

Recent clinical trials with IL-1β blockade have revealed an impressive and sustained reduction in recurrent attacks of gouty arthritis (1). Even with the use of allopurinol to reduce the systemic levels of uric acid and the anti-inflammatory properties of colchicine, there is no dearth of patients with recurrent episodes of painful gouty arthritis poorly controlled with these regimens who require intermittent courses of glucocorticoids. Thus, the success of IL-1β blocking therapies is a welcome addition for treating refractory gouty arthritis in these patients. Despite the availability of several widely used TNFα blocking therapies for rheumatoid arthritis and other autoimmune diseases, there is a paucity of reports that blocking TNFα provides an effective reduction in gout severity. One explanation for the lack of clinical trials of TNFα blockade in gout attacks is reluctance of subjecting these patients to suppression of host-defense by TNFα blockers. An alternative explanation is that efficacy of TNFα blockade in refractory gout is less than expected. One study reports a weak response with rather high doses of infliximab (2). There are also few publications on monosodium urate (MSU) crystals inducing TNFα from human and mouse cells unless co-stimulated with endotoxins. Therefore, IL-1β blockade may be used for inducing long-term remissions in refractory patients and replace glucocorticoids. If IL-1β blockade becomes the standard of care in refractory gout, it would be consistent with the unique role of IL-1β in the pathogenesis of auto-inflammatory diseases.
In this issue of Arthritis and Rheumatism, Joosten et al provide experimental data that causally explain the clinical events that consistently precede flares of gout and how these relate to the production of IL-1β. Despite several reports that the addition of MSU crystals to mononuclear phagocytes induces IL-1β secretion (3, 4), MSU as a sole activator of active IL-1β is inconsistent with the clinical reality of gouty attacks. For example, why do less than 10% of persons with hyperuricemia and deposits MSU crystals in joints develop the disease and the remainder do not? Why do acute flares of gout often occur in the middle of the night? Why do patients receiving anti-tumor therapies have high uric acid levels but no clinical signs of gout? Why do persons with hyperuricemia have attacks of gout when they diet and lose weight? Although the associations of food and alcohol consumption with attacks of gout have been known for many centuries, how such life-style events relate to IL-1β activity has not been explored. Thus published studies that show MSU crystals alone induce active IL-1β requires a careful re-evaluation to be consistent with the clinical associations of gout attacks. This editorial does not attempt to review the association of gout attacks with the consumption of food and alcohol. Instead, it draws attention to the study by Joosten and co-workers published in this issue (5), which goes a long way in providing the basis for the production of IL-1β by MSU and concludes that one needs more than MSU to trigger an attack.
First, the Joosten study confirms that pure MSU crystals per se do not induce IL-1β from primary peripheral blood mononuclear cells (PBMC) isolated from healthy donors (6) and extends these observations to mouse peritoneal macrophages. In order to relate the association with dietary intake of fatty foods, free fatty acids (FFA) of increasing lengths were also added to human or mouse macrophages and similar to MSU alone, did not result in the secretion of IL-1β. However, the combination of MSU plus FFA induced the release of high levels of IL-1β into the supernatants of either PBMC or mouse macrophages. The study identified the eighteen carbon fatty acid (stearic acid) as the most effective lipid for stimulating IL-1β in combination with MSU crystals.
The evaluation of studies adding exogenous stimuli to human PBMC for cytokine production must assess the role of TLR ligands, particularly endotoxin (lipopolysaccahride, LPS), as these are unusually potent inducers of IL-1β in the picomolar range. In the Joosten studies (5) and others (6), there was no production of IL-1β using MSU crystals added to human PBMC or mouse macrophages in vitro or in vivo. Although some have reported that MSU crystals alone induce IL-1β from macrophages (3, 4), others have not been able to demonstrate this response unless a small amount of bacterial endotoxin is present as a “priming agent” (3). In fact, there is a specific synergism of MSU crystals with LPS for the production of IL-1β (6). Moreover, the concept that MSU crystals per se induce IL-1β is inconsistent with that fact that there is no inflammation where there is ample presence of urate crystal (tophi). Moreover, reports of MSU crystals inducing IL-1β do not explain why the attacks are precipitated with intake of large amounts of food. The Joosten study builds on the concept that MSU crystals needs a second signal in inducing active IL-1β. However, from a clinical perspective, a second signal from LPS is hardly relevant to gout. There is usually no infection that triggers the flare of gout in susceptible persons. On the other hand, free fatty acids are relevant to gout. Indeed, one signal is provided by the high levels of MSU crystals in susceptible patients and a second signal comes from a rise in FFA following and episode of overnutrition. Neither MSU crystals nor FFA alone induce the release of biologically active IL-1β; both are needed.
As shown in Figure 1, the synovial macrophage is already stimulated with MSU crystals secondary to the hyperuricemia. Then a rise in serum lipids provides the trigger for IL-1β synthesis, processing and release, thus precipitating the inflammatory flare of the attacks. The two-signal concept for the secretion of IL-1β is hardly a new concept and has a firm foundation in understanding the molecular mechanisms for synthesis and release of the active cytokine. Unlike IL-1α, the IL-1β precursor is not constitutively present in blood monocytes or tissue macrophages but requires a stimulus to be induced. Although many cytokines are translated soon after transcription, this is not the case with IL-1β. For example, freshly obtained human blood monocytes adhering to glass or plastic transcribe large amounts of IL-1β mRNA without any significant translation (7). Naturally generated or recombinant complement C5a also results in transcription without translation (8). The first signal is sometimes termed “priming” and drives gene expression for the IL-1β precursor. Unless a second signal is provided, the mRNA is degraded or the poly-adenylated mRNA falls off the ribosome soon after initiation of translation (9). The signal for completion of ribosomal translation can come from various endogenous sources. Low picomolar concentrations of IL-1 itself provide a second signal for rapid translation of the IL-1β precursor (7-9). It is now clear that saturated FFA also provide a signal for translation. In the case of IL-1β production in gout, MSU crystals likely provide the transcriptional priming signal and FFA the second signal. The role of TLR in this scenario comes from the ability of TLR2 to recognize FFA (5).
Molecular mechanisms in flares of gouty arthritis.
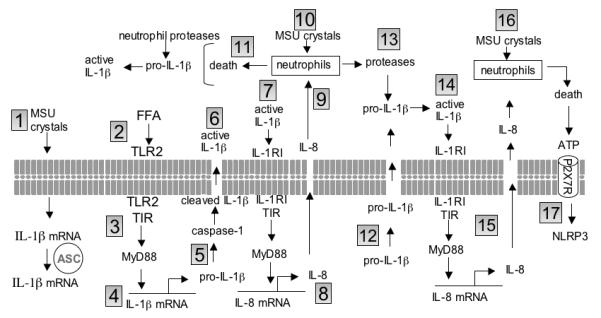
1. MSU crystals “prime” synovial cell expression of IL-1β mRNA via ASC. 2. FFAs trigger TLR2. 3. The Toll IL-1 Receptor (TIR) domain recruits MyD88. 4. Translation of the IL-1β precursor (pro-IL-1β). 5. Pro-IL-1β is cleaved by caspase-1. 6. Cleaved IL-1β is secreted from the cell (active IL-1β). 7. Active IL-1β binds to its receptor (IL-1RI) and recruits MyD88 via TIR domain. 8. Gene expression and synthesis of the chemokine IL-8. 9. IL-8 induces the influx of neutrophils into the joint space. 10. MSU crystals activate neutrophils. 11. Neutrophil cell death releases proteases and pro-IL-1β. Neutrophil elastase and proteinase-3 cleave pro-IL-1β close to the caspase-1 site and generate active IL-1β. 12. Pro-IL-1β enters the extracellular space via loss in membrane integrity. 13. MSU crystals activate neutrophils release proteases; cleavage of extracellular pro-IL-1β with the generation of active IL-1β (as in 11). 14. Active IL-1β triggers its receptor. 15. MyD88 is recruited to the TIR domain and induces IL-8. IL-8 induces neutrophil infiltration (as in 9). 16. MSU-mediated neutrophil death results in the release of ATP and ATP activates the P2X7 receptor. 17. ATP trigger of the P2X7 receptor drives potassium out of the cells and brings about the oligomerization of NLRP3.
One of the strengths of the Joosten studies can be found in the data from knee joints of mice injected with MSU or FFA. As of this writing, there appears to be one report on the effect of MSU crystals injected into a joint of experimental pigs (10). Most investigators study MSU crystal responses in primary as well as cell line lines. In vivo, MSU crystals are commonly injected into the peritoneal cavity or a skin pouch, both methods being rather different from the responses of the synovium to inflammatory agents. Similar to the observations in human PBMC and mouse macrophages, MSU crystals alone was not inflammatory as there was no joint swelling or induction of neutrophil infiltration following even high doses of MSU in the joints. The C18 FFA alone was also without effect but the combination of C18 plus MSU crystals induced joint swelling, neutrophil infiltration and cytokines and chemokines. Both the IL-1β precursor and the processed form were elevated. Next, given this validated and clinically relevant in vivo model, mice deficient in three molecular pathways for IL-1β processing and secretion were evaluated. Not unexpectedly, mice deficient in caspase-1 or ASC exhibited markedly reduced synovial inflammation in response to the MSU-C18 combination, and in mice deficient in ASC, histological examination of the joints revealed near complete protection. However, mice deficient in NLRP3 responded with same inflammatory responses as did wild-type mice. Other studies have also observed an NLRP3-independent role of IL-1β processing (11). As discussed below, the role of NLR3 may require TLR4 for activation of caspase-1.
In freshly obtained human PBMC, MSU crystals clearly synergizes with LPS via the TLR4 for the production and processing of the IL-1β (6), demonstrating that MSU crystals requires a second signal via a TLR. Since saturated FFA are inflammatory, how does FFA provide the TLR signal for MSU-induced inflammation? FFA appears to signal via TLR2 and not TLR4. These findings are consistent with those of others in that TLR2 recognizes FFA’s as endogenous ligands (12). The role of FFA’s has also been studied in the context of type 2 diabetes. FFA induces IL-1β, IL-6 and IL-8 in insulin-producing human islets and IL-1β in mouse islets (13). Moreover, high glucose concentrations enhanced IL-1β-inducing properties of FFA-induced in human islets. Blocking the IL-1RI with the IL-1R antagonist (IL-1Ra) strongly inhibited FFA-mediated expression of IL-1β and chemokines in human and mouse islets. Similar to inflammation by MSU injected into the peritoneal cavity (3), the FFA-induced IL-1β and chemokine production was dependent on MyD88 (13).
In freshly isolated human blood monocytes, the rate limiting step in IL-1β processing and secretion is at the level of transcription/translation and not at the level of caspase-1. Caspase-1 is constitutively active in blood monocytes, even before the cells are cultured (14). As such once the IL-1β precursor is synthesized, processing and secretion takes place. On the other hand, in macrophages derived from monocytes, caspase-1 is not active and requires a signal from ATP. Thus, in tissue macrophages three signals are required, once for transcription and one for translation and one for activation of caspase-1, the latter being the role of the inflammasome. Endotoxins provides the first two signals in the blood monocyte as well as in the macrophage. It is not uncommon that a low concentration of LPS is used to “prime” cells before another stimulant (such as MSU) is added to induce IL-1β. But in the case of gout, LPS as a priming agent is clinically irrelevant. One can conclude from the Joosten study that MSU is the priming agent by inducing gene expression for IL-1β and fatty acids provides a uniquely clinically relevant translational signal (See Figure 1). A similar mechanism likely takes place in the production of IL-1β in type 2 diabetes. Here, high concentrations of glucose provide a priming signal and fatty acids the second signal.
Unexpectedly, the production of joint inflammation by the combination of MSU crystals plus C18 was not observed in NALP3 deficient mice. This finding is different from that reported by others and raises the issue of the presence of LPS in MSU preparations. Nevertheless, in mice deficient in ASC, there was an impressive reduction in MSU/FFA inflammation. In fact, the lack of inflammation in the ASC deficient mice was observed at the level of gene expression for IL-1β. One interpretation is that ASC is needed for an upstream effect on IL-1β gene expression and not just as the required co-factor for NALP3 activation of caspase-1. In mice deficient in caspase-1, a reduction in joint inflammation was reduced, although not to the extent as was observed in mice deficient in ASC. Thus the reduction in IL-1β gene expression in ASC deficient mice would explain the lack of inflammation more than a reduced activation of caspase-1.
Since neutrophils dominate the inflammation of gouty arthritis in humans, the role of the neutrophil needs to be considered. As shown in Figure 1, cell death of neutrophils provides a wealth of possibilities for inflammation. For the synovial macrophage, dead neutrophils provide a source of ATP and other small molecules for activating caspase-1. Neutrophils also provide a source proteinase-3, which can process the IL-1β precursor into an active cytokine (15). The gouty attack is likely triggered by over nutrition with FFA providing the second signal in MSU primed cells, followed by the secretion of active IL-1β, which in turn, induces IL-8 and the infiltration of neutrophils. Large numbers of neutrophils augment the inflammation by providing enzymes and ATP, which induces more active IL-1β. Overall, the studies by Joosten and colleagues go along way to clarify the mechanisms by which IL-1β is produced in flares of gouty arthritis and mechanisms that are thoroughly linked to endogenous signals with considerable clinical validity. Thus gout is likely an auto-inflammatory disease and like others, uniquely IL-1β mediated.
Footnotes
Supported by AI-15614 from the National Institutes of Health
References
- 1.Terkeltaub R, Sundy JS, Schumacher HR, Murphy F, Bookbinder S, Biedermann S, et al. The interleukin 1 inhibitor rilonacept in treatment of chronic gouty arthritis: results of a placebo-controlled, monosequence crossover, non-randomised, single-blind pilot study. Ann Rheum Dis. 2009;68(10):1613–7. doi: 10.1136/ard.2009.108936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fiehn C, Zeier M. Successful treatment of chronic tophaceous gout with infliximab (Remicade) Rheumatol Int. 2006;26(3):274–6. doi: 10.1007/s00296-005-0617-7. [DOI] [PubMed] [Google Scholar]
- 3.Chen CJ, Shi Y, Hearn A, Fitzgerald K, Golenbock D, Reed G, et al. MyD88-dependent IL-1 receptor signaling is essential for gouty inflammation stimulated by monosodium urate crystals. J Clin Invest. 2006;116(8):2262–71. doi: 10.1172/JCI28075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440(7081):237–41. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 5.Joosten LA, Netea MG, Myloni E, Koenders MI, Malireddi-Subbarao RK, Giamarellos-Bourboulis EJ, et al. Fatty acids engagement with TLR2 drive IL-1beta producion via inflammasome acitvation by urate crystals in gouty arthritis. Arthr Rheumat. 2010 in press. [Google Scholar]
- 6.Giamarellos-Bourboulis EJ, Mouktaroudi M, Bodar E, van der Ven J, Kullberg BJ, Netea MG, et al. Crystals of monosodium urate monohydrate enhance lipopolysaccharide-induced release of interleukin 1 beta by mononuclear cells through a caspase 1-mediated process. Ann Rheum Dis. 2009;68(2):273–8. doi: 10.1136/ard.2007.082222. [DOI] [PubMed] [Google Scholar]
- 7.Schindler R, Clark BD, Dinarello CA. Dissociation between interleukin-1β mRNA and protein synthesis in human peripheral blood mononuclear cells. J Biol Chem. 1990;265:10232–10237. [PubMed] [Google Scholar]
- 8.Schindler R, Gelfand JA, Dinarello CA. Recombinant C5a stimulates transcription rather than translation of IL-1 and TNF; cytokine synthesis induced by LPS, IL-1 or PMA. Blood. 1990;76:1631–1638. [PubMed] [Google Scholar]
- 9.Kaspar RL, Gehrke L. Peripheral blood mononuclear cells stimulated with C5a or lipopolysaccharide to synthesize equivalent levels of IL-1β mRNA show unequal IL-1β protein accumulation but similar polyribosome profiles. J Immunol. 1994;153:277–286. [PubMed] [Google Scholar]
- 10.Chapman PT, Yarwood H, Harrison AA, Stocker CJ, Jamar F, Gundel RH, et al. Endothelial activation in monosodium urate monohydrate crystal-induced inflammation: in vitro and in vivo studies on the roles of tumor necrosis factor alpha and interleukin-1. Arthritis Rheum. 1997;40(5):955–65. doi: 10.1002/art.1780400525. [DOI] [PubMed] [Google Scholar]
- 11.Kolly L, Karababa M, Joosten LA. Inflammatory role of ASC. 2009 doi: 10.4049/jimmunol.0802173. [DOI] [PubMed] [Google Scholar]
- 12.Nguyen MT, Favelyukis S, Nguyen AK, Reichart D, Scott PA, Jenn A, et al. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J Biol Chem. 2007;282(48):35279–92. doi: 10.1074/jbc.M706762200. [DOI] [PubMed] [Google Scholar]
- 13.Boni-Schnetzler M, Boller S, Debray S, Bouzakri K, Meier DT, Prazak R, et al. Free fatty acids induce a proinflammatory response in islets via the abundantly expressed interleukin-1 receptor I. Endocrinology. 2009;150(12):5218–29. doi: 10.1210/en.2009-0543. [DOI] [PubMed] [Google Scholar]
- 14.Netea MG, Nold-Petry CA, Nold MF, Joosten LA, Opitz B, van der Meer JH, et al. Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood. 2009;113(10):2324–35. doi: 10.1182/blood-2008-03-146720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Joosten LA, Netea MG, Fantuzzi G, Koenders MI, Helsen MM, Sparrer H, et al. Inflammatory arthritis in caspase 1 gene-deficient mice: Contribution of proteinase 3 to caspase 1-independent production of bioactive interleukin-1beta. Arthritis Rheum. 2009;60(12):3651–3662. doi: 10.1002/art.25006. [DOI] [PMC free article] [PubMed] [Google Scholar]