

Abstract
The identification and application of druggable pockets of targets play a key role in in silico drug design, which is a fundamental step in structure-based drug design. Herein, some recent progresses and developments of the computational analysis of pockets have been covered. Also, the pockets at the protein–protein interfaces (PPI) have been considered to further explore the pocket space for drug discovery. We have presented two case studies targeting the kinetic pockets generated by normal mode analysis and molecular dynamics method, respectively, in which we focus upon incorporating the pocket flexibility into the two-dimensional virtual screening with both affinity and specificity. We applied the specificity and affinity (SPA) score to quantitatively estimate affinity and evaluate specificity using the intrinsic specificity ratio (ISR) as a quantitative criterion. In one of two cases, we also included some applications of pockets located at the dimer interfaces to emphasize the role of PPI in drug discovery. This review will attempt to summarize the current status of this pocket issue and will present some prospective avenues of further inquiry.
Electronic supplementary material
The online version of this article (doi:10.1208/s12248-012-9426-6) contains supplementary material, which is available to authorized users.
KEY WORDS: computer-aided drug design, ISR, pocket, SPA
INTRODUCTION
The binding of a protein to many molecules occurring at different binding pockets of a protein’s surface represents its various biochemical functions. These diverse pockets of a protein are particularly useful for target-based drug discovery as the detection of these binding pockets is considered as a premise of structure-based drug design. The most important task of a drug designer is to search for small drug-like molecules blocking these pockets on particular proteins related to some diseases. Furthermore, the identification and characterization of these binding pockets is of critical importance to understand the nature of molecular recognition and to enable functional annotation for orphan proteins (1).
Here, we reviewed the recent advances and applications with respect to the study of binding pockets, including binding pocket identification, binding pocket characterization, binding pocket druggability prediction, comparing different binding pockets, as well as binding pocket flexibility. Due to the ever-increasing concerns of pockets at the protein–protein interface (PPI), we also highlighted recent progress in characterizing PPI in drug discovery. The pocket characterization issue has been a very active field of research over the past two decades. There are many excellent reviews to address the binding pockets problem (1–5). Here, we only reviewed some applications of computational analysis methods of binding pockets. Additionally, we ended this review with two case studies to illustrate the applications of some of current methods.
Pocket Identification
The identification of binding pockets is considered as an initial step for structure-based drug discovery, after which a more rigorous description of the pocket is sought (4). Intuitively, pockets are surface concavities of proteins where a substrate might bind, whereas the concept of “druggable” pockets refers to target proteins where small drug-like molecules have been shown to bind (6–10). Other descriptions/definitions of binding pockets include novel binding site centric chemical space (4), the establishment of relationships across different target class (11), static pockets, transient pockets, dynamic pockets (12,13), monomeric pockets, as well as multimeric interfacial pockets (14–16). In summary, our categorization, description, and understanding of binding pockets is quickly evolving and is paving the way to the development of novel therapeutics and improved treatment of human disease.
During the past two decades, along with advances in our descriptions of binding pockets, many methods have been developed to improve their detection and quantitative characterization, more than 30 different methods to date. They can be mostly classified into two categories: geometry-based and energy-based (for a detailed overview, see refs. 4,5).
Geometry-Based Methods
Binding pockets for ligands are usually clefts or cavities of a protein. Laskowski et al. (17) have found that the ligand is bound in the largest cleft in over 83% of the single-chain enzymes. Thus, in many cases, the likely binding sites of an enzyme can be identified solely according to geometrical criteria. The conclusion is the underlying basis of geometry-based methods, and many have subsequently been developed (for a recent review, see ref. 5).
Recently, Voss and Gerstein (18) have released the Voss Volume Voxelator (3V) web server, which can help researchers investigate the volumes of large macromolecules, especially the internal volumes. In contrast to CAVER (19), 3V requires no starting point. Furthermore, users can fine-tune the radius of the probe to adapt to the size of any structure and its channels. However, the CAVER method can be used to detect channels on molecular dynamics (MD) trajectories or conformational ensembles of a protein.
Huang and colleagues have further developed their previous work (20) by incorporating four more methods with improved predictive success. The new method MetaPocket 2.0 (21) exhibits a better performance over the previous version for the common datasets. Specifically, it correctly predicts >74% drug binding sites on protein targets from the newly constructed dataset at the top 3 prediction.
Energy-Based Methods
There is no rule without an exception and not all binding sites are the largest pockets or clefts of a protein. Energy-based methods of binding pocket identification have addressed this problem using changeable probes to detect the different binding pockets (5). There are some energy-based methods that have been developed to identify and characterize the binding pockets (for a recent review, see ref. 4).
Laurie and Jackson (22) have proposed an approach to identify the regions of a protein usually corresponding to binding sites according to the interaction energy between the protein and a simple van der Waals probe. Recently, Ghersi and Sanchez (5) have emphasized the advantages of energy-based methods over geometry-based ones. These energy-based methods can provide maximal flexibility to discriminate different types of binding sites using the various chemical probes. And the authors described the important characteristics of the combined tools (EasyMIFs and SiteHound) and provided case studies for use with these tools to illustrate binding site identification and characterization. Recently, Yingjie et al. (23) have released the SiteComp sever, which performs detailed ligand binding site analysis with respect to the contributions of individual residues with a pocket as well as the identification of subsites.
Schmidtke et al. (24) have compared the performance of energy-based methods with geometry-based ones. For the holo structures, all four algorithms including SiteFinder (25), fpocket (26), ICM-PocketFinder (27), and SiteMap (28) correctly identified around 95% of pockets, though they performed with varying success when presented with the apo structures.
In summary, geometry-based methods have some advantages over energy-based methods, such as their computational efficiency, their insensitivity to the input, as well as their computational tractability (24). However, geometry-based methods are not suitable to discriminate different types of binding sites, and these methods tend to fail where the largest pockets did not correspond to the binding sites. Undoubtedly, each approach has advantages and disadvantages, and the final decision on how to implement a method or which method to be applied depends on the purpose at hand.
Recently, other approaches to identifying binding pockets have been proposed. Fukunishi and Nakamura (29) have developed a new approach called MolSite to predict the binding sites of proteins by molecular docking. The MolSite method was applied to 89 bound and 20 unbound structures for testing and could correctly identify binding sites in 80–99% of the cases, when only the single top-ranked site was considered. Ngan et al. (30) have described a binding site identification method called FTSite. This algorithm does not require any evolutionary or statistical information but can correctly identify the binding sites in over 94% of apo proteins from commonly used datasets applied by other methods.
Pocket Characterization
Proteins interact with many molecules via the diverse binding pockets to fulfill their biological function. In order to understand the physicochemical principles underlying these interactions, a thorough analysis of the binding pockets should be a precondition for further study. In addition, shape and chemical complementarity are the determinant factors of molecular interaction and recognition. Then, following the first step in identifying and predicting binding pockets, the detailed analysis and characterization of these pockets will further contribute to understanding molecular recognition and designing optimal ligands with both high affinity and specificity. To date, many properties and descriptors of binding pockets have been developed and refined to characterize the pockets.
Accurately characterizing the binding pockets is the cornerstone of pocket analysis. But there are still no gold standards to delineate pockets of interest (4). Herein, we cover only some applications of some of the various factors of binding pockets rather than providing a broad overview of all available properties, and most properties of binding pockets have been reviewed in detail in Henrich et al. (3) and Pérot et al. (4).
Seco et al. (31) have incorporated hydrophobic and hydrophilic interactions into MD simulations by the mixture of isopropyl alcohol and water molecules. The authors can identify the likely binding sites of the protein through the modified MD simulations and find the desolvated regions. Due to the intrinsic property of the isopropyl alcohol probe containing hydrophilic and hydrophobic parts, this current approach can characterize the polar and nonpolar properties of molecules by estimating the maximal affinity. Søndergaard et al. (32) have made further predictions concerning the pKa shifts relating to ligand binding site residues and the corresponding ionizable ligand groups in the PROPKA3.1 according to the improved empirical rules, which are the extensions of the rules from the PROPKA3.0 software package (33). The authors have included the predictions of ligand pKa value in the context of multiligand complexes, with covalently coupled intraligand and noncovalently coupled interligand interactions. In addition, a novel algorithm has been proposed to identify the knotty pKa predictions concerning noncovalently coupled ionizable groups. The new version 3.1 of PROPKA will contribute to better delineating and characterizing the binding pockets according to the prediction of pKa property.
Pocket Druggability Prediction
The assessment and prediction of protein druggability has been a hot topic in the pharmaceutical community for the past decade. Due to the high attrition rate and failure rates in drug discovery and development (34), the pharmaceutical industry has an urgent need to prioritize suitable drug targets based on solid and validated criteria in early stages of drug discovery. Hopkins and Groom (9) made the first review on chemical tractability by defining the term “druggability” as the ability of modulating a protein related to a specific disease by small molecules. Although druggability is still a debatable concept (35), it is currently a hot topic in the review literature (1,3,4,9,36).
After the characterization of binding pockets of a typical druggable target using the diverse physicochemical and geometric descriptors or properties favoring binding with high affinity and specificity, the next step is to examine whether the identified pocket is druggable. By assessing these properties or descriptors in the context of the known druggable targets, the quantitative criteria of druggability will be trained out to build a model for further druggability predictions. Most approaches to predicting druggability conform to the previously mentioned flow (28,37–41). For example, Volkamer et al. (41) have developed a druggability metric called DoGSiteScorer considering both the global properties of pockets and local similarities shared between pockets. They found that the trained support vector machine model from global descriptors can correctly identify the druggability of a target of interest. But global properties are subject to binding site flexibility and the way pockets are defined. Therefore, they proposed a subpocket prediction approach taking local pocket properties into account in terms of a nearest neighbor search. Then, the authors further confirmed that DoGSiteScorer can provide qualitatively and quantitatively valuable data for druggability assessment according to global and local measures. Furthermore, they also found that size, shape, and hydrophobicity as the global pocket descriptors are indeed important to automatically predict druggability. Additionally, incorporating subpocket properties for the assessment of druggability is of particular importance to subpocket similarity detection. Recently, Perola and Herman (42) have developed a novel algorithm to discriminate the characterized binding pockets of protein targets. After a comparative analysis between the binding pockets of 60 targets, where approved drugs are bound, and a collection of 440 ligand binding pockets, the authors obtained a set of simple rules covering five key properties (volume, depth, enclosure, percentage of charged residues, and hydrophobicity) to assess the chemical tractability (druggability) of prospective targets. Furthermore, a preferred property space has been proposed based on the previously mentioned five properties. Different from some other methods (28,31,35,37,39,41,43) in pursuit of the specific measure of druggability, the derived “druggability rules” are simple and physically interpretable.
Currently, there are still no gold standards for characterizing binding pocket properties and what constitutes a pocket (4). Likewise, there is no unique definition of druggability and its measure (1). However, these did not hamper the applications of “druggability”. Palomo et al. (44) have applied the fpocket algorithm (39) to carry out an extensive search for druggable sites on glycogen synthase kinase 3 in pursuit of the allosteric potential binding sites for further drug discoveries.
Pocket Similarity
The hypothesis that homologous proteins typically have similar sequences, three-dimensional (3D) structures, and biological functions has promoted the elucidation of biochemical functions of newly characterized proteins. However, comparison of the overall protein sequences, folds, and structures tend to fail to address the problem. Proteins with dissimilar sequences or structures can also show similar biological functions. As a rule, not all residues on a protein’s surface are involved in molecular recognition; rather, binding occurs at the binding pockets (45). So, focusing on the comparison between local binding sites of a protein rather than global sequences, structures, and folds provides an extra opportunity to detect similarities of binding sites among remotely relative, even heterogeneous, proteins for biofunctional annotations.
Unearthing similarities between binding pockets should also facilitate a fuller understanding of the chemical basis for side effects. Several approaches and databases have been developed for the functional annotation of proteins and detection of similarity among binding pockets (see Table 2 in ref. 4). For example, the SitesBase database (46) can be used to study the functions of the protein and generalize the pharmacophores from the known small molecules. Furthermore, the database contains the precalculated similarities between protein–ligand binding sites for easy retrieval and the further classifications of binding sites.
As in the case of pockets druggability prediction, pocket similarity prediction methods also conform to a basic flow: identifying the binding pocket feature, scanning the similar pockets, and assessing them (1). These methods use various features to represent the binding pockets, from the intuitive atom, residue, surface dots, etc., to abstract structural or numerical descriptors of binding sites. Herein, we present only some representative methods and the databases to illustrate the applications for future drug discovery (for a detailed overview of methods, see ref. 1).
Yeturu and Chandra (47) have developed the PocketMatch program to compare the binding sites of proteins, which employs the lists of sorted distances to encode the shape and chemical nature of the sites of interest. The final similarity score for pairs of sites derives from these lists aligned by an incremental alignment approach. The sequence order–independent profile–profile alignment (SOIPPA) (48) translates the concrete 3D structures of proteins into the abstract 3D graphs encoding the geometric and evolutionary information of binding sites. These graph representations are then aligned using a sequence order–independent alignment method for binding site comparisons. Subsequently, this approach was continuously adopted for drug discovery (49–51).
Recently, Sael and Kihara (52) have developed the Patch-Surfer approach to predicting the binding of a ligand to a query protein. A binding pocket is considered to consist of various segmented surface patches represented by a set of properties, such as geometric shape, electrostatic potential, hydrophilicity or hydrophobicity, etc. Then, these properties of surface patches are described using the 3D Zernike descriptor. The following comparisons between two binding pockets are performed according to a modified weighted bipartite matching algorithm.
The databases generated along with the binding site comparison are expected to be useful for the annotation of protein functions and rapid drug screening against targets related to some diseases. And these databases can also provide valuable information that will help determine possible side effects of the drugs which can bind to multiple targets. In drug discovery, finding a potential chemical entity binding to a target of interest with high affinity and specificity is the core issue of drug design. An appropriate and traditional approach is to retrieve a set of related binding sites from homologous and nonhomologous targets. Currently, these binding sites can be easily and rapidly retrieved from the binding site databases (see Table 2 in ref. 4). Then, one can discriminate the differences of binding affinity modulated by ligands binding for high specificity.
Meslamani et al. (53) have released the sc-PDB database, which is an archive of binding sites with functional annotations based on the PDB database. It currently contains 3D structures of 9,877 protein–ligand complexes (entries) consisting of 3,034 different proteins and 5,339 different ligands in total. The selected complexes are chosen according to their corresponding properties, such as ligand molecular weight, chemical structure, buried surface area, etc. This database includes the comprehensive information for proteins, ligands, and binding modes of complexes. In addition, the sc-PDB can be used to perform classifications of targets by binding site similarity. This will facilitate ligand-based and structure-based drug design.
The PoSSuM database (54) houses a much larger number of known binding sites compared to SitesBase (46). Additionally, this database also contains potential ligand binding regions predicted by the GHECOM program (55). As of March 28, 2012, PoSSuM has stored 3,361,043 known and putative binding sites from the PDB database. These authors have developed an ultrafast method (56) based on molecular fingerprinting (57) to perform the exhaustive similarity search for over one million binding sites in a reasonable time frame. The ligand binding sites are first encoded as feature vectors according to their physicochemical and geometric properties. Then, a fast neighbor search method called SketchSort (58) is employed to enumerate similar pairs in a huge number of binding sites. Finally, over 24 million similar pairs of binding sites were discovered based on the current binding sites stored in this database.
Furthermore, Medvedeva et al. (59) have extended the scope of binding sites into the gene field and developed a new database, SitEx, which includes information on positions of functional site amino acid in the exon structure of the encoding eukaryotic genes. The authors have studied the relationship of the exon–intron structure of the genes to the protein functional sites by searching the sequential–structural similarity among polypeptides encoded by single exons. This resource incorporates projection of protein domains, functional sites, and exon boundaries on proteins and coding gene sequence. Currently, the database includes 9,994 functional sites from 2,021 different proteins.
Pocket Flexibility
Putting protein flexibility into structure-based drug design is a challenging task. Likewise, a vexing problem in pocket characterization is dealing with a protein’s inherent flexibility. How to account for the dynamic behavior of a protein in the pocket field is of utmost importance to pocket-based drug design. Although some bound complexes still satisfy the “lock and key” model and Luque and Freire (60) have revealed that the local regions with low structural stability and regions with high stability coexist in the binding sites, it is not always convincing to rely on a single rigid conformation of a protein for further study. Eyrisch and Helms (61) have found that the binding pockets can frequently last some lifetimes in the context of protein flexibility. However, these binding pockets have not been identified in the crystal structures of the unbound protein, whereas the native binding sites were clearly identified from the bound structures when the corresponding ligand was manually removed. Based on induced fit or conformational selection theory (62), the interplay between the protein and the ligand will significantly change the characteristics of the binding sites upon binding. Hence, the collections regarding the conformations of proteins will be useful for the analysis and prediction of protein binding pockets. These conformations can be derived from experimental technologies including crystallography and NMR or computational methods, e.g., MD, normal mode analysis (NMA), and graph-theoretical approaches (12,13).
Some valuable protocols (63,64) have developed in order to deal with the issue of flexibility. They usually generate conformational ensembles using MD and select some relevant conformations according to a set of criteria for further study using virtual screening or molecular docking. These methodologies are useful for capturing transient binding pockets or transient potential binding sites. Eyrisch and Helms (61) have proposed the EPOSBP approach to investigating the flexibility of pockets, which identifies the transient pockets on a sequence of MD frames using the geometry-based PASS pocket detection algorithm (65) and clusters these pockets using the generated pocket-lining atoms (PLA) and tracks their dynamic behaviors in a conformational ensemble. Recently, Schmidtke et al. (66) have released a free open source tool called MDpocket, allowing easy extraction of binding pockets and gas migration channels from conformational ensembles extracted from MD trajectories. Compared with EPOSBP, MDpocket represents the pockets using a continuous descriptor rather than splitting them into subpockets according to clustering rules. However, the most important drawback of MDpocket is the initial superimposition. Such a superimposition frequently results in errors on conformational ensembles. Thus, local structural alignment is more acceptable when using the MDpocket package. Craig et al. (67) have also reported a novel approach called PocketAnalyzerPCA; for the selection of the representative pockets, the authors tried to address the problem of how to prune conformation ensembles of a protein down to a subset with the substantially distinct binding pockets. Remarkably, Kufareva et al. (68) have successfully constructed the Pocketome database, which is regarded as an encyclopedia concerning the experimental conformational ensembles of druggable binding sites. The Pocketome includes a detailed classification of the binding pockets within each conformational ensemble. The current release of the Pocketome encyclopedia (August 2011) contains 988 entries. This comprehensive resource will tremendously facilitate progress in the fields of binding pocket characterization, molecular dockings, as well as virtual screenings.
Pockets at the PPI
PPI are becoming fascinating and promising targets as a consequence of their pivotal role in a large number of biological pathways and networks related to diseases. Drug discovery targeting PPI has become a hot topic in the scientific and industrial communities (69). However, compared with proteins, the applicability of PPI to drug design is hindered by the intrinsic properties of protein–protein interaction interfaces. The binding site of a protein often corresponds to the large or deep pocket on the protein surface, whereas most PPIs are intrinsically disordered and shallow, and these PPIs are mostly not continuous regions consisting of multiple “hot spots”. Although the identification of these noncontiguous “hot spots” will significantly contribute to the understanding of protein–protein interactions, those insignificant-seeming non-hot spots may be associated with specificity. Furthermore, another hurdle is the more extensive range of structural flexibility of PPIs relative to single protein binding pockets. Some reviews have provided detailed insights into the PPI (4,14,70–72). Herein, we only give a brief overview of recent efforts directed towards PPIs for drug discovery.
Recently, Gao and Skolnick (16) have reported the distribution of ligand binding pockets at PPI, assisting in understanding the underlying interplay between PPI and protein–ligand binding pockets. The authors scanned 1,611 representative protein–protein complexes to detect the potential ligand binding pockets. The results show that these identified ligand binding pockets are mostly located within 6 Å of PPI. Ligands are spatially closer to the PPI compared with a random surface patch from the same solvent-accessible surface area. The authors further investigated the ligand distribution around domain–domain interfaces in 1,416 nonredundant representative two-domain protein structures and obtained similar results. This study gives a key underlying formation mechanism of the ligand binding pocket, which lies in the packing surrounding PPI or domain–domain interfaces. This work provides a clue to the detection and identification of ligand binding pockets. Mysinger et al. (73) have performed comparative virtual screening against the homology model and the newly released crystal structure of CXCR4, a typical PPI target, for novel PPI inhibitors, respectively. The different performances of two virtual screenings demonstrate the significant influence of structures on structure-based drug design. The results show that only one antagonist with low specificity and high similarity with known ligands was identified using the homology model, whereas four compounds with novel scaffolds and high specificity were discovered based on the crystal structure. This further confirmed that the crystal structure as a starting point for structure-based drug design is more reliable and useful for PPI discovery. And among the four novel compounds, one compound with 306 nM has a ligand efficiency of 0.36. Thus, it rationalizes structure-based attempts to discover leads for chemokine G protein-coupled receptors. Gautier et al. (74) have performed virtual screening against the vascular endothelial growth factor receptor (VEGFR) D2 domain, which has a very flat PPI and is a promising target for antiangiogenic treatments. The authors have identified 20 active compounds that each has an IC50 in the micromolar range and a common thiophene unit. Further investigation revealed that the most active compound can effectively inhibit the VEGF-induced VEGFR-1 transduction pathways. The findings suggest that this optimal hit may provide a promising chemical scaffold constituting a potent probe against cancer and other VEGFR-1-dependent diseases; these results support structure-based PPI drug discovery efforts even for very flat interfaces.
Case Study: Targeting the Binding Pockets on Ras Protein Surface and Interfaces for Lead Generation
The release of the complete 3D crystal structure of H-Ras “state 1” with two stable surface pockets (75) provides new insights into the underlying mechanism for the state transition of the Ras protein and facilitates structure-based drug design. The structural determination of the complexes (1wq1:Ras-GAPs (76); 1nvw:Ras-GEFs (77); 1gua:Ras-Raf (78); 1he8:Ras-PI3K (79); 1lfd:Ras-RalGDS (80)) also support the development of PPI inhibitors targeting their interfaces. In our previous study, we had performed some investigations regarding the Ras protein using computer-aided drug design (CADD), which focused on one Ras-GTP intermediate state (a transient surface pocket of the H-Ras protein) for the potential inhibitors (81). In this review, we further investigated the Ras-GTP “inactive” state 1 targeting a series of transient states (surface pockets) of H-Ras protein through the ensemble-based virtual screening method for potential inhibitors. Furthermore, due to Ras protein’s key role in the signaling pathway, modulating its biological functions by blocking pockets at the interfaces will aid in finding PPI inhibitors. Thus, we have also performed a series of virtual screenings against these pockets. We have presented the corresponding schemes of two case studies in this review (Fig. 1).
Fig. 1.
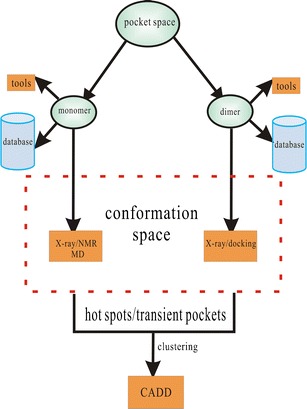
The scheme of two case studies in this review
Case Study: Targeting the Potential Allosteric Pockets on Src Kinase for Lead Generation
The notorious flexibility of kinase enormously hampers the development of structure-based drug design against the whole kinase family. Currently, the ensemble-based virtual screening technology taking the conformation changes into account was proposed to attempt to address the problem of protein flexibility (63,64). Shan et al. (82) have performed comprehensive MD to capture the process of ligands binding to the native binding site of Src kinase, and a potential allosteric site was found during the process of binding. The discovery of this novel allosteric site opens up an exciting new avenue for the discovery of novel Src kinase inhibitors. Furthermore, information concerning the dynamics of the ATP-binding site and the allosteric site gives us a clue to probe into the problem of pocket flexibility. With the aim of identifying multitarget or multipocket drugs, we employed two ensemble-based virtual screenings to find hits against the ATP-binding site and the allosteric site concurrently.
METHODS
Construction of the Focused Library for Ras and Src Kinase Protein
For the Ras protein, compounds in the ZINC (83) database using the Tanimoto score of 0.8 as a threshold for the potential inhibitor (81) were selected, and de novo design based on the scaffold of the potential inhibitor (see Fig. 6 in ref. 81) has been carried out using the autogrow package (84) according to the default parameters to find its derivatives. The ADME/Tox Filtering was carried out via the online FAF-Drugs2 tool (85). Finally, we have obtained 90 chemical entities for the subsequent screenings. Under the same protocol as the Ras system, for the native binding site and the potential allosteric binding site (one pocket labeled “a” in ref. 82), we have also performed de novo design using the autogrow package to obtain the PP1 derivatives. We also got the chemical compounds similar to the PP1 according to the same criterion as Ras protein. The 25 chemical entities have been chosen for further calculations.
Generation of Conformational Ensembles for the Flexible Pockets
In the Ras protein case study, we applied the EN.NMA approach developed by Rueda et al. (86) to generate conformational ensembles. The method is simple to use without any a priori knowledge concerning the structures of interest. Additionally, this method is fast and generally represents the equilibrium dynamics of various structures without any further refinements. The crystal structure of the H-RAS (1XCM (87)) was used as a template. One hundred frames were obtained for the following pocket analysis.
For Src kinase, MD is more suitable to the Src kinase system possessing apparently tremendous conformational changes. The MD trajectories of the Src–PP1 system have been obtained from Shaw’s group (82), for which they have applied the all-atom model MD to capture the binding process of PP1 binding to the ATP-binding site. In this case study, we have chosen two types of conformations (according to Fig. 2A in ref. 82): (1) where the PP1 has been bound to the ATP-binding site steadily and (2) where the PP1 was located in the predicted allosteric site.
Flexible Pocket Analysis
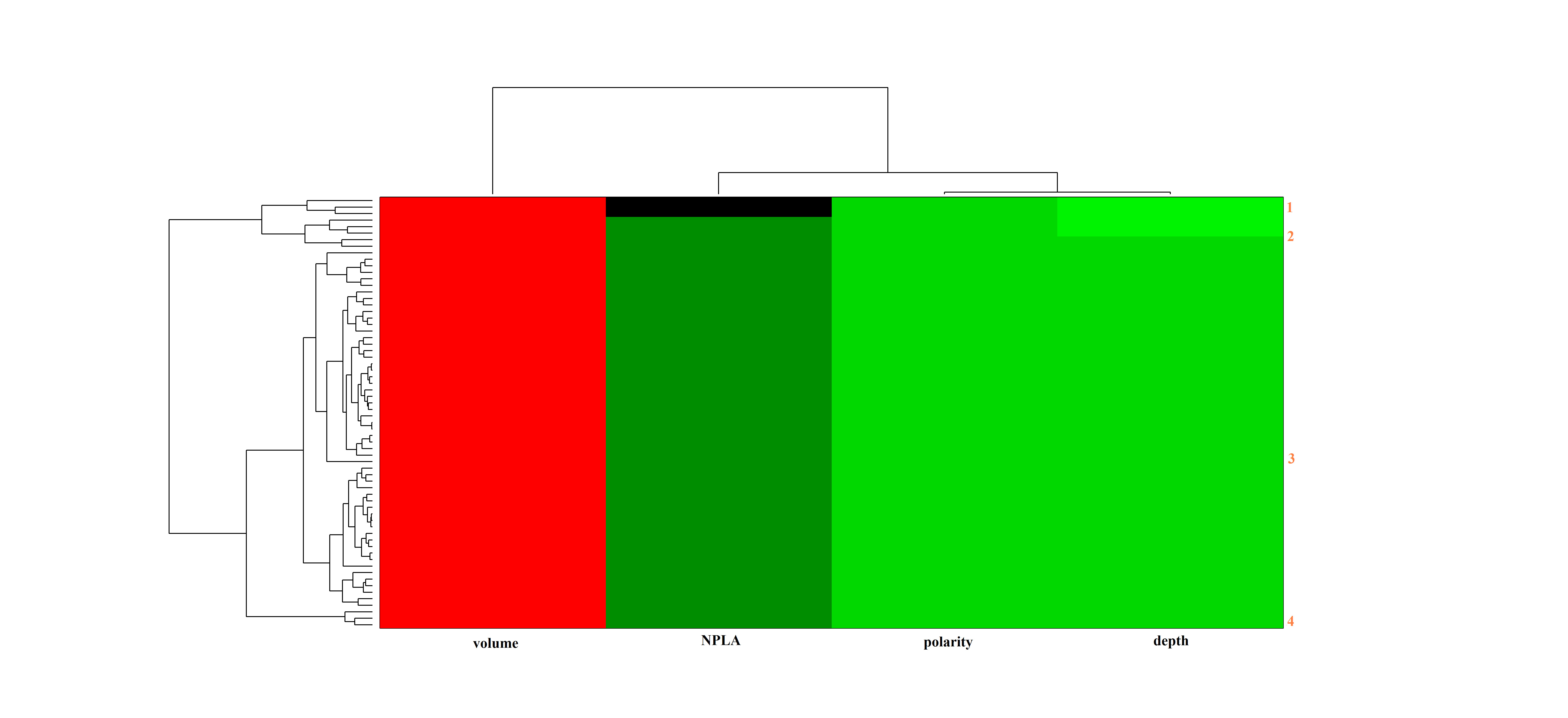
As previously mentioned, we have reviewed some packages for the analysis and detection of the transient pockets on static structures or conformational ensembles of a protein. Here, we applied the EPOSBP method to complete the task, some geometric and physicochemical pocket properties (volume, polarity, and depth) are calculated for each conformation. Two output files, the “patch file” and the “pocket-lining atom (PLAs)” are then generated, the former is used to calculate the pocket volume and identify the PLAs and the classification of the binding pocket of each conformation are performed based on the latter (PLAs). The resultant analysis result will contain the information concerning the properties and clusters of binding pocket in the conformation ensembles. Instead of clustering by conformation, we have carried out the clustering by pocket. Hierarchical clustering of the specific pocket ensembles based on the corresponding properties of binding pockets, such as volume and depth, was performed using MATLAB’s Clustergram algorithm (88,89). Thus, we can achieve the aim of reducing the conformational ensembles into a subset according to the pockets, which contain the representative pockets for the subsequent calculations.
Ensemble-Based Virtual Screenings for the Kinetic Pockets of Ras and Src Kinase Protein
Next, we have performed a series of virtual screenings against the transient pockets located between switch I and GTP in the Ras protein extracted from the obtained conformational ensembles. The GNP and the cofactor Mg2+ were retained for the screenings; and the water molecules interacting with the magnesium ion were also retained. The other cofactors, water and sugars, were discarded. The side chains and termini protons were assigned using the corresponding protonation states at pH 7.0 as a template. The initial preparation regarding the conformations and the cofactors retained is determined by the AutoDock Tools (90). All docking calculations were performed with the AutoDock package (91) to sample the conformation space of the ligands for further specificity and affinity (SPA) score and intrinsic specificity ratio (ISR) calculations. The compounds from the focused library were docked into each of the four representative pockets of the ensembles generated by simulation tools.
The Autogrid, with 60 × 60 × 60 grid size and value of 0.375 Å spacing centered on the special position in the binding pocket, was prepared using the AutoDock tools (90). Dockings were performed based on the empirical free energy function (the AutoDock4.2 score function) and Lamarckian genetic algorithm for sampling. Molecular modeling was carried out based on the following parameters: the population size of 100 and the energy evaluation of 100,000 per run with the maximum number of generations of 27,000. The other parameters were set at default values. The number of docking runs was 1,000. The docking results were evaluated according to the predicted binding energies. And the subsequent cluster analysis based on the root mean square deviation (RMSD) value of <2.0 Å was carried out. The same protocols have been carried out on the Src kinase system targeting the native site and the potential allosteric site.
PPI-Based Virtual Screening for the Pockets at PPI for the Ras Protein
In order to discover the potential PPI inhibitors, the PPI involving SOS and GAPs as well as the primary downstream factors Raf, RalGDS, and PI3K were chosen to construct the target library. Fuller et al. (14) indicate that the methods of binding pockets identification will be a promising tool for drug design and discovery targeting the PPI in the context of known structures. So, the potential pockets at the interfaces involving five proteins have been detected by MetaPocket (20,21), which have currently included eight different pocket detection methods for higher accuracy (see the “Pocket Identification” section), with the centers of the pockets predicted by the different methods shown as colored spheres. The series of virtual screenings have been performed under the same protocols as the ensemble-based virtual screening part, making interactions between the different binding pockets located on the interface regions and the focused chemical library constructed previously. The downstream effectors (RalGDS, PI3K, and Raf) interact with the conserved region of the Ras protein through the Ras binding domain. However, the interactions involving GAP and SOS occur at different regions of the Ras protein surface.
Rescoring and Specificity Evaluation for Hits
Although affinity has been quantified and studied intensively, the quantification of specificity is far less addressed. We developed a novel scoring function called SPA to quantify specificity in addition to affinity (92). Each sampled docking pose was rescored with the SPA scoring function to predict the corresponding affinity and the native structure pose discriminating against others. SPA is based on our energy landscape theory of biomolecular recognition, in particular, docking, with the aim of simultaneously optimizing the ISR and the affinity predicted.
Conventional definition of specificity is the discrimination of a ligand against all available receptors. In other words, in order to judge whether or not a compound is specific to a receptor target, one has to explore the whole universe of receptors in order to see whether the affinity of this ligand–receptor pair has a discrimination against all the others. This is impossible to realize in practice since not all the receptor proteins are known and not all the known ones have determined structures. Our approach is based on a thought experiment (see Fig. 2). Imagine we connect all the receptors by linkers (for example, connecting the N terminus of a protein and the C terminus of another by glycines), and then the whole universe of receptors now becomes one giant protein. The discrimination of ligand binding to a specific receptor against the rest of the others becomes the discrimination of ligand binding to a specific (native) pocket or binding site against the rest of the other binding sites. Therefore, under the assumption that the receptor protein is large enough, specificity can be quantified by discrimination of the affinity of the native pose against the rest of the other non-native binding poses of this ligand binding to this receptor. We term this specificity as intrinsic specificity to differentiate this specificity from the conventional definition of specificity. We see under the assumption of receptor protein being large, the two definitions are equivalent. The quantitative issue is how large the protein should be in order to see the approximate equivalence. Since the protein folds have been estimated to be on the order of a thousand, the actual number of interactions of the ligand with the receptors is finite. In other words, one does not have to go through the whole universe of proteins to quantify specificity. Or in other words, a large but finite size protein may already contain most of the interactions encountered for ligand binding. Obviously, searching for all the binding sites or poses of a particular finite size protein for quantifying specificity is far easier than searching for the whole universe of the receptors for quantifying specificity in the conventional way. For this, we have tested the ligand binding with Cox2 enzyme receptors. The preliminary results show the strong correlation between conventional specificity and intrinsic specificity.
Fig. 2.
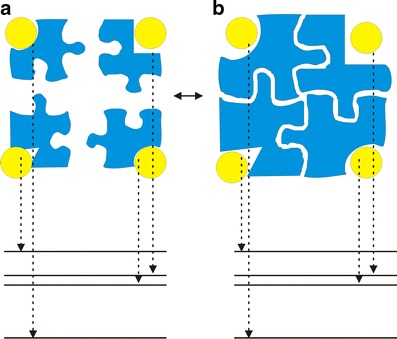
Illustration of the relationship between intrinsic specificity and conventional specificity as well as the corresponding energy spectrum. a The conventional definition of specificity is the difference(s) or discrimination(s) in affinity of the target receptor against other receptors binding to the same ligand; b the definition of intrinsic specificity is the difference(s) or discrimination(s) in binding energies of native (lowest) binding mode or site against other non-native binding modes (pockets) for a ligand binding to a receptor. The giant receptor here can be considered as the combination of many smaller receptors connected by certain linkers. When the giant receptor is large enough to cover all the possible ligand–protein interactions, the definition of intrinsic specificity is equivalent to the definition of conventional specificity. The receptors are colored blue; the yellow ball represents the ligand
Intrinsic specificity can be quantified by a dimensionless ratio, called ISR. It was defined as 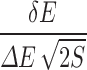 . According to the energy landscape theory, the conformation or the pose of the ligand–receptor complex with the lowest binding energy is considered as the native one (in practice, there will be an ensemble of native binding states) and the binding energies of the rest of the non-native conformations or binding modes are statistically distributed. Then, δE is defined as the energy gap between the conformation of the native binding complex and the average of the non-native ones, ΔE is the energy dispersion or the square root of the variance of the non-native conformations, and S corresponds to the configurational entropy. A large ISR corresponds to a high discrimination of the native conformation against the others, therefore a high intrinsic specificity. As mentioned, we have demonstrated that the intrinsic specificity correlates with the conventional specificity. Therefore, ISR provides a quantitative measure of specificity independent of the prior knowledge regarding all the other receptors (the receptor universe) against a specific drug.
. According to the energy landscape theory, the conformation or the pose of the ligand–receptor complex with the lowest binding energy is considered as the native one (in practice, there will be an ensemble of native binding states) and the binding energies of the rest of the non-native conformations or binding modes are statistically distributed. Then, δE is defined as the energy gap between the conformation of the native binding complex and the average of the non-native ones, ΔE is the energy dispersion or the square root of the variance of the non-native conformations, and S corresponds to the configurational entropy. A large ISR corresponds to a high discrimination of the native conformation against the others, therefore a high intrinsic specificity. As mentioned, we have demonstrated that the intrinsic specificity correlates with the conventional specificity. Therefore, ISR provides a quantitative measure of specificity independent of the prior knowledge regarding all the other receptors (the receptor universe) against a specific drug.
The SPA scoring function developed by us based on the intrinsic specificity previously discussed shows the best performance against 16 other popular scoring functions on both affinity prediction and the ability to reproduce the X-ray crystal pose. We demonstrated that it is possible to classify the small molecule compounds into the marked drugs and the random small molecule library, even into the selective drugs and the nonselective ones only using the SPA score function alone. Thus, the SPA can be employed as a new score function for lead compound discovery. Then, the compounds ranked as the top 5 with SPA were selected out to build a hit library for each binding pocket. Filtering for specificity by ISR (93,94) was carried out for screening the number of hits for further testing. We are applying SPA with the quantification of both affinity and specificity to several other targets for uncovering the lead compounds.
RESULTS
For the Ras protein, firstly, the top5 results were selected out for the individual virtual screening. Our aim is to find the multitarget drugs affecting multiple relevant targets in a parallel fashion. In this case, the multiple targets include the Ras protein and the relevant proteins along the signaling pathway and biological process related to Ras. Thus, we have obtained two multitarget hits (Fig. S1) against the transient binding pockets of multiple conformations of the Ras protein and the five predicted binding pockets at the interfaces involving SOS, GAPs, and downstream effectors, namely, PI3K, RalGDS, and Raf, with high affinity and specificity. Herein, we have rescored the decoys generated by the AutoDock package using the SPA score for affinity and evaluated specificity by ISR. For the Src kinase system, according to the same protocol as the Ras protein, two hits (Fig. S2) have been discovered simultaneously targeting the ATP-binding site and the allosteric site predicted by the MD simulation. The experimental validations of these potential hits presented herein are currently underway.
DISCUSSION
Targeting the Kinetic Pockets on the Ras Protein for Lead Generation
Conformational changes in proteins modulated by ligands are a very common phenomenon; obviously, it can impact the characterizations of binding pockets. Handling pocket flexibility resulting from the conformational changes of a protein remains a formidable challenge (see the “Pocket Flexibility” section). Currently, a number of methods can be used to generate these diverse conformations of a protein, such as MD and NMA. In this case study, we have applied the NMA method to generate conformations of the loop, in which the surface pocket of interest is located (Fig. 3). In conformational selection theory (62), for the energy landscape explored by the unbound protein, the majority of conformations occupy the lowest energy states, except for the minor higher-energy states. Then, the various conformations are screened out by various binding partners among the conformational ensembles to fulfill the biological functions, whose binding sites undergo the corresponding changes to fit these partners. For the activated Ras protein with GTP binding, the NMR structures (95) without the corresponding protein partner did not present the low-frequency transient pocket (the potential intermediate bound state (87)); this experimental information further confirms this theory. So in order to sample the low-frequency neighboring states similar to the intermediate, maintaining this open conformation of the loop will largely reproduce the bound state. Although Eyrisch and Helms (96) have concluded that the conformations generated by NMA methods had the smaller pockets compared with MD simulations, the NMA method, which can generate much smaller RMSD from the experimental structures than the MD methods, is more suitable for this system. The calculated RMSD values based on the NMA conformations were nearly constant (Fig. 3), which enables the binding pocket to be available to biological functions. On the contrary, as a result of the enhanced conformational sampling of the tCONCOORD (97), the method generating the pockets comparable to those observed in MD methods is not suitable for this system, it generated many different conformations of this loop from the NMR or crystal structures frequently circumventing the intermediate state (data have not been presented).
Fig. 3.
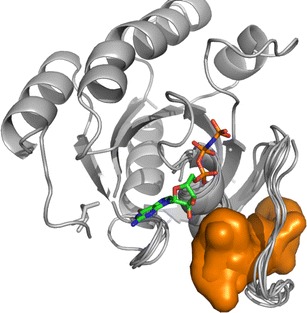
Potential ligand binding sites identified on the Ras conformational ensembles. The site is colored orange, and the GTP is shown as ball and stick model
Additionally, selection of the relevant conformations among a pool of conformations suitable for the subsequent virtual screening is another complication. Cavasotto et al. (98) have found that the large number of normal modes for generating the conformations is debatable. However, if the normal modes only involving the local regions, such as pockets, are applied, the NMA method is acceptable with the least number of normal modes. Furthermore, Sperandio et al. (64) have also proposed a new protocol to attempt to address the problem of generation and selection of conformations. In this protocol, the authors chose the relevant conformation according to the properties of global conformations and local binding pockets. So, in the case study, we employed the EN.NMA method to generate the conformations in the context of the binding pocket, which are structurally related to the crystal conformation (Fig. 3). Next, we explored the characteristics of binding pockets to cluster the pockets of conformations to select out the representative conformations for the virtual screenings (Fig. 4). This step was carried out using the EPOSBP method, which performs the detection of binding pockets in each conformation of the conformational ensembles by the program PASS (65) and calculates the properties of binding pockets, such as volume, polarity, and depth, for clustering. As previously mentioned, we have primarily introduced three methods attempting to address problems of the dynamical behavior of binding pockets, including the EPOSBP, PocketAnalyzerPCA, and MDpocket. Among these methods, only EPOSBP maps the binding pockets to a set of concrete PLAs. However, the identified pockets with the other two methods are mapped to a grid representation. MDpocket can detect the diverse transient binding pockets, such as druggable pockets and big external pockets; however, according to the various sets of parameters, to obtain the 3D structures of the conformations containing the transient binding pockets of interest corresponding to the calculated grid points, further conversions are needed but inconvenient. EPOSBP is enough to deal with specific pockets, though it does not use a continuous pocket representation. Likewise, an advantage of PocketAnalyzerPCA compared with EPOSBP is that it directly applies the pocket shape descriptors rather than a set of atomic coordinates to represent the pockets. It can avoid some common problems arising from the methods based on the atomic coordinates, but using the outputs of PocketAnalyzerPCA as the starting points of structure-based drug design also needs the technically detailed explanations. So in the case studies, we applied the EPOSBP to perform the pocket analysis. Considering few properties tracked in the EPOSBP, we added the number of pocket-lining atoms (NPLA) as a rough descriptor for the pocket size. The analysis and clustering allowed us to choose four different binding pockets, which were shown in Fig. 5, respectively. The four pockets as the starting points provide the structural information for the subsequent virtual screening.
Fig. 4.
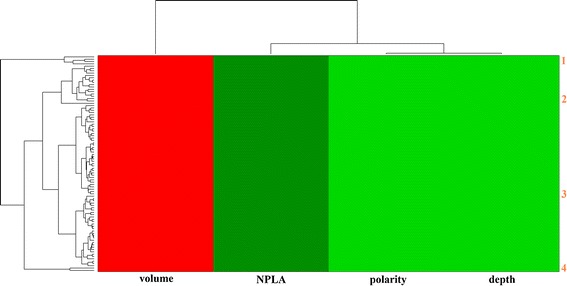
The heat map clustering of the binding pocket on the Ras protein. The horizontal axis labels are colored by properties of pockets (red for volume, green for depth, polarity, and NPLA; for further details, see the “Methods” section). Major pockets are indicated by the orange labels and corresponding marginal dendrograms
Fig. 5.
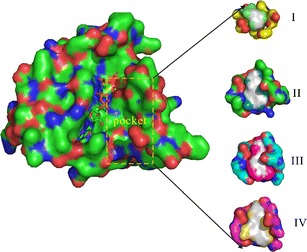
The binding pockets of four representative conformations of the Ras protein. The negative image of the pockets formed by the probes and the PLAs are shown as colored surface on the right; the Ras protein is shown as surface model containing the GTP (ball and stick model) and the cofactor Mg2+ (green dot) as well as the water molecules interacting with the magnesium ion (red dot) on the left. The orange rectangle indicates the positioning of these binding pockets on the Ras protein
The focused library we built was docked against these identified pockets in ensemble-based virtual screening using the AutoDock package (91). All compounds were able to bind to all four pockets. Then rescoring was performed to re-rank the chemical compounds using SPA. Herein, one thing to point out is that we applied SPA (92) based on the energy landscape for the following re-scorings. Many score functions have been developed in the past two decades (for a detailed overview of the comparisons for score functions beyond the scope of this review, see ref. 81). The compounds ranked in the top 5 were selected for the evaluation of specificity to further reduce the hits’ size.
Targeting the Pockets at PPI for Ras Protein for Multitarget Leads
Protein–protein interactions play a key role in drug discovery, and the study concerning the underlying mechanism of their modulation by ligands remains challenging. The detection and characterization of binding sites located on the PPI is of upmost importance in structure-based drug design. The methods of identifying these binding pockets can provide valuable information for subsequent drug design efforts, such as molecular docking method and virtual screening technique. In the case study related to the Ras protein, we have covered the PPI-based virtual screenings against the pockets at the different interfaces involving the key players in the Ras-related pathway and Ras cycling (Fig. 6). Some pockets were detected using MetaPocket (20,21) and were shown in Fig. 6 as colored spheres, with the pockets located at the interfaces surrounded by green circles. Herein, it should be stressed that the MetaPocket (here, MetaPocket 2.0) method was applied to detect the pockets at the interfaces with a twofold purpose. First, it is a simple online tool to identify pockets and only needs the PDB structures or PDB IDs as inputs. It outputs the standard PDB files and can be directly downloaded. The computational performance of MetaPocket is fast (10 to 30 s), even if all predictions are from eight different prediction methods. Second, its pocket prediction performance is good enough (see the “Pocket Identification” section), allowing us to consider it as the first choice. MetaPocket 2.0 outperforms the previous MetaPocket 1.0 and eight individual prediction methods (for detailed data, see Table 1 or 2 of ref. 21).
Fig. 6.
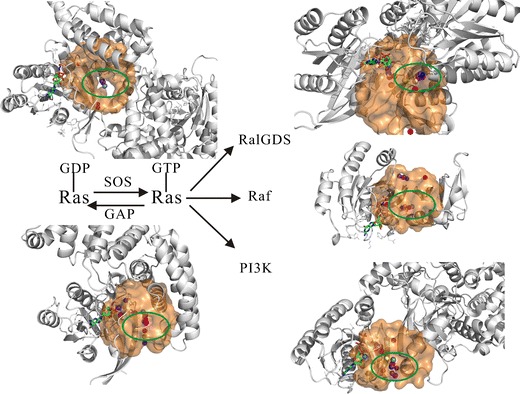
The binding pockets at the interfaces of Ras proteins with SOS, GAPs, RalGDS, Raf, and PI3K, respectively. The cycling between the active and inactive states of the Ras protein controlled with GEF (SOS) and GAP proteins is described, then the complexes containing five proteins previously mentioned are shown as a cartoon model (grey) and placed at the corresponding positions. The GTP on the Ras protein as an indicator is shown as ball and stick model, the centers of predicted pockets are labeled using the colored spheres (for further details, see the “Methods” section), the centers of predicted pockets at the interfaces are highlighted (green circle)
The focused library we built was also docked against these identified pockets in PPI-based virtual screening using the AutoDock package (91). Due to the intrinsic properties of PPIs (see the “Pockets at the PPI” section), these compounds bind to the different binding pockets in each of the PPIs (Fig. S3) with different binding modes, but the docking poses are all within a 6Å distance from protein interfaces. This is in line with the conclusion in ref. 16.
With the advent of systems biology, a new way of looking at drugs in the form of holism is blooming and pathway-based and network-based drug discovery has become the mainstream. Drug design efforts focusing simultaneously on several targets related to a pathway or networks for multitarget drugs are attracting more and more the attention of big pharma. In the case study related to Ras, with the aim of finding multitarget leads with high affinity and specificity, we employed ensemble-based virtual screening and PPI-based virtual screening against the kinetic pockets of Ras and the static pockets at the PPIs, respectively. Finally, we have theoretically identified two compounds, which are expected to modulate the interconverting conformations between “inactive” state 1 and “active” state 2 of activated Ras and to disrupt the interfaces involving the key players in the Ras-related pathway and Ras cycling (Fig. 6).
Targeting the Kinetic Pockets of ATP-Binding and Allosteric Binding Sites of Src Kinase for Multitarget Leads
The identification of putative allosteric sites is still a major challenge, though steady progress in this field is paving the way for drug discovery. Allosteric sites may provide an alternative way to disrupt protein function, especially in cases where the active sites of related proteins are almost identical, such as within the kinase family, which is highly conserved at the ATP-binding site. This potential allosteric site presents a promising opportunity for further drug design efforts. Shan et al. (82) have carried out MD simulations to capture the process of ligand binding to the target in great detail. We have obtained the corresponding trajectories for further study. As mentioned in the Ras case, MD can be used to generate the conformational ensembles. In this case study, we chose the corresponding frames for constructing the focused conformational ensembles to address the allosteric sites flexibility problem. The putative allosteric site and ATP-binding site are shown in Fig. 7. Compared with the allosteric site, the ATP-binding site is deeper. We have also selected out the four representative pockets concerning the ATP-binding site and the allosteric site using the same protocol as the one in the Ras case study, respectively (Figs. S4 and S5; Figs. 8 and 9).
Fig. 7.
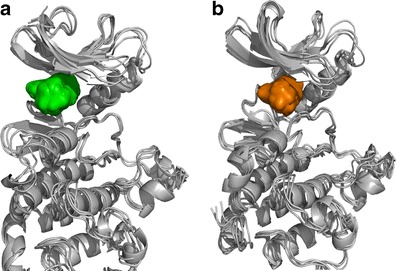
Potential ligand binding sites identified on the Src kinase conformational ensembles. The allosteric site is colored green, and the native ATP-binding site is colored orange. From the structural point of view, the allosteric site can be considered as a periphery of the ATP-binding site
Fig. 8.
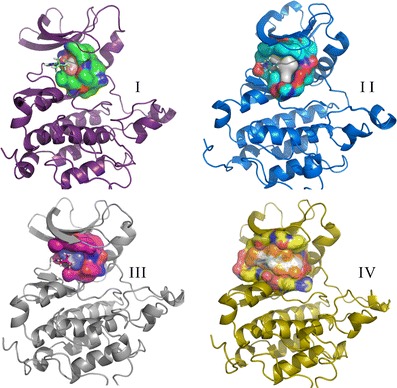
The ATP-binding sites of four representative conformations of the Src kinase. The Src protein is shown as a cartoon model, the negative image of the pockets and PLAs are shown as colored surfaces, and the corresponding PP1 is shown as a ball and stick model
Fig. 9.
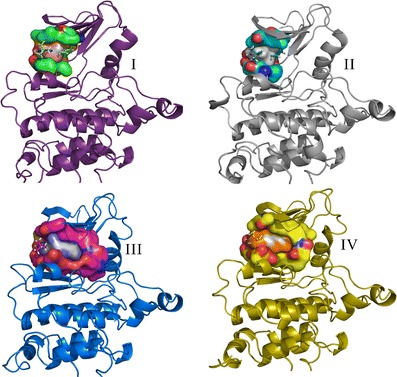
The allosteric sites of four representative conformations of the Src kinase. The Src protein is shown as a cartoon model, the negative image of the pockets and PLAs are shown as colored surfaces, and the corresponding PP1 is shown as a ball and stick model
The focused library we built for the Src kinase was docked against the ATP-binding site and the predicted allosteric site in ensemble-based virtual screening using the AutoDock package (91), respectively. All compounds target the same pockets in each of the binding sites. Actually, the allosteric site is a neighboring pocket of the ATP-binding site, which can be considered as the ATP-binding site peripheral regions. Thus, it facilitates further chemical modifications (such as fragment growing) based on the discovered leads targeting the ATP-binding site or this allosteric site (Fig. 7).
In the case study related to Src kinase, along the same line as the Ras case, we employed ensemble-based virtual screening against the kinetic ATP-binding site and the allosteric site of Src kinase, respectively, aiming to find the hits that can bind to two pockets concurrently, with high affinity and specificity. Finally, two compounds were initially identified by targeting the allosteric site and the ATP-binding site of the Src kinase protein (Fig. S6).
It is worthwhile to point out that targeting the PPI to modulate the biochemical process is also an application of allostery to some extent, so the pockets at the PPI may provide us a diverse and potential source for finding the allosteric pockets.
CONCLUSIONS AND FUTURE PERSPECTIVES
The binding pocket issue is a hot field of research with immense potential. In this review, we have attempted to cover the key efforts regarding the characterization of binding pockets applied to CADD, such as the identification of binding pockets, the comparison of binding pockets, the characterization of binding pockets, the druggability prediction of binding pockets, as well as to pocket flexibility. Functional annotation and identification of binding pockets are of great importance in the prediction of unknown protein functions and in drug discovery. The detailed analysis of binding pockets can give us the insight needed to optimize leads for higher affinity and specificity. Comprehensive understanding of the pocket space will also be useful to identify and avoid serious side effects and any undesirable “promiscuity” on a system scale. Apart from the self-development of pocket characterization, the pocket space can provide a platform to integrate many methods from several research fields to address a host of problems endemic in drug discovery. Many significant efforts have been devoted to describing pocket flexibility and to applying mathematical modeling to sieve the representative conformations from a large set. Additionally, targeting the pockets at the PPI is becoming a more prominent focus in the field of drug discovery. Undoubtedly, this current review is noncomprehensive and has possibly missed many important investigations regarding the pocket issue, but the recent progress in the area of pockets is encouraging and should prompt the drug designer to explore this interesting topic.
ELECTRONIC SUPPLEMENTARY MATERIAL
Below is the link to the electronic supplementary material.
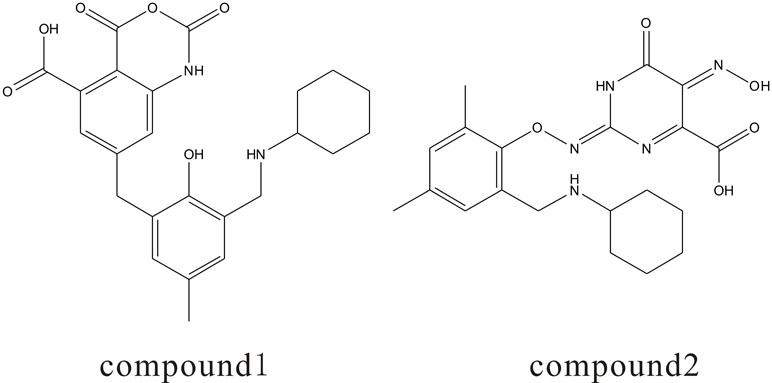{kind=link}
The molecular structures of two candidate compounds for Ras protein. (JPEG 45 kb)
{kind=link}
The molecular structures of two candidate compounds for Src kinase. (JPEG 41 kb)
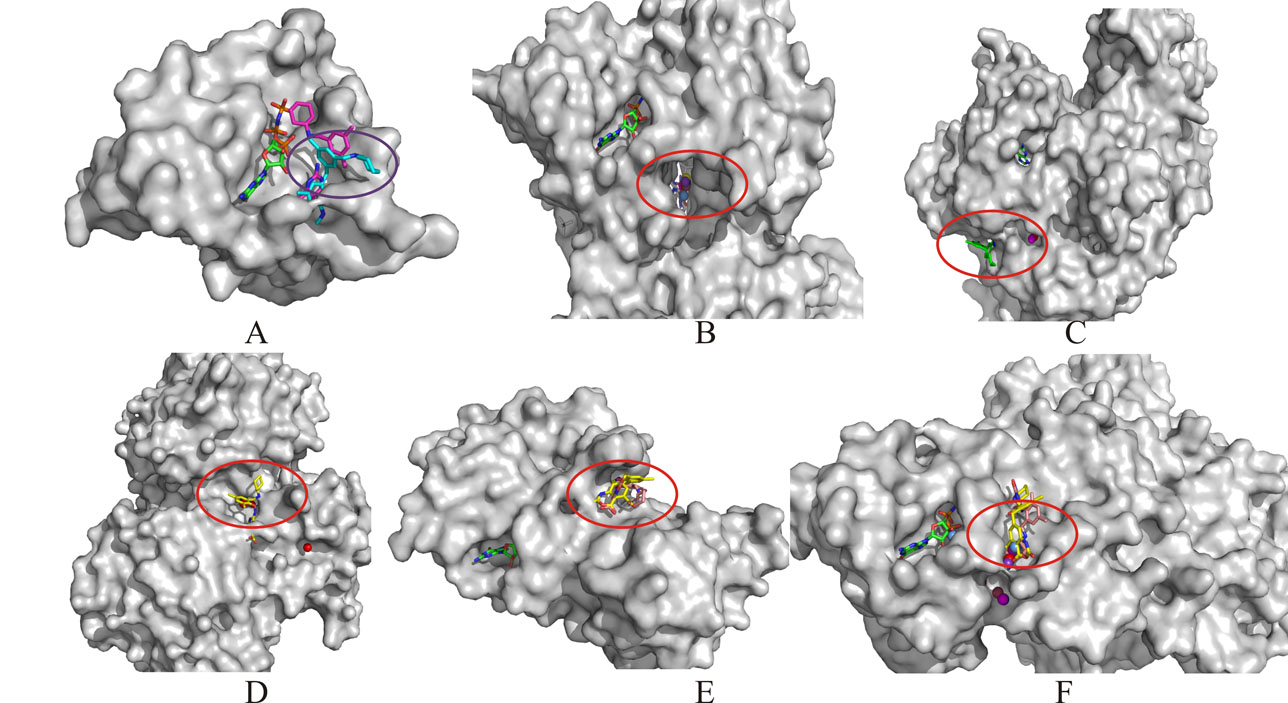{kind=link}
The positions of the candidates in the binding pockets on the Ras protein and interfaces mentioned above are shown respectively. The surfaces of the Ras and five complex structures are shown and colored grey, the GTP, compound 1 and compound 2 are all shown as ball and stick model, A: the binding pocket (purple circle) on the Ras protein with compound 1, 2; B: the binding pocket (red circle) at the interface between Ras protein and Sos protein with compound 1, 2; C: the binding pocket (red circle) at the interface between Ras protein and GAP with compound 2; D: the binding pocket (red circle) at the interface between Ras protein and RalGDS with compound 2; E: the binding pocket (red circle) at the interface between Ras protein and Raf with compound 1, 2; F: the binding pocket (red circle) at the interface between Ras protein and PI3K with compound 1, 2. (JPEG 187 kb)
{kind=link}
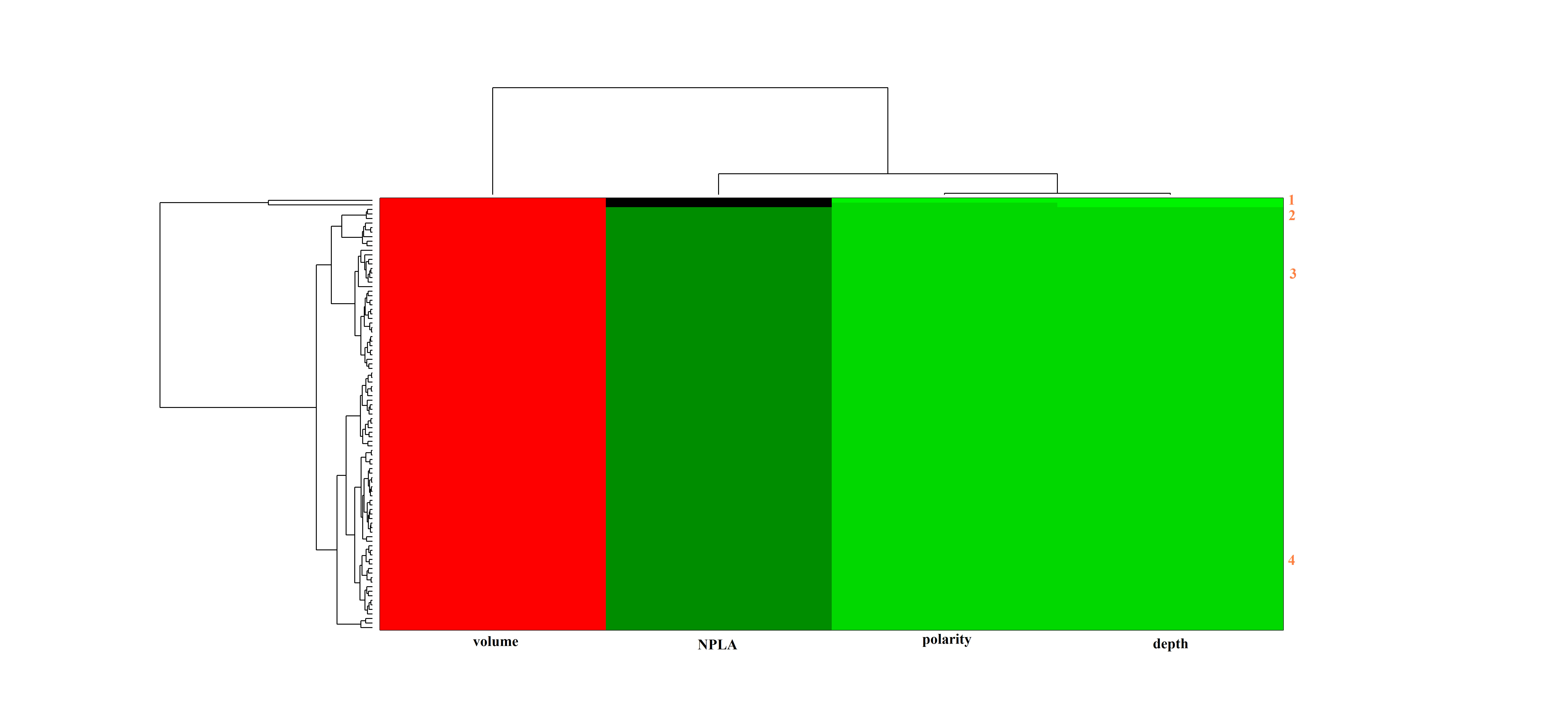
The heat map clustering of the ATP-binding site on the Src kinase (see also Figure 7). (JPEG 788 kb)
{kind=link}
The heat map clustering of the allosteric site on the Src kinase (see also Figure 7). (JPEG 768 kb)
{kind=link}
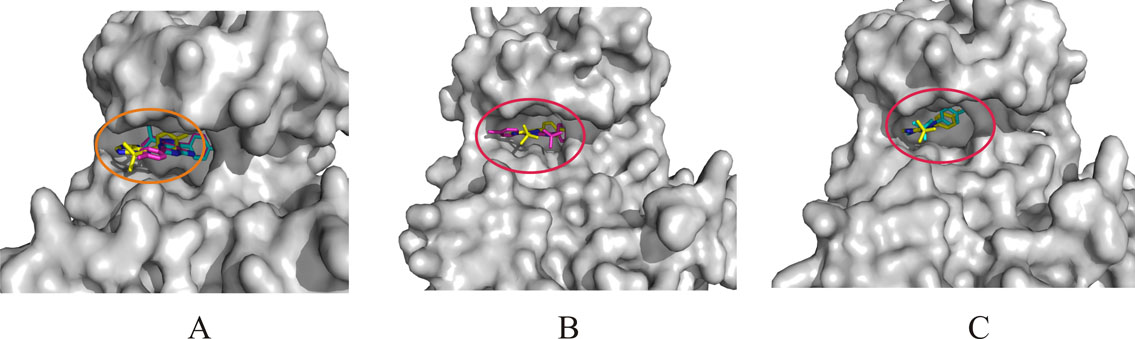
The positions of the candidates in the binding pockets on the Src kinase are shown respectively. The surface of the Src kinase structure is shown and colored grey, the PP1 (yellow), compound 4 (magenta) and compound 3 (cyan) are all shown as ball and stick model, A: the allosteric site (orange circle) with compound 3 and compound 4; B: the ATP-binding site (red circle) with compound 4; C: the ATP-binding site (red circle) with compound 3. (JPEG 83 kb)
ACKNOWLEDGMENTS
JW thanks the National Science Foundation for the support. XLZ and EKW are supported by the National Natural Science Foundation of China (grants 11174105 and 21190040) and the 973 project 2009CB930100 and 2010CB933600. JW thanks David E. Shaw for providing the valuable trajectories for the Src kinase, and XLZ thanks Xiakun Chu for analyzing these trajectories. XLZ thanks Jeremy Adler for proofreading the manuscript.
Contributor Information
Erkang Wang, Phone: +86-431-85262003, FAX: +86-431-85689711, Email: ekwang@ciac.jl.cn.
Jin Wang, Phone: +1-631-6321185, FAX: +1-631-6327960, Email: jin.wang.1@stonybrook.edu.
REFERENCES
- 1.Nisius B, Sha F. Structure-based computational analysis of protein binding sites for function and druggability prediction. J Biotechnol. 2011;159:123–34. doi:10.1016/j.jbiotec.2011.12.005. [DOI] [PubMed]
- 2.Laurie ATR, Jackson RM. Methods for the prediction of protein–ligand binding sites for structure-based drug design and virtual screening. Curr Protein Pept Sci. 2006;7:395–406. doi: 10.2174/138920306778559386. [DOI] [PubMed] [Google Scholar]
- 3.Henrich S, Salo-Ahen O, Huang B, Rippmann F, Cruciani G, Wade RC. Computational approaches to identifying and characterizing protein binding sites for ligand design. J Mol Recognit. 2010;23:209–219. doi: 10.1002/jmr.984. [DOI] [PubMed] [Google Scholar]
- 4.Pérot S, Sperandio O, Miteva MA, Camproux A-C, Villoutreix BO. Druggable pockets and binding site centric chemical space: a paradigm shift in drug discovery. Drug Discov Today. 2010;15(15–16):656–667. doi: 10.1016/j.drudis.2010.05.015. [DOI] [PubMed] [Google Scholar]
- 5.Ghersi D, Sanchez R. Beyond structural genomics: computational approaches for the identification of ligand binding sites in protein structures. J Struct Funct Genom. 2011;12(2):109–117. doi: 10.1007/s10969-011-9110-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vajda S, Guarnieri F. Characterization of protein–ligand interaction sites using experimental and computational methods. Curr Opin Drug Discov Dev. 2006;9:354–362. [PubMed] [Google Scholar]
- 7.An J, Totrov M, Abagyan R. Comprehensive identification of “druggable” protein ligand binding sites. Genome Inform. 2004;15:31–41. [PubMed] [Google Scholar]
- 8.Keller TH, Pichota A, Yin Z. A practical view of ‘druggability’. Curr Opin Chem Biol. 2006;10:357–361. doi: 10.1016/j.cbpa.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 9.Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov. 2002;1:727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 10.Hajduk PJ, Huth JR, Tse C. Predicting protein druggability. Drug Discov Today. 2005;10:1675–1682. doi: 10.1016/S1359-6446(05)03624-X. [DOI] [PubMed] [Google Scholar]
- 11.Kellenberger E, Schalon C, Rognan D. How to measure the similarity between protein ligand binding sites? Curr Comput Aided Drug Des. 2008;4:209–220. doi: 10.2174/157340908785747401. [DOI] [Google Scholar]
- 12.McCammon JA. Target flexibility in molecular recognition. Biochim Biophys Acta. 2005;1754:221–224. doi: 10.1016/j.bbapap.2005.07.041. [DOI] [PubMed] [Google Scholar]
- 13.Cozzini P, Kellogg G, Spyrakis F, Abraham D, Costantino G, Emerson A, et al. Target flexibility: an emerging consideration in drug discovery and design. J Med Chem. 2008;51:6237–6255. doi: 10.1021/jm800562d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fuller JC, Burgoyne NJ, Jackson RM. Predicting druggable binding sites at the protein–protein interface. Drug Discov Today. 2009;14(3–4):155–161. doi: 10.1016/j.drudis.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 15.Metz A, Pfleger C, Kopitz H, Pfeiffer-Marek S, Baringhaus KH, Gohlke H. Hot spots and transient pockets: predicting the determinants of small-molecule binding to a protein–protein interface. J Chem Inf Model. 2012;52:120–133. doi: 10.1021/ci200322s. [DOI] [PubMed] [Google Scholar]
- 16.Gao M, Skolnick J. The distribution of ligand-binding pockets around protein–protein interfaces suggests a general mechanism for pocket formation. Proc Natl Acad Sci. 2012;109:3784–3789. doi: 10.1073/pnas.1117768109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laskowski RA, Luscombe NM, Swindells MB, Thornton JM. Protein clefts in molecular recognition and function. Protein Sci. 1996;5:2438–2452. doi: 10.1002/pro.5560051206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Voss NR, Gerstein M. 3V: cavity, channel and cleft volume calculator and extractor. Nucleic Acids Res. 2010;38(Web Server issue):W555–W562. doi: 10.1093/nar/gkq395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Petrek M, Otyepka M, Banas P, Kosinova P, Koca J, Damborsky J. CAVER: a new tool to explore routes from protein clefts, pockets and cavities. BMC Bioinforma. 2006;7:316. doi: 10.1186/1471-2105-7-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang B. MetaPocket: a meta approach to improve protein ligand binding site prediction. OMICS. 2009;13:325–330. doi: 10.1089/omi.2009.0045. [DOI] [PubMed] [Google Scholar]
- 21.Zhang ZM, Li Y, Lin BY, Schroeder M, Huang BD. Identification of cavities on protein surface using multiple computational approaches for drug binding site prediction. Bioinformatics. 2011;27(15):2083–2088. doi: 10.1093/bioinformatics/btr331. [DOI] [PubMed] [Google Scholar]
- 22.Laurie AT, Jackson RM. Q-SiteFinder: an energy-based method for the prediction of protein–ligand binding sites. Bioinformatics. 2005;21:1908–1916. doi: 10.1093/bioinformatics/bti315. [DOI] [PubMed] [Google Scholar]
- 23.Yingjie L, Yoo S, Sanchez R. SiteComp: a server for ligand binding site analysis in protein structures. Bioinformatics. 2012;28(8):1172–1173. doi: 10.1093/bioinformatics/bts095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmidtke P, Souaille C, Estienne F, Baurin N, Kroemer RT. Large-scale comparison of four binding site detection algorithms. J Chem Inf Model. 2010;50(12):2191–2200. doi: 10.1021/ci1000289. [DOI] [PubMed] [Google Scholar]
- 25.Labute P, Santavy M. Locating binding sites in protein structures. Montreal: Chemical Computing Group, Inc. http://www.chemcomp.com/journal/sitefind.htm (2001). Accessed on 30 June 2010.
- 26.Le GV, Schmidtke P, Tuffery P. Fpocket: an open source platform for ligand pocket detection. BMC Bioinforma. 2009;10:168. doi: 10.1186/1471-2105-10-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.An J, Totrov M, Abagyan R. Pocketome via comprehensive identification and classification of ligand binding envelopes. Mol Cell Proteomics. 2005;4:752–761. doi: 10.1074/mcp.M400159-MCP200. [DOI] [PubMed] [Google Scholar]
- 28.Halgren TA. Identifying and characterizing binding sites and assessing druggability. J Chem Inf Model. 2009;49:377–389. doi: 10.1021/ci800324m. [DOI] [PubMed] [Google Scholar]
- 29.Fukunishi Y, Nakamura H. Prediction of ligand-binding sites of proteins by molecular docking calculation for a random ligand library. Protein Sci. 2011;20:95–106. doi: 10.1002/pro.540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ngan C-H, David RH, Brandon Z, Laurie EG, Dima K, Sandor V. FTSite: high accuracy detection of ligand binding sites on unbound protein structures. Bioinformatics. 2012;28:286–287. doi: 10.1093/bioinformatics/btr651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seco J, Luque J, Barril X. Binding site detection and druggability index from first principles. J Med Chem. 2009;52:2363–2371. doi: 10.1021/jm801385d. [DOI] [PubMed] [Google Scholar]
- 32.Søndergaard CR, Olsson MHM, Rostkowski M, Jensen JH. Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values. J Chem Theory Comput. 2011;7(7):2284–2295. doi: 10.1021/ct200133y. [DOI] [PubMed] [Google Scholar]
- 33.Olsson MH, Søndergaard CR, Rostkowski M, Jensen JH. PROPKA3: consistent treatment of internal and surface residues in empirical pKa predictions. J Chem Theory Comput. 2011;7(2):525–537. doi: 10.1021/ct100578z. [DOI] [PubMed] [Google Scholar]
- 34.Bains W. Failure rates in drug discovery and development: will we ever get any better? Drug Discov World. 2004;5(4):9–18.
- 35.Sheridan RP, Maiorov VN, Holloway MK, Cornell WD, Gao YD. Drug-like density: a method of quantifying the “bindability” of a protein target based on a very large set of pockets and drug-like ligands from the Protein Data Bank. J Chem Inf Model. 2010;50:2029–2040. doi: 10.1021/ci100312t. [DOI] [PubMed] [Google Scholar]
- 36.Joanna O. Determining druggability. Nat Rev Drug Discov. 2007;6(3):187. doi: 10.1038/nrd2275. [DOI] [Google Scholar]
- 37.Hajduk PJ, Huth JR, Fesik SW. Druggability indices for protein targets derived from NMR-based screening data. J Med Chem. 2005;48:2518–2525. doi: 10.1021/jm049131r. [DOI] [PubMed] [Google Scholar]
- 38.Krasowski A, Muthas D, Sarkar A, Schmitt S, Brenk R. DrugPred: a structure-based approach to predict protein druggability developed using an extensive nonredundant data set. J Chem Inf Model. 2011;51:2829–2842. doi: 10.1021/ci200266d. [DOI] [PubMed] [Google Scholar]
- 39.Schmidtke P, Barril X. Understanding and predicting druggability. A high-throughput method for detection of drug binding sites. J Med Chem. 2010;53:5858–5867. doi: 10.1021/jm100574m. [DOI] [PubMed] [Google Scholar]
- 40.Weisel M, Proschak E, Kriegl JM, Schneider G. Form follows function: shape analysis of protein cavities for receptor-based drug design. Proteomics. 2009;9:451–459. doi: 10.1002/pmic.200800092. [DOI] [PubMed] [Google Scholar]
- 41.Volkamer A, Kuhn D, Grombacher T, Rippmann F, Rarey M. Combining global and local measures for structure-based druggability predictions. J Chem Inf Model. 2012;52:360–372. doi: 10.1021/ci200454v. [DOI] [PubMed] [Google Scholar]
- 42.Perola E, Herman L. Development of a rule-based method for the assessment of protein druggability. J Chem Inf Model. 2012;52(4):1027–1038. doi: 10.1021/ci200613b. [DOI] [PubMed] [Google Scholar]
- 43.Cheng AC, Coleman RG, Smyth KT, Cao Q, Soulard P, Caffrey DR, et al. Structure-based maximal affinity model predicts small-molecule druggability. Nat Biotechnol. 2007;25:71–75. doi: 10.1038/nbt1273. [DOI] [PubMed] [Google Scholar]
- 44.Palomo V, Soteras I, Perez DI, Perez C, Gil C, Campillo NE, et al. Exploring the binding sites of glycogen synthase kinase 3. Identification and characterization of allosteric modulation cavities. J Med Chem. 2011;54(24):8461–8470. doi: 10.1021/jm200996g. [DOI] [PubMed] [Google Scholar]
- 45.Martin J. Beauty is in the eye of the beholder: proteins can recognize binding sites of homologous proteins in more than one way. PLoS Comput Biol. 2010;6:e1000821. doi: 10.1371/journal.pcbi.1000821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gold ND, Jackson RM. SitesBase: a database for structure-based protein–ligand binding site comparisons. Nucleic Acids Res. 2006;34:D231–D234. doi: 10.1093/nar/gkj062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yeturu K, Chandra N. PocketMatch: a new algorithm to compare binding sites in protein structures. BMC Bioinforma. 2008;9:543. doi: 10.1186/1471-2105-9-543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xie L, Bourne PE. Detecting evolutionary relationships across existing fold space, using sequence order-independent profile–profile alignments. Proc Natl Acad Sci U S A. 2008;105:5441–5446. doi: 10.1073/pnas.0704422105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xie L, Evangelidis T, Xie L, Bourne PE. Drug discovery using chemical systems biology: weak inhibition of multiple kinases may contribute to the anti-cancer effect of nelfinavir. PLoS Comput Biol. 2011;7(4):e1002037. doi: 10.1371/journal.pcbi.1002037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kinnings SL, Liu N, Buchmeier N, Tonge PJ, Xie L, Bourne PE. Drug discovery using chemical systems biology: repositioning the safe medicine Comtan to treat multi-drug and extensively drug resistant tuberculosis. PLoS Comput Biol. 2009;5(7):e1000423. doi: 10.1371/journal.pcbi.1000423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xie L, Li J, Xie L, Bourne PE. Drug discovery using chemical systems biology: identification of the protein–ligand binding network to explain the side effects of CETP inhibitors. PLoS Comput Biol. 2009;5(5):e1000387. doi: 10.1371/journal.pcbi.1000387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sael L, Kihara D. Detecting local ligand-binding site similarity in nonhomologous proteins by surface patch comparison. Proteins Struct Funct Bioinforma. 2012;80(4):1177–1195. doi: 10.1002/prot.24018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meslamani J, Rognan D, Kellenberger E. sc-PDB: a database for identifying variations and multiplicity of ‘druggable’ binding sites in proteins. Bioinformatics. 2011;27(9):1324–1326. doi: 10.1093/bioinformatics/btr120. [DOI] [PubMed] [Google Scholar]
- 54.Ito J-I, Tabei Y, Shimizu K, Tsuda K, Tomii K. PoSSuM: a database of similar protein–ligand binding and putative pockets. Nucleic Acids Res. 2012;40(Database issue):D541–D548. doi: 10.1093/nar/gkr1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kawabata T. Detection of multiscale pockets on protein surfaces using mathematical morphology. Proteins Struct Funct Bioinforma. 2010;78(5):1195–1211. doi: 10.1002/prot.22639. [DOI] [PubMed] [Google Scholar]
- 56.Ito J, Tabei Y, Shimizu K, Tomii K, Tsuda K. PDB-scale analysis of known and putative ligand binding sites with structural sketches. Proteins. 2011;80:747–763. doi: 10.1002/prot.23232. [DOI] [PubMed] [Google Scholar]
- 57.Yin S, Proctor EA, Lugovskoy AA, Dokholyan NV. Fast screening of protein surfaces using geometric invariant fingerprints. Proc Natl Acad Sci U S A. 2009;106:16622–16626. doi: 10.1073/pnas.0906146106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tabei Y, Uno T, Sugiyama M, Tsuda K. Single versus multiple sorting for all pairs similarity search. The 2nd Asian Conference on Machine Learning (ACML2010). 2010.
- 59.Medvedeva I, Demenkov P, Kolchanov N, Ivanisenko V. SitEx: a computer system for analysis of projections of protein functional sites on eukaryotic genes. Nucleic Acids Res. 2012;40(Database issue):D278–D283. doi: 10.1093/nar/gkr1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Luque I, Freire E. Structural stability of binding sites: consequences for binding affinity and allosteric effects. Proteins. 2000;(Suppl. 4):63–71 [DOI] [PubMed]
- 61.Eyrisch S, Helms V. Transient pockets on protein surfaces involved in protein–protein interaction. J Med Chem. 2007;50:3457–3464. doi: 10.1021/jm070095g. [DOI] [PubMed] [Google Scholar]
- 62.Wlodarski T, Zagrovic B. Conformational selection and induced fit mechanism underlie specificity in noncovalent interactions with ubiquitin. Proc Natl Acad Sci U S A. 2009;106:19346–19351. doi: 10.1073/pnas.0906966106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rueda M, Bottegoni G, Abagyan R. Recipes for the selection of experimental protein conformations for virtual screening. J Chem Inf Model. 2010;50:186–193. doi: 10.1021/ci9003943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sperandio O, Mouawad L, Pinto E, Bruno OV, Perahia D, Miteva MA. How to choose relevant multiple receptor conformations for virtual screening: a test case of Cdk2 and normal mode analysis. Eur Biophys J. 2010;39:1365–1372. doi: 10.1007/s00249-010-0592-0. [DOI] [PubMed] [Google Scholar]
- 65.Brady GP, Stouten PFW. Fast prediction and visualization of protein binding pockets with PASS. J Comput Aided Mol Des. 2000;14:383–401. doi: 10.1023/A:1008124202956. [DOI] [PubMed] [Google Scholar]
- 66.Schmidtke P, Bidon-Chanal A, Luque FJ, Barril X. MDpocket: open-source cavity detection and characterization on molecular dynamics trajectories. Bioinformatics. 2011;27(23):3276–3285. doi: 10.1093/bioinformatics/btr550. [DOI] [PubMed] [Google Scholar]
- 67.Craig IR, Pfleger C, Gohlke H, Essex JW, Spiegel K. Pocket-space maps to identify novel binding-site conformations in proteins. J Chem Inf Model. 2011;51(10):2666–2679. doi: 10.1021/ci200168b. [DOI] [PubMed] [Google Scholar]
- 68.Kufareva I, Ilatovskiy AV, Abagyan R. Pocketome: an encyclopedia of small-molecule binding sites in 4D. Nucleic Acids Res. 2012;40(1):D535–D540. doi: 10.1093/nar/gkr825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein–protein interfaces. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 70.Wanner J, Fry DC, Peng ZW, Roberts J. Druggability assessment of protein–protein interface. Future Med Chem. 2011;3(16):2021–2038. doi: 10.4155/fmc.11.156. [DOI] [PubMed] [Google Scholar]
- 71.Leis S, Schneider S, Zacharias M. In silico prediction of binding sites on protein. Curr Med Chem. 2010;17:1550–1562. doi: 10.2174/092986710790979944. [DOI] [PubMed] [Google Scholar]
- 72.Jubb H, Higueruelo AP, Winter A, Blundell TL. Structural biology and drug discovery for protein–protein interactions. Trends Pharmacol Sci. 2012;33(5):241–248. doi: 10.1016/j.tips.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 73.Mysinger MM, Weiss DR, Ziarek JJ, Gravel S, Doak AK, Karpiak J, et al. Structure-based ligand discovery for the protein–protein interface of chemokine receptor CXCR4. Proc Natl Acad Sci U S A. 2012;109(14):5517–5522. doi: 10.1073/pnas.1120431109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gautier B, Miteva MA, Goncalves V, Huguenot F, Coric P, Bouaziz S. Targeting the proangiogenic VEGF-VEGFR protein–protein interface with drug-like compounds by in silico and in vitro screening. Chem Biol. 2011;18(12):1631–1639. doi: 10.1016/j.chembiol.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 75.Shima F, Ijiri Y, Muraoka S, Liao J, Ye M, Araki M, et al. Structural basis for conformational dynamics of GTP-bound Ras protein. J Biol Chem. 2010;285(29):22696–22705. doi: 10.1074/jbc.M110.125161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Scheffzek K, Ahmadian MR, Kabsch W, Wiesmüller L, Lautwein A, Schmitz F, et al. The Ras-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science. 1997;277(5324):333–338. doi: 10.1126/science.277.5324.333. [DOI] [PubMed] [Google Scholar]
- 77.Margarit SM, Sondermann H, Hall BE, Nagar B, Hoelz A, Pirruccello M, et al. Structural evidence for feedback activation by Ras.GTP of the Ras-specific nucleotide exchange factor SOS. Cell. 2003;112(5):685–695. doi: 10.1016/S0092-8674(03)00149-1. [DOI] [PubMed] [Google Scholar]
- 78.Nassar N, Horn G, Herrmann C, Block C, Janknecht R, Wittinghofer A. Ras/Rap effector specificity determined by charge reversal. Nat Struct Biol. 1996;3(8):723–729. doi: 10.1038/nsb0896-723. [DOI] [PubMed] [Google Scholar]
- 79.Pacold ME, Suire S, Perisic O, Lara-Gonzalez S, Davis CT, Walker EH, et al. Crystal structure and functional analysis of Ras binding to its effector phosphoinositide 3-kinase gamma. Cell. 2000;103(6):931–943. doi: 10.1016/S0092-8674(00)00196-3. [DOI] [PubMed] [Google Scholar]
- 80.Huang L, Hofer F, Martin GS, Kim SH. Structural basis for the interaction of Ras with RalGDS. Nat Struct Biol. 1998;5(6):422–426. doi: 10.1038/nsb0698-422. [DOI] [PubMed] [Google Scholar]
- 81.Zheng X, Liu ZJ, Li D, Wang EK, Wang J. Rational drug design: the search for Ras protein hydrolysis intermediate conformation Inhibitors with both affinity and specificity. Curr Pharm Des. 2012; in press. [DOI] [PubMed]
- 82.Shan Y, Kim ET, Eastwood MP, Dror RO, Seeliger MA, Shaw DE. How does a drug molecule find its target binding site? J Am Chem Soc. 2011;133(24):9181–9183. doi: 10.1021/ja202726y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Irwin JJ, Shoichet BK. ZINC—a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45:177–182. doi: 10.1021/ci049714+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Durrant JD, Amaro RE, McCammon JA. AutoGrow: a novel algorithm for protein inhibitor design. Chem Biol Drug Des. 2009;73(2):168–178. doi: 10.1111/j.1747-0285.2008.00761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lagorce D, Maupetit J, Baell J, Sperandio O, Tuffery P, Miteva MA, et al. The FAF-Drugs2 server: a multistep engine to prepare electronic chemical compound collections. Bioinformatics. 2011;27(14):2018–2020. doi: 10.1093/bioinformatics/btr333. [DOI] [PubMed] [Google Scholar]
- 86.Rueda M, Bottegoni G, Abagyan R. Consistent improvement of cross-docking results using binding site ensembles generated with elastic network normal modes. J Chem Inf Model. 2009;49(3):716–725. doi: 10.1021/ci8003732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ford B, Skowronek K, Boykevisch S, Bar-Sagi D, Nassar N. Structure of the G60A mutant of Ras. J Biol Chem. 2005;280:25697–25705. doi: 10.1074/jbc.M502240200. [DOI] [PubMed] [Google Scholar]
- 88.Bar-Joseph Z, Gifford DK, Jaakkola TS. Fast optimal leaf ordering for hierarchical clustering. Bioinformatics. 2001;17(Suppl 1):S22–S29. doi: 10.1093/bioinformatics/17.suppl_1.S22. [DOI] [PubMed] [Google Scholar]
- 89.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.ADT/AutoDockTools. http://autodock.scripps.edu/resources/adt/index_html.
- 91.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19:1639–1662. doi: 10.1002/(SICI)1096-987X(19981115)19:14<1639::AID-JCC10>3.0.CO;2-B. [DOI] [Google Scholar]
- 92.Yan Z, Wang J. Specificity quantification of biomolecular recognition and its implication for drug discovery. Sci Rep. 2012;2:309. doi: 10.1038/srep00309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang J, Zheng X, Yang Y, Drueckhammer D, Yang W. Quantifying intrinsic specificity: a potential complement to affinity in drug screening. Phys Rev Lett. 2007;99:1981011–1981014. doi: 10.1103/PhysRevLett.99.198101. [DOI] [PubMed] [Google Scholar]
- 94.Wang J, Verkhivker GM. Energy landscape theory, funnels, specificity, and optimal criterion of biomolecular binding. Phys Rev Lett. 2003;90:188101–188104. doi: 10.1103/PhysRevLett.90.188101. [DOI] [PubMed] [Google Scholar]
- 95.Araki M, Shima F, Yoshikawa Y, Muraoka S, Ijiri Y, Nagahara Y, et al. Solution structure of the state 1 conformer of GTP-bound H-Ras protein and distinct dynamic properties between the state 1 and state 2 conformers. J Biol Chem. 2011;286(45):39644–39653. doi: 10.1074/jbc.M111.227074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Eyrisch S, Helms V. What induces pocket openings on protein surface patches involved in protein–protein interactions? J Comput Aided Mol Des. 2009;23(2):73–86. doi: 10.1007/s10822-008-9239-y. [DOI] [PubMed] [Google Scholar]
- 97.Seelinger D, Haas J, de Groot BL. Geometry-based sampling of conformational transitions in proteins. Structure. 2007;15:1482–1492. doi: 10.1016/j.str.2007.09.017. [DOI] [PubMed] [Google Scholar]
- 98.Cavasotto CN, Kovacs JA, Abagyan RA. Representing receptor flexibility in ligand docking through relevant normal modes. J Am Chem Soc. 2005;127:9632–9640. doi: 10.1021/ja042260c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The molecular structures of two candidate compounds for Ras protein. (JPEG 45 kb)
The molecular structures of two candidate compounds for Src kinase. (JPEG 41 kb)
The positions of the candidates in the binding pockets on the Ras protein and interfaces mentioned above are shown respectively. The surfaces of the Ras and five complex structures are shown and colored grey, the GTP, compound 1 and compound 2 are all shown as ball and stick model, A: the binding pocket (purple circle) on the Ras protein with compound 1, 2; B: the binding pocket (red circle) at the interface between Ras protein and Sos protein with compound 1, 2; C: the binding pocket (red circle) at the interface between Ras protein and GAP with compound 2; D: the binding pocket (red circle) at the interface between Ras protein and RalGDS with compound 2; E: the binding pocket (red circle) at the interface between Ras protein and Raf with compound 1, 2; F: the binding pocket (red circle) at the interface between Ras protein and PI3K with compound 1, 2. (JPEG 187 kb)
The heat map clustering of the ATP-binding site on the Src kinase (see also Figure 7). (JPEG 788 kb)
The heat map clustering of the allosteric site on the Src kinase (see also Figure 7). (JPEG 768 kb)
The positions of the candidates in the binding pockets on the Src kinase are shown respectively. The surface of the Src kinase structure is shown and colored grey, the PP1 (yellow), compound 4 (magenta) and compound 3 (cyan) are all shown as ball and stick model, A: the allosteric site (orange circle) with compound 3 and compound 4; B: the ATP-binding site (red circle) with compound 4; C: the ATP-binding site (red circle) with compound 3. (JPEG 83 kb)