased expression of the iNOS, NOX4, DUOX1 and the NADPH component p22phox genes combined with the decrease or lack of change in anti-oxidant gene (SOD and thioredoxin) and protein (glutathione) expression in the exposed rats are all consistent with the induction of significant oxidative stress, particularly in brain of the animals.
In contrast to the tendency for CS to decrease the risk of Parkinson’s disease, results of cohort studies have shown there to be an increased risk of Alzheimer’s disease among cigarette smokers (Almeida et al., 2002; Cataldo et al., 2010). This is the case despite the fact that nicotine and cotinine has been shown to slow formation of amyloid fibrils in vitro (Liu and Zhao, 2004; Ono et al., 2002; Salomon et al., 1996). However, the risk of the Parkinson’s disease developing in former smokers is higher than for individuals who never smoked and less than for current smokers (Tanaka et al., 2010; Thacker et al., 2007). This supports the likelihood for there being a protective component present in CS smoke that, when removed, allows for damage to occur and to progress uninhibited. Potential candidates for the agents responsible for this protective effect are nicotine and it metabolite cotinine, which have been found to protect SHSY5Y cells from toxicity otherwise induced by the dopamine metabolite 6-hydroxydopamine (Riveles et al., 2008). Another is a constituent of cigarette smoke identified as 2,3,6-trimethyl-1,4-naphthoquinone, which was found to inhibit monoamine oxidase A and B activity and to protect against damage to striatal neurons induced by the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) in the mouse model of Parkinson’s disease (Castagnoli et al., 2001).
In lungs of smokers there has been shown to be increased numbers of bronchial alveolar macrophages that secrete increased levels of lytic enzymes and free radicals (Sopori, 2002a). More than 4,000 chemicals and toxic substances which can potentially cause serious and permanent negative health consequences have been identified in CS (Brunnemann and Hoffmann, 1991). It is not practical to analyze them all individually for these effects. However, among those that have been identified is bacterial endotoxin, which has been found to be present on cigarette tobacco and filter tips as well as in smoke produced by the cigarettes (Hasday et al., 1999). Both acute and chronic proinflammatory responses associated with CS exposure may be induced by polycyclic hydrocarbons that are present in cigarette smoke (Tomaki et al., 2007). In addition, SIRT1, a protein deacetylase that increases TNF-α production by deacetylation of RelA/p65 at lysine-310, is decreased in the presence of oxidative stress (Rajendrasozhan et al., 2008), which, in this report, was shown to be increased by CS.
The Lewis rat strain, which was used for the studies reported here, has for decades been used for studies of experimental autoimmune CNS disease. Immunizing the rats either with preparations of whole myelin or myelin peptides results in the induction of experimental allergic encephalomyelitis (EAE) (McFarlin et al., 1975). This disorder can be also induced by inoculating non-immunized rats with sensitized T cells obtained from immunized animals. The inflammatory response in EAE involves both innate and adaptive responses which result in the migration of activated T cells from blood into the central nervous system. The immune involvement of the nervous system leads to the subsequent development of a marked pathological change, including a reactive astrocytosis, an increase in production of reactive oxygenated species and markers of oxidative stress (Bernstein and Miller, 2010; Toft-Hansen et al., 2011). It is possible that in humans the described effects from cigarette smoke could interact with genetic and environmental factors, such as EBV infection or vitamin D deficiency (Ascherio and Munger, 2007; Simon et al., 2010), that have been linked with an increased risk of MS to enhance the propensity for developing the disease. Similarly, these potential interactions could at least in part explain the more rapid disease progression and an observed increased risk of treatment failure that has been also observed for individuals with MS who smoke cigarettes (D’hooghe et al., 2011; Healy et al., 2009; Pittas et al., 2009).
In summary, this study is the first to comprehensively examine effects of CS on specific mediators of immune responses and oxidative stress in the rat brain. In this report a number of potential explanations for the observed inflammatory and other cellular responses are presented. Though supported by studies performed by previous investigators, firm conclusions related to such responses require the support of information from detailed mechanistic studies. Another limitation of the studies presented here is that they do not address the possible more acute responses as well as the longer term consequences to the brain, which can provide information regarding how the various responses might be coordinated and inter-related. A better understanding of these neuroimmune effects of cigarette smoke will likely assist in the development of effective strategies for treating smoking related diseases of the nervous system.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Almeida OP, Hulse GK, Lawrence D, Flicker L. Smoking as a risk factor for Alzheimer’s disease: contrasting evidence from a systematic review of case-control and cohort studies. Addiction. 2002;97:15–28. doi: 10.1046/j.1360-0443.2002.00016.x. [DOI] [PubMed] [Google Scholar]
- Ascherio A, Munger KL. Environmental risk factors for multiple sclerosis. Part II: Noninfectious factors. Ann Neurol. 2007;61:504–513. doi: 10.1002/ana.21141. [DOI] [PubMed] [Google Scholar]
- Baumann RJ, Jameson HD, McKean HE, Haack DG, Weisberg LM. Cigarette smoking and Parkinson disease: 1. Comparison of cases with matched neighbors. Neurology. 1980;30:839–843. doi: 10.1212/wnl.30.8.839. [DOI] [PubMed] [Google Scholar]
- Bernstein AI, Miller GW. Oxidative signaling in experimental autoimmune encephalomyelitis. Toxicol Sci. 2010;114:159–161. doi: 10.1093/toxsci/kfq012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunnemann KD, Hoffmann D. Analytical studies on tobacco-specific N-nitrosamines in tobacco and tobacco smoke. Crit Rev Toxicol. 1991;21:235–240. doi: 10.3109/10408449109017910. [DOI] [PubMed] [Google Scholar]
- Castagnoli KP, Steyn SJ, Petzer JP, Van der Schyf CJ, Castagnoli N., Jr Neuroprotection in the MPTP Parkinsonian C57BL/6 mouse model by a compound isolated from tobacco. Chem Res Toxicol. 2001;14:523–527. doi: 10.1021/tx000224v. [DOI] [PubMed] [Google Scholar]
- Cataldo JK, Prochaska JJ, Glantz SA. Cigarette smoking is a risk factor for Alzheimer’s Disease: an analysis controlling for tobacco industry affiliation. J Alzheimers Dis. 2010;19:465–480. doi: 10.3233/JAD-2010-1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Cowan MJ, Hasday JD, Vogel SN, Medvedev AE. Tobacco smoking inhibits expression of proinflammatory cytokines and activation of IL-1R-associated kinase, p38, and NF-kappaB in alveolar macrophages stimulated with TLR2 and TLR4 agonists. J Immunol. 2007;179:6097–6106. doi: 10.4049/jimmunol.179.9.6097. [DOI] [PubMed] [Google Scholar]
- Churg A, Dai J, Tai H, Xie C, Wright JL. Tumor necrosis factor-alpha is central to acute cigarette smoke-induced inflammation and connective tissue breakdown. Am J Respir Crit Care Med. 2002;166:849–854. doi: 10.1164/rccm.200202-097OC. [DOI] [PubMed] [Google Scholar]
- D’hooghe MB, Haentjens P, Nagels G, De KJ. Alcohol, coffee, fish, smoking and disease progression in multiple sclerosis. Eur J Neurol. 2011 doi: 10.1111/j.1468-1331.2011.03596.x. [DOI] [PubMed] [Google Scholar]
- D’Hulst AI, Vermaelen KY, Brusselle GG, Joos GF, Pauwels RA. Time course of cigarette smoke-induced pulmonary inflammation in mice. Eur Respir J. 2005;26:204–213. doi: 10.1183/09031936.05.00095204. [DOI] [PubMed] [Google Scholar]
- De Boer ML, Hu J, Kalvakolanu DV, Hasday JD, Cross AS. IFN-gamma inhibits lipopolysaccharide-induced interleukin-1 beta in primary murine macrophages via a Stat1-dependent pathway. J Interferon Cytokine Res. 2001;21:485–494. doi: 10.1089/10799900152434358. [DOI] [PubMed] [Google Scholar]
- De DX, Wang D, Many MC, Costagliola S, Libert F, Vassart G, Dumont JE, Miot F. Cloning of two human thyroid cDNAs encoding new members of the NADPH oxidase family. J Biol Chem. 2000;275:23227–23233. doi: 10.1074/jbc.M000916200. [DOI] [PubMed] [Google Scholar]
- Gebel S, Gerstmayer B, Bosio A, Haussmann HJ, Van ME, Muller T. Gene expression profiling in respiratory tissues from rats exposed to mainstream cigarette smoke. Carcinogenesis. 2004;25:169–178. doi: 10.1093/carcin/bgg193. [DOI] [PubMed] [Google Scholar]
- Geiszt M, Kopp JB, Varnai P, Leto TL. Identification of renox, an NAD(P)H oxidase in kidney. Proc Natl Acad Sci USA. 2000;97:8010–8014. doi: 10.1073/pnas.130135897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng Y, Savage SM, Johnson LJ, Seagrave J, Sopori ML. Effects of nicotine on the immune response. I Chronic exposure to nicotine impairs antigen receptor-mediated signal transduction in lymphocytes. Toxicol Appl Pharmacol. 1995;135:268–278. doi: 10.1006/taap.1995.1233. [DOI] [PubMed] [Google Scholar]
- Grandinetti A, Morens DM, Reed D, MacEachern D. Prospective study of cigarette smoking and the risk of developing idiopathic Parkinson’s disease. Am J Epidemiol. 1994;139:1129–1138. doi: 10.1093/oxfordjournals.aje.a116960. [DOI] [PubMed] [Google Scholar]
- Harman SM, Liang L, Tsitouras PD, Gucciardo F, Heward CB, Reaven PD, Ping W, Ahmed A, Cutler RG. Urinary excretion of three nucleic acid oxidation adducts and isoprostane F(2)alpha measured by liquid chromatography-mass spectrometry in smokers, ex-smokers, and nonsmokers. Free Radic Biol Med. 2003;35:1301–1309. doi: 10.1016/j.freeradbiomed.2003.07.003. [DOI] [PubMed] [Google Scholar]
- Hasday JD, Bascom R, Costa JJ, Fitzgerald T, Dubin W. Bacterial endotoxin is an active component of cigarette smoke. Chest. 1999;115:829–835. doi: 10.1378/chest.115.3.829. [DOI] [PubMed] [Google Scholar]
- Healy BC, Ali EN, Guttmann CR, Chitnis T, Glanz BI, Buckle G, Houtchens M, Stazzone L, Moodie J, Berger AM, Duan Y, Bakshi R, Khoury S, Weiner H, Ascherio A. Smoking and disease progression in multiple sclerosis. Arch Neurol. 2009;66:858–864. doi: 10.1001/archneurol.2009.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakel RJ, Townsend JA, Kraft AD, Johnson JA. Nrf2-mediated protection against 6-hydroxydopamine. Brain Res. 2007;1144:192–201. doi: 10.1016/j.brainres.2007.01.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JA, Johnson DA, Kraft AD, Calkins MJ, Jakel RJ, Vargas MR, Chen PC. The Nrf2-ARE pathway: an indicator and modulator of oxidative stress in neurodegeneration. Ann NY Acad Sci. 2008;1147:61–69. doi: 10.1196/annals.1427.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna AK, Xu J, Uber PA, Burke AP, Baquet C, Mehra MR. Tobacco smoke exposure in either the donor or recipient before transplantation accelerates cardiac allograft rejection, vascular inflammation, and graft loss. Circulation. 2009;120:1814–1821. doi: 10.1161/CIRCULATIONAHA.108.840223. [DOI] [PubMed] [Google Scholar]
- Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- Liu Q, Zhao B. Nicotine attenuates beta-amyloid peptide-induced neurotoxicity, free radical and calcium accumulation in hippocampal neuronal cultures. Br J Pharmacol. 2004;141:746–754. doi: 10.1038/sj.bjp.0705653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarlin DE, Hsu SC, Slemenda SB, Chou FC, Kibler RF. The immune response against myelin basic protein in two strains of rat with different genetic capacity to develop experimental allergic encephalomyelitis. J Exp Med. 1975;141:72–81. doi: 10.1084/jem.141.1.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moerloose KB, Pauwels RA, Joos GF. Short-term cigarette smoke exposure enhances allergic airway inflammation in mice. Am J Respir Crit Care Med. 2005;172:168–172. doi: 10.1164/rccm.200409-1174OC. [DOI] [PubMed] [Google Scholar]
- Ono K, Hasegawa K, Yamada M, Naiki H. Nicotine breaks down preformed Alzheimer’s beta-amyloid fibrils in vitro. Biol Psychiatry. 2002;52:880–886. doi: 10.1016/s0006-3223(02)01417-8. [DOI] [PubMed] [Google Scholar]
- Pittas F, Ponsonby AL, van der Mei IA, Taylor BV, Blizzard L, Groom P, Ukoumunne OC, Dwyer T. Smoking is associated with progressive disease course and increased progression in clinical disability in a prospective cohort of people with multiple sclerosis. J Neurol. 2009;256:577–585. doi: 10.1007/s00415-009-0120-2. [DOI] [PubMed] [Google Scholar]
- Rajendrasozhan S, Yang SR, Kinnula VL, Rahman I. SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;177:861–870. doi: 10.1164/rccm.200708-1269OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riveles K, Huang LZ, Quik M. Cigarette smoke, nicotine and cotinine protect against 6-hydroxydopamine-induced toxicity in SH-SY5Y cells. Neurotoxicology. 2008;29:421–427. doi: 10.1016/j.neuro.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins DS, Shirazi Y, Drysdale BE, Lieberman A, Shin HS, Shin ML. Production of cytotoxic factor for oligodendrocytes by stimulated astrocytes. J Immunol. 1987;139:2593–2597. [PubMed] [Google Scholar]
- Royal W, 3rd, Zhang L, Guo M, Jones O, Davis H, Bryant JL. Immune activation, viral gene product expression and neurotoxicity in the HIV-1 transgenic rat. J Neuroimmunol. 2012;247:16–24. doi: 10.1016/j.jneuroim.2012.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon AR, Marcinowski KJ, Friedland RP, Zagorski MG. Nicotine inhibits amyloid formation by the beta-peptide. Biochemistry. 1996;35:13568–13578. doi: 10.1021/bi9617264. [DOI] [PubMed] [Google Scholar]
- Simon KC, van der Mei IA, Munger KL, Ponsonby A, Dickinson J, Dwyer T, Sundstrom P, Ascherio A. Combined effects of smoking, anti-EBNA antibodies, and HLA-DRB1*1501 on multiple sclerosis risk. Neurology. 2010;74:1365–1371. doi: 10.1212/WNL.0b013e3181dad57e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobczak A, Golka D, Szoltysek-Boldys I. The effects of tobacco smoke on plasma alpha- and gamma-tocopherol levels in passive and active cigarette smokers. Toxicol Lett. 2004;151:429–437. doi: 10.1016/j.toxlet.2004.03.010. [DOI] [PubMed] [Google Scholar]
- Sopori ML, Kozak W. Immunomodulatory effects of cigarette smoke. J Neuroimmunol. 1998;83:148–156. doi: 10.1016/s0165-5728(97)00231-2. [DOI] [PubMed] [Google Scholar]
- Sopori ML, Kozak W, Savage SM, Geng Y, Soszynski D, Kluger MJ, Perryman EK, Snow GE. Effect of nicotine on the immune system: possible regulation of immune responses by central and peripheral mechanisms. Psychoneuroendocrinology. 1998;23:189–204. doi: 10.1016/s0306-4530(97)00076-0. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Miyake Y, Fukushima W, Sasaki S, Kiyohara C, Tsuboi Y, Yamada T, Oeda T, Miki T, Kawamura N, Sakae N, Fukuyama H, Hirota Y, Nagai M. Active and passive smoking and risk of Parkinson’s disease. Acta Neurol Scand. 2010;122:377–382. doi: 10.1111/j.1600-0404.2010.01327.x. [DOI] [PubMed] [Google Scholar]
- Thacker EL, O’Reilly EJ, Weisskopf MG, Chen H, Schwarzschild MA, McCullough ML, Calle EE, Thun MJ, Ascherio A. Temporal relationship between cigarette smoking and risk of Parkinson disease. Neurology. 2007;68:764–768. doi: 10.1212/01.wnl.0000256374.50227.4b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toft-Hansen H, Fuchtbauer L, Owens T. Inhibition of reactive astrocytosis in established experimental autoimmune encephalomyelitis favors infiltration by myeloid cells over T cells and enhances severity of disease. Glia. 2011;59:166–176. doi: 10.1002/glia.21088. [DOI] [PubMed] [Google Scholar]
- Tomaki M, Sugiura H, Koarai A, Komaki Y, Akita T, Matsumoto T, Nakanishi A, Ogawa H, Hattori T, Ichinose M. Decreased expression of antioxidant enzymes and increased expression of chemokines in COPD lung. Pulm Pharmacol Ther. 2007;20:596–605. doi: 10.1016/j.pupt.2006.06.006. [DOI] [PubMed] [Google Scholar]
- Wong GH, Bartlett PF, Clark-Lewis I, Battye F, Schrader JW. Inducible expression of H-2 and Ia antigens on brain cells. Nature. 1984;310:688–691. doi: 10.1038/310688a0. [DOI] [PubMed] [Google Scholar]