

Abstract
“For-cause” inspections are initiated during the review of bioequivalence (BE) data submitted to Abbreviated New Drug Applications when possible scientific misconduct and study irregularities are discovered. We investigated the common reasons for initiating “for-cause” inspections related to the clinical, analytical, and dissolution study sites associated with BE studies. This information may help the pharmaceutical industry to understand the root causes of compliance failures in BE studies and help them to improve compliance with FDA’s regulations, thereby facilitating more rapid approval of safe and effective generic drugs.
KEY WORDS: bioequivalence, FDA, inspection
INTRODUCTION
Studies to establish bioequivalence (BE) of a product are submitted to the FDA’s Center for Drug Evaluation and Research (CDER) in support of abbreviated new drug applications (ANDAs) and their post-approval supplements (1). To be deemed therapeutically equivalent to the corresponding reference-listed drug (RLD), a generic product must be pharmaceutically equivalent and bioequivalent to the RLD (2).
Bioequivalence is defined as the absence of a significant difference in the rate and extent to which the active ingredient or active moiety in pharmaceutical equivalents or pharmaceutical alternatives becomes available at the site of drug action when administered at the same molar dose under similar conditions in an appropriately designed study (2,3). There are several in vivo and in vitro approaches available to demonstrate bioequivalence. The most common used approach is the in vivo pharmacokinetic bioequivalence studies that compare the systemic exposure profile of a test drug product to that of a reference drug product. For ANDA bioequivalence submissions that contain the results of in vivo studies, the four major study report components are the following: bioanalytical methodology, clinical study reports, statistical analysis and in vitro dissolution testing (4). During the bioequivalence study review process, deficiencies related to these components can be identified and recommendations to correct the deficiencies are communicated to the applicants. If any substantive information suggesting scientific misconduct, major human subject protection violations, or compromised BE data is discovered during the bioequivalence study review process, a “for-cause” inspection of the dissolution, analytical and/or clinical facilities in which the bioequivalence studies were conducted will be requested by the FDA reviewer, through FDA’s Office of Scientific Investigations (OSI). In response to this request, the OSI will then schedule an inspection of the facilities where the suspected misconduct was investigated. OSI inspections are conducted to ensure that the rights, safety, and welfare of the human study subjects have been protected, and to verify compliance with Title 21 of the Code of Federal Regulations (CFR), Part 320, Bioavailability and Bioequivalence Requirements (3). Such processes assist in ensuring the integrity and reliability of the bioequivalence studies submitted to the FDA.
The objective of this investigation is to identify the common reasons that FDA conducts “for-cause” inspections on clinical, analytical, and dissolution study sites associated with bioequivalence studies of generic drug products, and to summarize these inspection outcomes. It is hoped that publication of the findings of this study will assist with identifying the root causes of compliance failures in bioequivalence studies of generic drug products, so that these problems may be prevented from occurring in the future, thus facilitating approval of only high-quality, safe, and effective generic drugs.
METHODS
Internal FDA databases were searched to determine the reasons for ANDA bioequivalence study-related “for-cause” inspections from January 2003 to December 2011 (total of 9 years). The applications included bioequivalence studies of various dosage forms including solid oral dosage forms, transdermal drug delivery systems, medicated chewing gums, and suspensions for oral or parenteral delivery. Incidents of “for-cause” inspections were analyzed according to (1) categories of inspection reasons; (2) type of inspection sites, i.e., clinical, analytical, and dissolution sites; (3) number of inspections requested each year; and (4) outcomes of inspections.
The common reasons for requesting “for-cause” inspections are categorized as follows (Table I):
- Data integrity and validity issues: this category is further subdivided into the following subcategories (Table II):
-
1.1.Inspection requested to ensure data accuracy: the study results are suspected to be biased, potentially resulting in an invalid conclusion of bioequivalence.
-
1.2.Discrepancy between the sponsor and other information available to FDA: the FDA reviewer identified significant differences in the firm’s bioequivalence study outcomes, i.e., PK parameters, or dissolution results when compared with other data available to FDA, and such differences were unjustifiable.
-
1.3.Protocol deviations: the firm did not ensure that the study was conducted according to the investigational plan.
-
1.4.Inadequate method validation: the study lacks adequate validation study data, for example, missing a cross-validation study.
-
1.5.Inconsistent or conflicting information in the submission: the reviewers observed discrepancies in the firm’s report that were unexplainable.
-
1.1.
Unacceptably large number of re-analyzed samples: a high percentage of study samples were repeated in the bioequivalence studies and the applicant could not provide satisfactory documentation to justify these repeats.
Prior adverse inspection history of the inspected site: a previous inspection of a site raised data integrity issues of the studies conducted at this site. The inspection was requested to determine whether similar objectionable study practices were used for the studies under review.
Inadequate documentation: the firm did not maintain adequate and accurate documents that were critical for bioequivalence determination.
Improper study design and conduct: examples of this category included insufficient number of control subjects in a re-dosing study or inappropriate selection of the QC concentrations in analytical studies.
“Others”: deficiencies which did not fall into one of the above defined categories. Examples of “others” include inspections related to studies with severe adverse events, and/or study validation in question.
Table I.
Categories of Reasons for FDA to Request “For-Cause” Inspections from 2003 to 2011
| Categories of reasons | Number of inspections requested | Percentage, % | |
|---|---|---|---|
| 1 | Data integrity and validity issues | 55 | 60.4 |
| 2 | Unacceptably large number of re-analyzed samples | 7 | 7.7 |
| 3 | Prior adverse inspection history of the inspected site | 11 | 12.1 |
| 4 | Inadequate documentation | 9 | 9.9 |
| 5 | Improper study design and conduct | 4 | 4.4 |
| 6 | “Others” | 5 | 5.5 |
| Total | 91 | 100 | |
Table II.
Reasons to Request “For-Cause” Inspections Under “Data Integrity and Validity Issues” Category
| Reasons under “data integrity and validity issues” category | Number of inspections requested | Percentage, % | |
|---|---|---|---|
| 1.1 | Inspection requested to ensure data accuracy | 34 | 61.8 |
| 1.2 | Discrepancy between the sponsor and other information available to FDA | 2 | 3.6 |
| 1.3 | Protocol deviations | 5 | 9.1 |
| 1.4 | Inadequate method validation | 8 | 14.5 |
| 1.5 | Inconsistent/conflicting information in the submission | 6 | 10.9 |
| Total | 55 | 100 | |
RESULTS AND DISCUSSION
A total of 90 “for-cause” inspections were identified from January 2003 to December 2011. These inspections are associated with 66 ANDA applications. During this time frame, FDA has received 7,207 ANDAs. Thus, the rate of requests for “for-cause” inspections was about 0.9% for all ANDAs submitted from 2003 to 2011. Please note that in addition to “for-cause” inspections, the FDA also conducts “Routine” inspections, which were initiated in the absence of concerns about misconduct or as a follow-up to a complaint. Generally, “Routine” inspections evaluate studies supporting regulatory submissions such as New Drug Applications, ANDAs, and Investigational New Drug Applications. Evaluation of “Routine” inspections is not within the scope of this paper.
In general, a typical pharmacokinetic bioequivalence study involves a clinical site where subjects are dosed and blood samples are collected, and an analytical site where blood samples are analyzed to determine drug concentration. There are also clinical endpoint bioequivalence studies which may involve multiple clinical sites. For certain dosage forms, such as tablets and capsules, the ANDA sponsor also conducts comparative dissolution tests to support demonstration of bioequivalence. Among the 90 “for-cause” inspections, 54 were requested for analytical sites (Fig. 1). These were generally based on concerns regarding the accuracy and reliability of the bioanalytical methods. Thirty-three inspections were requested for clinical sites. These inspections were performed to determine if the clinical studies were appropriately conducted and that the rights, safety, and welfare of the human subjects were protected. There were three inspections requested for dissolution study sites. These dissolution site inspections were requested because the FDA reviewers observed discrepancies between the dissolution results submitted by ANDA sponsor and information available to the agency from other applications with regard to dissolution of the RLD, as well as inconsistencies in the ANDA sponsor’s dissolution testing report.
Fig. 1.

Categorization of “for-cause” inspections based on study sites from 2003 to 2011
We subsequently analyzed the common reasons for requesting these “for-cause” inspections. We found that the most common reason the FDA requests “for-cause” inspections was related to “data integrity and validity issues”, constituting 60.4% of the total number of inspections requested (Table I). The second most common reason for requesting a “for-cause” inspection was for those sites which had prior adverse inspection histories. In this case, the FDA reviewer intended to examine whether similar objectionable study practices relating to data integrity issues, identified in a prior FDA inspection, were also applicable to the studies currently under review. This reason occupied 12.1% of the total requested inspections. “Inadequate documentation” was also found to be a serious issue, i.e., 9.9% of all inspections. For example, if the sponsor does not maintain adequate and accurate case histories for study subjects, and the FDA reviewer determines that such incompleteness may compromise the study outcome, the reviewer may request an inspection for clarification and assurance. Besides these top three reasons, “for-cause” inspections due to other reasons were also examined. These reasons were related to study design, study conduct, standard procedures, data reporting and sample reanalysis inadequacies identified in the applications (See Table I). For certain reasons that are not obviously assignable, we classified it as “Others”. Examples belonging to this category included a case where one subject died in the fasting bioequivalence study. The FDA reviewer examined the sponsor’s protocols and medical records, but found no evidence of gross negligence or medical mismanagement. The reviewer yet remained concern about the occurrence of a death and the serious adverse events. Therefore, a “for-cause” inspection was requested to obtain further verification of clinical trial and medical documentation, investigator qualifications, and appropriateness of Standard Operating Procedures (SOPs) for evaluating and managing safety issues and adverse events in study participants.
We further scrutinized the details for the reason under “data integrity and validity issues”. Under this category, the top most reason for inspection requests was to ensure the data accuracy of the bioequivalence study sponsors submitted. For example, a BE reviewer had noticed that drug concentrations for a particular subject were below the limit of quantitation at all sampling time points in the BE study. The inspection was then requested to verify the adequacy of the firm’s procedures at the clinical site to assure subject dosing, as well as to confirm that there were no other analytical deficiencies that could invalidate the results of the bioequivalence studies. “Inadequate method validation” appears as the second most occurring issue under this category. Furthermore, if an FDA reviewer finds inconsistent or conflicting information in the sponsor’s submissions which are not explainable, an inspection is requested. Besides the reasons mentioned above, a small portion of inspections were requested for potential noncompliance issues and failures such as protocol deviations, and unexplainable discrepancies observed between the sponsor and other information available to FDA (See Table II).
It should be noted that for many inspections included in this study, the inspections were not requested just for a single issue. Generally, a systemic problem based on multiple issues with the study report likely caused the FDA reviewer’s concern regarding the validity of the study, thus leading to an inspection. For example, an FDA reviewer found that numerous reassays of the same samples were reassayed for different reasons, and the sponsor did not provide adequate justification for these reassays. Furthermore, when checking into the details of the analytical report, it was found that the sponsor’s analytical study appeared inconsistent, incoherent, and invalid. These reasons together prompted the reviewer’s decision to request a “for-cause” inspection. Tables III and IV provide some typical examples of reasons of “for-cause” inspections identified by the FDA reviewers.
Table III.
Examples of Reasons for FDA to Request “For-Cause” Inspections
| “For-cause” inspection reason | Examples | |
|---|---|---|
| 1 | Data integrity and validity issues | See Table IV |
| 2 | Unacceptably large number of re-analyzed samples | A large number of sample runs were interrupted and/or repeated for the analytes in the fasting and fed studies; The sponsor has not provided satisfactory documentation to justify these interruptions and repeats. Also, a large number of samples were reintegrated for the analytes but the sponsor did not provide adequate justification for these reintegrations |
| 3 | Prior adverse inspection history of the inspected site | Another inspection of this site raised integrity issues of many subjects’ study samples |
| 4 | Inadequate documentation | The sponsor did not maintain adequate and accurate case histories in progress notes for study subjects |
| 5 | Improper study design and conduct | Insufficient number of control subjects in re-dosing study, and lack of SOP in effect at the time of the study related to the conditions that warrant the performance of an outlier test. In addition, analytical deficiencies consist of inappropriate selection of the QC concentrations |
| 6 | “Others” | An inspection of the clinical facility was requested given the known severity of the adverse events of a particular drug and the hospitalization of a study subject. |
Table IV.
Examples of Reasons for “For-Cause” Inspections for “Data Integrity and Validity Issues” Category
| “For-cause” inspection reason | Examples | |
|---|---|---|
| 1.1 | Inspection requested to ensure data accuracy | Inspection was requested to verify that drug concentrations for Subject X were below the limit of quantitation at all sampling time points in the BE study; to verify the adequacy of the firm’s procedures at the clinical site to assure subject dosing, as well as to confirm that there are no other analytical deficiencies that could invalidate the results of the BE studies |
| 1.2 | Discrepancy between the sponsor and FDA in-house data | The AUC0-t, AUC∞, and Cmax parameter values obtained for the analyte are much deviated from those observed in other FDA in-house sources |
| 1.3 | Protocol deviations | The sponsor did not ensure that the investigation was conducted according to the investigational plan |
| 1.4 | Inadequate method validation | Lack of cross-validation study data for the interested analytes |
| 1.5 | Inconsistent/conflicting information in the submission | The sponsor provided conflicting study dates which impact the storage time and stability of the subjects’ samples |
In terms of the number of “for-cause” inspections requested per year for ANDA submissions, there appears to be no clear trend. In 2003, only two “for-cause” inspections were requested, whereas in 2010, there were 23 “for-cause” inspections (Fig. 2). The average “for-cause” inspections requested over the 9-year study period are ten inspections per year.
Fig. 2.
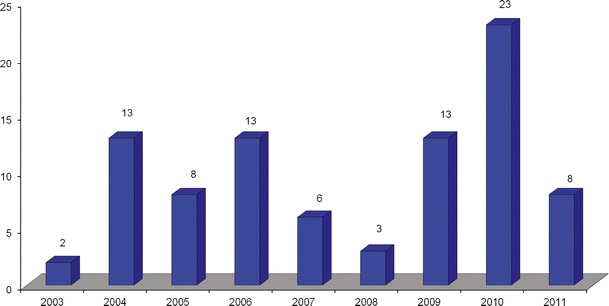
Number of “for-cause” inspections requested in each year from 2003 to 2011
Inspection outcome decisions fall into one of three categories: no action indicated (NAI), voluntary action indicated (VAI), and official action indicated (OAI). NAI means that no objectionable conditions or practices were found during an inspection, or the objectionable conditions found do not justify further regulatory action. VAI means that objectionable conditions or practices were found that represent departures from the regulations but the applicant subsequently corrected these conditions or practices by voluntary action. OAI means that regulatory and/or administrative actions were recommended in response to findings of objectionable conditions/practices (5). As shown in Fig. 3, of the “for-cause” inspections examined in this study, 15 were classified as NAI, 53 as VAI, and 15 as OAI. Figure 3 also shows that seven inspections were pending inspection outcomes. From this data, it is concluded that over 75% of “for-cause” inspections requested by FDA reviewers were later identified to have potential issues (i.e., VAI) or serious problems (i.e., OAI) as determined by the inspections conducted by Office of Compliance. These bioequivalence studies would then require a certain level of corrective action, official or voluntary, from the sponsors.
Fig. 3.
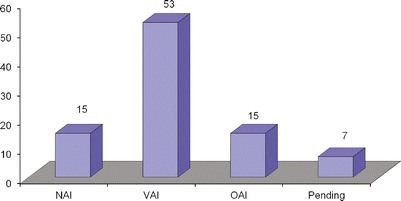
Type of outcomes of “for-cause” inspections from 2003 to 2011
CONCLUSION
This investigation identifies common reasons for requesting “for-cause” inspections on clinical, analytical, and dissolution study sites associated with bioequivalence studies submitted in ANDAs. Inspections with outcomes of official and voluntary action may delay the approval of sponsors’ applications and, therefore, should be avoided. It is hoped that publication of this information will suggest ways to improve compliance with FDA’s regulations, thereby facilitating more rapid approval of safe and effective drugs.
Acknowledgments
Disclaimer
This article reflects the views of the author and should not be construed to represent FDA’s views or policies.
References
- 1.U.S. Food and Drug Administration. Guidance for Industry: Bioavailability and bioequivalence studies for orally administered drug products—general considerations. 2003. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm070124.pdf. Accessed 8 Aug 2012.
- 2.Approved products with therapeutic equivalence evaluations. 31st edition. Washington, DC: US Department of Health and Human Services, Public Health Service, Food and Drug Administration, Center for Drug Evaluation and Research, Office of Pharmaceutical Science, Office of Generic Drugs. 2009. http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/UCM071436.pdf. Accessed 8 Aug 2012.
- 3.USFDA. Title 21 of the Code of Federal Regulations (CFR), Part 320. Bioavailability and Bioequivalence Requirements.
- 4.Liu Q, Davit B, Cherstniakova S, Dandamudi S, Walters J, Lee C, Raines K, Ren K, Williamson L, Conner D. Common deficiencies with bioequivalence submissions in abbreviated new drug applications assessed by FDA. AAPS J. 2011. doi:10.1208/s12248-011-9312-7. [DOI] [PMC free article] [PubMed]
- 5.FDA/ORA Bioresearch Monitoring Information Page, http://www.fda.gov/ICECI/EnforcementActions/BioresearchMonitoring/default.htm. Accessed 8 Aug 2012.