

Abstract
Practical food effect predictions and assessments were described using in silico, in vitro, and/or in vivo preclinical data to anticipate food effects and Biopharmaceutics Classification System (BCS)/Biopharmaceutics Drug Disposition Classification System (BDDCS) class across drug development stages depending on available data: (1) limited in silico and in vitro data in early discovery; (2) preclinical in vivo pharmacokinetic, absorption, and metabolism data at candidate selection; and (3) physiologically based absorption modeling using biorelevant solubility and precipitation data to quantitatively predict human food effects, oral absorption, and pharmacokinetic profiles for early clinical studies. Early food effect predictions used calculated or measured physicochemical properties to establish a preliminary BCS/BDDCS class. A rat-based preclinical BCS/BDDCS classification used rat in vivo fraction absorbed and metabolism data. Biorelevant solubility and precipitation kinetic data were generated via animal pharmacokinetic studies using advanced compartmental absorption and transit (ACAT) models or in vitro methods. Predicted human plasma concentration–time profiles and the magnitude of the food effects were compared with observed clinical data for assessment of simulation accuracy. Simulations and analyses successfully identified potential food effects across BCS/BDDCS classes 1–4 compounds with an average fold error less than 1.6 in most cases. ACAT physiological absorption models accurately predicted positive food effects in human for poorly soluble bases after oral dosage forms. Integration of solubility, precipitation time, and metabolism data allowed confident identification of a compound’s BCS/BDDCS class, its likely food effects, along with prediction of human exposure profiles under fast and fed conditions.
Electronic supplementary material
The online version of this article (doi:10.1208/s12248-012-9419-5) contains supplementary material, which is available to authorized users.
KEY WORDS: absorption modeling, BCS/BDDCS, food effect prediction, human PBPK model, oral bioavailability
INTRODUCTION
The effect of food on drug absorption can be mediated through various mechanisms, including enhancement in drug solubility, change of gastrointestinal (GI) pH and mobility, delayed stomach emptying, increased bile salt concentration, or direct interactions with the drug (1, 2). Food effects may significantly alter the systemic availability of orally dosed drugs which can impact pharmacological responses or safety margins (3–5). Fleisher, Wu, and Benet used the Biopharmaceutics Classification System (BCS; 6) to predict the direction and change in the extent of drug exposure affected by food (1, 7). The Biopharmaceutics Drug Disposition Classification System (BDDCS; 8, 9) which classifies compounds by solubility and metabolism has been applied for anticipating food effects (7). However, the BDDCS is apparently less frequently used by pharmaceutical scientists (8), possibly due to its perceived complexity. The BDDCS system assigns a drug classification using human in vivo drug metabolism data in lieu of human intestinal permeability to demonstrate that the extent of absorption is greater than 90% (7). Recently, Benet et al. utilized in silico/in vitro physicochemical parameters (e.g., LogP) to assign BDDCS class and probability of metabolism for new chemical entities (NCEs; 10). A BDDCS class could be used to identify likely food effects in early drug discovery stages. However, concentration–time profiles for clinical trials could not be predicted (e.g., Cmax as peak plasma concentration or area under the curve (AUC) as area under the plasma concentration time curve) after various dosage forms. The BCS/BDDCS system may only provide a rough prediction for high-fat meal effects for drug products with limited formulation optimization. In reality, the effects of a meal on drug absorption and systemic exposure can be formulation dependent, and may be exacerbated with higher doses. The prediction of whether orally dosed drug product will show a food effect in human can be challenging, especially when an insoluble compound has undergone formulation optimization. In theory, a solubility-optimized formulation can convert a BCS class 2 drug to exhibit class 1 behavior when the dose number is reduced to less than 1 (6). For example, solid dispersions, nanocrystals, self-emulsifying drug delivery systems, and soluble cyclodextrin complexes have been successfully used to improve solubility for poorly water-soluble drugs (classes 2 or 4; 11). In such cases, predictions using the conventional BCS/BDDCS classes can fail or be inadequate. Traditionally, food effects have been assessed using dog in vivo studies with some success (12–15). However, the extent of food effect (changes on Cmax, AUC) observed in dogs may not always translate directly to the human situation, and mechanistic understanding can be limited without the use of mechanistic models (16).
Recently, there has been a growing interest to evaluate the in vivo drug product performance with simulated pharmacokinetic (PK) profiles using physiologically based (PB) absorption models that are combined with a PK disposition model (17). Now, the integration of in vitro, in silico, and in vivo data is greatly facilitated due to advances of physiologically based pharmacokinetic (PBPK) modeling tools, such as GastroPlus (18, 19), Simcyp (20, 21), and STELLA (22–24). Integration of in vitro dissolution/solubility data generated in biorelevant media (25, 26) with in silico simulation tools (18, 19, 27, 28) may complement or even substitute animal models for quantitative assessment of the food effects trends. Although universally predictive food effect models remain elusive, even with advanced in silico advanced compartmental absorption transit (ACAT) models, appropriate model selection, and parameterization of biopharmaceutical properties can enable pharmaceutical scientists to predict food effect risks successfully. Here, several case examples related to practical food effect predictions and analyses across all BCS/BDDCS classes, using in silico predictions, in vitro biorelevant precipitation/solubility data, in vitro transporter kinetics, and in vivo animal models are described for all drug development stages.
MATERIALS AND METHODS
Computer Hardware and Software
GastroPlus (version 7.0, Simulations Plus, Inc, CA, USA) or Simcyp simulator (v11, Simcyp Limited, Sheffield, UK) were run on a Lenovo computer with Intel® Core™ i5 processor. These programs enable the prediction of rate and extent of oral drug absorption from the gastrointestinal tract using ACAT or the advanced drug absorption and metabolism (ADAM) model based on an absorption model originally established by Yu and Amidon (29). With the input of physicochemical properties (e.g., solubility, permeability, LogP, pKa, particle sizes), dissolution rates for solid formulations, and systemic PK parameters human concentration time profiles can be generated.
General Strategy for Human Food Effect Predictions
A flow chart for the qualitative and/or quantitative evaluations of food effects suitable for early discovery, drug candidate selection, and clinical development is shown in Fig. 1.
Fig. 1.
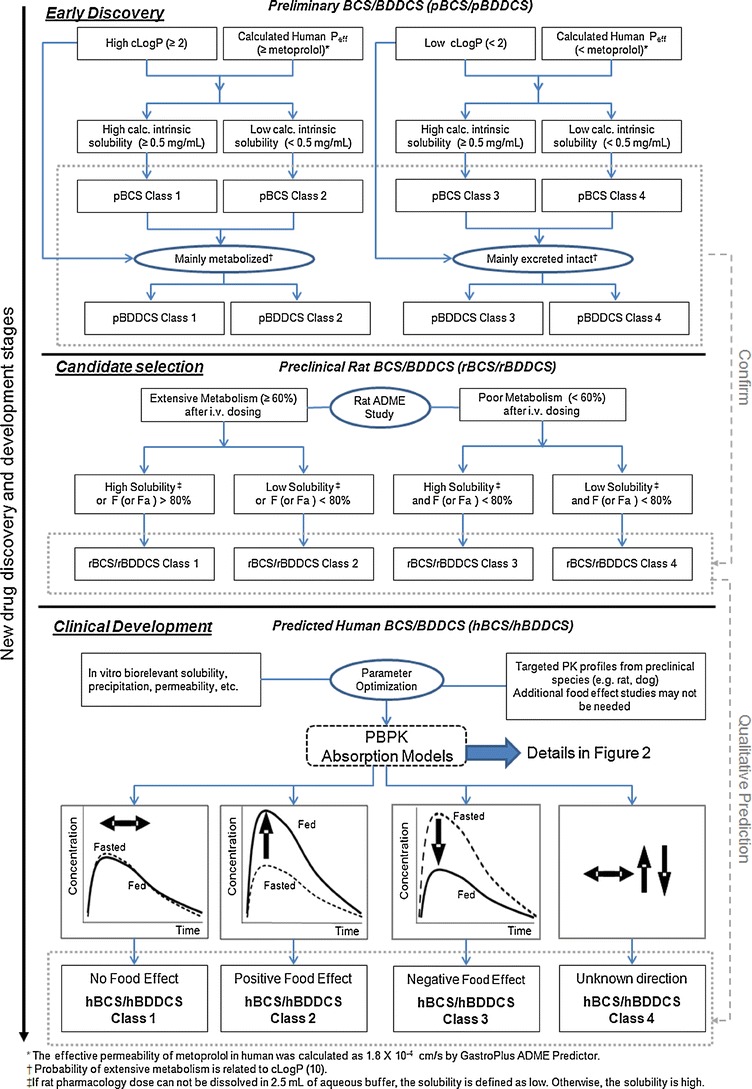
Flowchart for “practical” food effect predictions across BCS/BDDCS classes from early discovery through clinical development using in silico, in vitro, and preclinical in vivo data
During early discovery stages, readily available calculated in silico or in vitro parameters may be used to define a preliminary BCS/BDDCS (pBCS/pBDDCS). The pBDDCS can be used to identify a food effect, as in silico predicted parameters, e.g., the calculated log of the octanol/water partition coefficient (clog P), correlated with the probability of extensive metabolism for orally dosed drug (10). The pBCS/pBDDCS class defined by in silico/in vitro parameters can be confirmed once metabolism and extent of absorption have been characterized in rats; the most commonly used preclinical species (Fig. 1). When in vivo radiolabeled mass balance and excretion studies have been conducted, a preclinical BCS/BDDCS classification based on rat data (rBCS/rBDDCS) can be established. The pBCS/pBDDCS and rBCS/rBDDCS, if consistent, will provide a rational estimate of human BCS/BDDCS class (hBCS/hBDDCS) to qualitatively predict likely human food effects. The practical approach to qualitatively predict or assess food effects from early discovery through candidate selection stages is described in Table I for seven Novartis compounds across BCS/BDDCS classes. The predictability of food effects using BCS/BDDCS relies on appropriate classification using multiple parameters (e.g., fraction absorbed, permeability, dose number, transporter effects, drug metabolism after intravenous dosing) as shown in Fig. 1 and Table I. As the conventional four class BCS/BDDCS cannot provide complete mechanistic information, food effects of some drug products may be not be predicted correctly, e.g., when drug delivery systems have been designed to improve oral absorption. To overcome these limitations and to predict human exposure profiles, physiologically based absorption models have been used to link drug products properties with in vivo performance (3).
Table I.
In silico, In vitro, and In vivo Data to Predict the BCS/BDDCS Class of Seven Novartis Compounds
| Parameter | NVS732 | NVS406 | NVS562 | NVS701 | NVS001 | NVS169 | NVS113 |
|---|---|---|---|---|---|---|---|
| In silico preliminary BCS/BDDCS | |||||||
| Calculated log P, or LogDpH = 7.4 | 0.83 | 3.4 | 5.0 | 4.5 | 1.0 (log DpH = 7.4) | 5.0 | 4.6 |
| Calc. human perm., P eff (10−4 cm/s)a | 0.56 (low) | 2.47 (high) | 3.13 (high) | 1.34 (low) | 1.63 (low) | 0.21 (low) | 1.24 (low) |
| Calc. intrinsic solubility (mg/mL)b | 14.1 (high) | 0.0026 (low) | 0.0036 (low) | 0.0028 (low) | 6.77 (high) | 0.077 (low) | 0.11 (low) |
| Probability of extensive metabolism | Low | High | High | High | Low | High | High |
| pBCS/pBDDCS class | 3/3 | 2/2 | 2/2 | 4/2 | 3/3 | 4/2 | 4/2 |
| Rat BDDCS (ADME study with radiolabeled compounds) | |||||||
| Rat dose numberc | 2.8 | 1286 | 3214 | 857 | 0.007 | 6.59 | 51.4 |
| F a (%)d | 87 | 45 | >80 | 26–34 | 8.5 | 29 | 27 |
| Elimination pathway in Rat (% of dose recovered after an i.v. administration) | |||||||
| Urinary excretion of intact drug | 26.9 | 0.1 | 0.5e | 0.0 | 5.0 | 2.54 | < 0.1 |
| Biliary excretion + GI secretion | 8.6 | 9.7 | 99.5e | 15.3 | 77.7 | 15.4 | 35 |
| Percent metabolism | 64.3 | 90.2 | N/A | 84.7 | 17.3 | 81.3 | 65 |
| rBCS/rBDDCS class | 1 | 2 | 2 | 2 | 3 | 2 | 2 |
| Human BCS/BDDCS | |||||||
| Dose number for drug substances (DS)f | 0.013 | 600 | 1500 | 400 | 0.003 | 3.07, 0.51 (ME) | 24, 1.7 (SF) |
| F or F a (%), fasted condition | F: 85 | F: 20 (susp.) | F a: 50–97 (G + pred.) | F a < 30 (G + pred.) | F: ∼3.0 | F a >90 (ME) Simcyp pred. | F a > 92 (SF) G + pred. |
| Transporter effects | Weak P-gp substrate | P-gp substrate | Minimal P-gp effects | Weak P-gp substrate | Substrate for uptake transporters | P-gp substrate | Weak P-gp substrate |
| Pred. hBCS/BDDCS Classg | 1 | 2 | 2 | 2 | 3 | 2 | 2 |
| Pred. human food effects | No | Positive | Positive | Positive | Negative | No | No |
| Obs. human food effectsh | No | Positive | Positive | Positive | Negative | No | Negative |
| Obs. hBCS/hBDDCS Classg | 1 | 2 | 2 | 2 | 3 | 2i | 4j |
aHuman effective permeability was calculated by GastroPlus ADMET predictor. Metoprolol was used as a high permeability marker, thus calculated P eff > 1.8 × 10−4 cm/s indicate high permeability
bLowest water solubility (milligrams per milliliter) over the pH range 1–8 calculated by GastroPlus ADMET predictors. A value > 0.5 mg/mL was defined as high solubility
cDose number (rat) = human equivalent dose (milligrams per kilogram)/water solubility of drug substance (milligrams per milliliter)/maximum dose volume (10 mL/kg). The equivalent dose in rats is approximately 6 fold of the highest human dose tested in clinical food effects study
dFraction absorbed calculated based on the following equation (41). If non-radiolabeled compounds are used, F
a can be estimated by equation of “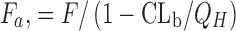 ”, where Q
H (hepatic blood flow) = 3.3 L/h/kg and CLb is the blood clearance
”, where Q
H (hepatic blood flow) = 3.3 L/h/kg and CLb is the blood clearance
eCompound related radioactivity recovered
fDose number (human) = dose (milligrams)/water solubility of drug substance or formulation (milligrams per milliliter)/maximum dose volume (250 mL). Water solubility of each compound is listed in Table II
gThe hBCS/BDDCS class is predicted based on estimated or observed human bioavailability for clinical service dosage form. The category of human BDDCS class for a compound can be formulation and dose dependent. The hBCS/BDDCS of a drug product is finalized or confirmed by the direction of observed human food effects
hThe observed magnitudes for clinical food effects for each compound were listed in Table III
iLack of food effect demonstrates potential class 1 behavior, due to low dose and optimized formulation
jNegative food effect may be the result of optimized solubility in fasted state
DS drug substance, ME microemulsion, SF solid formulation
Physiologically based models which integrate anatomical and physiological parameters of gastrointestinal tracts in preclinical species and humans, along with physicochemical drug product formulation properties, have been used to predict absorption and disposition (18–24, 30–32). These approaches have been applied during formulation development; yet their practical application in the prediction of human food effects has not been described extensively. Heimbach et al. reported the practical application of preclinical and clinical PK/PD modeling by integrating in vitro and preclinical in vivo data for the anticipation of human doses (30). Here, we used a similar strategy to predict potential food effect outcomes that can be conducted by both DMPK and formulation scientists. Briefly, the projection processes may include the following steps: (a) characterization of preclinical PK and related biopharmaceutical parameters under fasted and fed state; (b) correction interspecies differences; (c) scaling of preclinical in vitro and in vivo parameters to human situation; and (d) prediction of human PK profiles in presence or absence of meals.
Differences in physiological conditions between fasted and fed states can impact oral absorption and food effects (1). Therefore, physiological parameters, either obtained from the literature or from default ACAT values, must be carefully examined and selected. Solubility measured in biorelevant media can be more predictive for in vivo drug solubilization than solubility measured in aqueous buffers (33). In vivo dissolution in ACAT models can be calculated with modified Noyes–Whitney equations or be described by in vitro dissolution data (19). The latter approach predicted the dissolution process for formulations containing various excipients and complex matrices when in vitro and in vivo dissolution rate could be correlated (25, 26). Precipitation in the intestinal lumen can occur for poorly water soluble basic drugs at low gastric pH in the stomach (1, 22). With a meal, in vivo precipitation can be delayed due to a higher degree of supersaturation as the fed state pH is slightly lower and bile salt concentration is elevated (22). In the ACAT model, the precipitation process can be characterized by the precipitation time, which represents the time for drug particles to precipitate from solution when the local concentration exceeds the drug solubility. Direct measurements of in vivo precipitation time are impractical and difficult. Here, we describe how precipitation time can be estimated from physiologically based absorption models for BCS 2 drugs. An in vitro model for predicting the precipitation process of poorly soluble weak bases in the fasted and fed intestinal fluid has been reported, and a correlation between in vitro precipitation and in vivo absorption has been observed (34), suggesting that precipitation time could be estimated using in vitro methods. Alternatively, since dog studies are often used to predict human food effects (15), a method which estimates human precipitation time under fasted and fed condition by fitting PK data from dog food effect studies is proposed (Fig. 2).
Fig. 2.
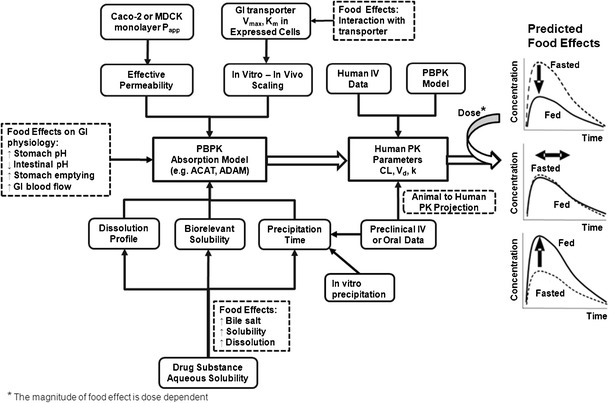
General flowchart for quantitative food effect prediction using physiologically based absorption modeling
For some drugs, absorptive transporters are involved in oral absorption. For a BCS class 3 drug which is a substrate for intestinal absorptive transporters, a “reverse pharmacokinetic” approach was used retrospectively to understand the underlying mechanism because the scaling factors of enzyme expression level from in vitro cell lines to the in vivo values in intestines remain unknown. In vitro Michaels–Menten kinetic data obtained from transfected expressed cells together with appropriated scaling factors (SF) that were optimized using existing clinical data under the fasted state are included in the PBPK model. When food inhibits absorptive transporters, SF can be optimized by fitting the observed fed PK profiles. This was done via increasing SF for the Michaels constant (Km) or decreasing SF for maximal velocity of drug uptake (Jmax). For BCS class 4 compounds, it is challenging to anticipate the direction of food effects, as food effects can be formulation and dose dependent as both solubility and permeability can be the rate limiting steps for absorption. For BCS 4 class drugs, our PBPK models obtained key absorption parameters (e.g., solubility and permeability) by fitting observed PK profiles from preclinical animal studies in which clinically relevant formulations had been administered prior to clinical trials. The optimized parameters were then used for prospective prediction of human food effects in human PBPK models. A general flow chart for quantitative food effect prediction using preclinical in vitro and in vivo data by physiologically based absorption modeling is shown in Fig. 2.
PBPK Modeling and Human Pharmacokinetics Predictions
Simulated human PK profiles were compared with observed data for each dose cohort from fasted and fed studies (Table III). Noncompartmental analysis was used to calculate PK parameters for predicted and observed human PK profiles using the WinNonlin Phoenix v6.1 (Pharsight, Sunnyvale, CA, USA). Area under the plasma concentration time curve (AUC) was calculated using trapezoidal calculation method, and peak plasma concentration (Cmax) was directly determined from the observed and predicted plasma concentration time curves. The magnitude of food effect was measured in terms of the fold changes of AUC and Cmax under fed state versus fasted state. The accuracy of prediction for food effect magnitude was evaluated by the fold error which has been published previously (Eq. 1; 33, 35–37). A prediction fold error that falls below 2 was defined as an accurate prediction.
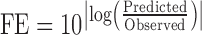 |
1 |
Table III.
Summary of Observed and Predicted Food Effects (Represented as Fold Difference) for Seven Novartis Compounds with Respect to Area Under the Plasma Concentration–Time Curve (AUC0−last) and Peak Plasma Concentration (C max)
| Compound | Modeling approacha | Dose (mg) | Formulations | Human C max ratio (fed/fasted) | Human AUC0−last ratio (fed/fasted) | Dog observed AUC (C max) Ratio (fed/fasted) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Predicted | Observed | Prediction fold error | Predicted | Observed | Prediction fold error | |||||
| NVS732 | Prospective | 100 | Tablet | 0.84 | 0.90 | 1.07 | 0.99 | 0.88 | 1.12 | |
| NVS406 | Prospective | 150 | Suspension | 4.84 | 6.11 | 1.26 | 3.98 | 4.35 | 1.09 | Suspension: 5.57 (4.62)d; liquid form: 1.30 (0.99)d |
| NVS406 | Prospective | 450 | Suspension | 7.54 | 6.12 | 1.23 | 6.86 | 4.34 | 1.58 | |
| NVS562 | Pro./Retro. | 50 | SEDDS | 0.82b, 0.88c | 0.85 | 1.04b, 1.04c | 0.97b, 0,94c | 1.01 | 1.04b, 1.08c | |
| NVS562 | Pro./Retro. | 50 | Tablet, salt | 1.48b, 1.77c | 1.60 | 1.08b, 1.11c | 1.57b, 1.70c | 1.59 | 1.01b, 1.07c | |
| NVS701 | Prospective | 200 | Capsule F1 | 1.57 | 1.71 | 1.09 | 1.71 | 1.55 | 1.10 | 2.04 (1.73)d |
| NVS701 | Prospective | 200 | Capsule F2 | 1.37 | 1.37 | 1.00 | 1.42 | 1.35 | 1.05 | 1.53 (1.39)d |
| NVS001 | Retrospective | 300 | Tablet | 0.28 | 0.28 | 1.00 | 0.27 | 0.31 | 1.15 | 0.14 (0.08)e |
| NVS169 | Prospective | 100 | ME | 0.96 | 0.52 | 1.84 | 1.14 | 1.01 | 1.13 | |
| NVS113 | Prospective | 30 | SF | 0.93 | 0.60 | 1.53 | 0.97 | 0.71 | 1.37 | 1.09 (1.03)d |
aProspective prediction and/or retrospective analysis are done to analyze the human food effects. In this study, if the parameters used in the models are not optimized using observed human PK data in the food effect study, the modeling approach is considered as prospective prediction. Retrospective analysis can be used to refine the model if parameters in the model are unknown or can not be well characterized from experiments
bPredicted with the mean precipitation time fitted against the in vivo plasma concentration–time curve
cPredicted with the mean precipitation time using in vitro dissolution/precipitation test
dResults obtained from dog studies
eResults obtained from primate studies
Models and Modeling Parameters Used
A PBPK model was established for each compound to simulate PK profile under fasted and fed condition, using available in vitro and in vivo parameters summarized in Table II for seven compounds. Table III lists the modeling approaches as “prospective prediction” or/and “retrospective analysis”. The GastroPlus default human or dog physiology (Opt-Log D model) or Simcyp default ADAM model in v11 were used, except for lower gastric pH values (pH = 2, default pH = 3–5) in pentagastrin-treated dogs. The compound’s PBPK disposition parameters were calculated directly from available intravenous PK profiles obtained from clinical studies or were projected from preclinical PK parameters and profiles (see Electronic Supplementary Material). For selected case studies, parameter sensitivity analyses were performed in two- or three-dimension response plots to explore the interaction between parameters and their influence on extent of absorption (Fa) under fasted and/or fed conditions.
Table II.
PBPK Input Modeling Parameters for Clinical Food Effect Prediction of Seven Novartis Compounds
| PBPK Model Parameter | NVS732 | NVS406 | NVS562 | NVS701 | NVS001 | NVS169 | NVS113 |
|---|---|---|---|---|---|---|---|
| Biopharmaceutical Properties | |||||||
| Acid/Base | Base | Base | Base | Base | Acid | Ampholyte | Ampholyte |
| MW (g/mol) | ∼300 | >350 | >500 | >500 | >600 | >600 | >600 |
| Measured Log D at pH 7.4 | 0.83 | 1.3 | 5.03 | 4.7 | 1.01 | 5.0 | 2.81 |
| pKaa | 7.4 | 3.7 | 5.0 | 3.5, 4.2, 6.2 | 8.4 | 5.8 (acid), 8.36 (base) | 4.8 (acid), 4.2 (base) |
| Formulations | Tablet | Suspension | (1) SEDDS, (2) Tablet | (1) Capsule F1,(2) Capsule F2 | Tablet | (1) DS, (2) ME | (1) DS, (2) SF |
| Obs. water solubility (mg/mL) | >30 | 0.003 | 0.0004 | 0.002 | 350 | (1) 0.13, (2) 0.78 (fitted)f | (1) 0.005, (2) 0.07 |
| SGF solubility at pH 1.2 (mg/mL) | 30 | 0.03 | (1)2.48, (2) 2.3 | 2 | 350 | (1) 0.65 | (1) 0.0010, (2) 0.011 |
| FaSSIF solubility at pH 6.0 | 6.38 | 0.004 | (1) 0.10, (2) 0.026 | (1) 0.0083, (2) 0.05 | N/A | (1) 0.23 | (1) 0.0085, (2) 0.5 |
| FeSSIF solubility at pH 5.4 | 6.87 | 0.10 | (1) 0.10, (2) 0.047 | (1) 0.022, (2) 0.09 | N/A | (1) 0.4 | (1) 0.045, (2) 1.0 |
| Caco-2 permeability (10−6 cm/s, A to B) | 1.5 | 17 | 2.0 | 5.3 | 0.015 | 0.5; 3.7 (passive)g | 2.4 |
| Effective Human permeabilityb(10−4 cm/s) | 0.81 | 3.0 | 0.95 | 1.71, 6.15 (dog) | 0.067 | 0.45, 1.78 (fitted) | 1.0, 3.70 (dog) |
| Particle radius (μm)c | 25 | 25 | 25 | 19 | 25 | Set as 1 | Set as 1 |
| Diffusion Coefficients (cm2/s) | 0.771 | 0.75 | 0.697 | 0.65 | 0.49 | 0.402 | 0.506 |
| Particle density (g/mL) | 1.2 | 1.2 | 1.2 | 1.2 | 1.2 | 1.2 | 1.2 |
| Precipitation time (s)—fasted | 900 | 900 | (1) 6,000, (2) 6,000 | (1) 1,800, (2) 2,500 | 900 | 900 | 900 |
| Precipitation time (s)—fed | 900 | 900 | (1) 3,500, (2) 6,000 | (1) 6,000, (2) 8,000 | 900 | 900 | 900 |
| Pharmacokinetic properties d | |||||||
| Human | 2-comp. fit-obs. | 3-comp. fit-obs. | 2-comp. fit-pred. | 2-comp. fit-pred. | 3-comp. fit-obs. | 2 -comp. fit-pred. | 2 -comp. fit-pred. |
| Preclinical: | N/A | N/A | N/A | 3-comp. fit (dog) obs. | N/A | 2-comp. fit (rat) obs. | 2-comp. fit (dog) obs. |
| Hepatic FPEe (%) | 10 | 3.5 | 1.5 | 10, 30 (dog) | 10 | 60, 95 (dog) | 2, 2 (dog) |
| Fraction unbound in plasma (f up) | 0.9 | 0.04 | 0.03 | 1.6, 0.9 (dog) | 0.5 | 0.136 | 0.006 |
| Blood to plasma partitioning ratio | 0.97 | 0.73 | 0.58 | 0.68, 0.8 (dog) | 0.83 | 0.62 | 0.57 |
| Clearance (L/h/kg) | 0.57 | 0.044 | 0.015 | 0.11, 0.54 (dog) | 0.128 | 0.38 | 0.018 |
| Volume of distribution (L/kg) | 1 | 2 | 3.39 | 2.1, 2.5 (dog) | 3.8 | 1.3 | 0.40 |
aExperimental pKa values were used for NVS732, NVS406, NVS562, NVS701, NVS001 and in silico calculated pKa values were used for NVS169 and NVS113
bThe values of effective human permeability were calculated from measured Caco-2 apparent permeability using the GastroPlus converter. A value >1.8 × 10−4 cm/s (metoprolol) indicates high human permeability. Mannitol, a low permeability marker has a value of 0.3 × 10−4 cm/s
cThe default value of 25 μm in GastroPlus built-in model was used as the particle radius if such information is not available. For NVS169 and NVS113, the particle radius was both set as 1 μm since oral solution or fine dispersion formulation were used respectively
dAll pharmacokinetic parameter listed are values for human unless annotated. For clearance and volume values listed are either predicted (pred.) via allometry or are observed (obs.) clinical data
eFirst-pass extraction (FPE = CLb/Q H) where Q H is the hepatic blood flow and CLb is the blood clearance
fBile salt dissolution model is not used in this model. The solubility is enhanced by the formulation and is assumed to be constant across the physiological pH range (1–8)
gThe apparent Caco-2 permeability was measured in the presence of a potent P-gp inhibitor and the efflux ratio (B- > A/A- > B) was reduced from 50- to 1.5-fold
Case Study 1: Food Effect Predictions for a BCS/BDDCS Class 1 Drug
NVS732 is a weak base with high solubility and moderate permeability (Tables II and III). The oral bioavailability (F) was moderate-to-high after dosing solutions with 94%, 64%, and 96%, in mice, rats, and dogs, respectively. Clinical food effect studies were conducted using a two-period, open label, single dose, and cross-over design in healthy volunteers. Subjects were randomized to receive single oral doses of 100 mg (fasted and fed). In this model, the default mean precipitation time of 900 s was used. The solubility of NVS732 was also measured in biorelevant media, i.e. simulated gastric fluid (SGF, pH 1.6 with 0.08 mM sodium taurocholate), FaSSIF (pH 6.5 with 3 mM sodium taurocholate), and FeSSIF (pH 5 with 10 mM sodium taurocholate), and the measured values were incorporated in the ACAT model.
Case Study 2: Food Effect Prediction using Biorelevant Solubility Data
NVS406 is a weak base with low, pH-dependent solubility and high permeability. In human, PK studies after IV dosing were conducted and CL and Vss were determined as 0.044 L/h/kg and 2.0 L/kg. NVS406 was mainly eliminated by hepatic metabolism and renal excretion was a minor elimination pathway (<10% of dose), regardless of the dosing route. A single dose of 150 or 450 mg was given as a coarse suspension formulation in fasted and fed condition in a clinical study. The default fasted or fed human ACAT physiology model was selected for the predictions. Solubility in different biorelevant media (SGF, FaSSIF, and FeSSIF) were measured and incorporated in ACAT model for the prediction of in vivo solubility in each gastrointestinal region.
Case Study 3: Food Effect Prediction using Two-Step Dissolution and Precipitation Data
NVS562 is a poorly soluble, lipophilic weak base (pKa = 5.0) with high calculated human effective permeability and high in vivo absorption (80%) in the rat (Table I). Formulations used in the clinical trials included a self-emulsifying solution (SEDDS) of the free base and a tablet of a stable salt. Solubility values for tablet or SEDDS formulations were measured in different biorelevant media (Table II). NVS562 dissolution and supersaturation behaviors were evaluated using a two-step dissolution method for each formulation. Initially, the drug release profile was investigated in 500 ml of simulated gastric fluid (pH 1.6) using the U.S. Pharmacopeia (USP) paddle method at a rotating speed of 50 rpm at 37°C. After 60 min, the prewarmed (37°C) FaSSIF or FeSSIF medium (2×, 500 mL) was added and drug started to precipitate. A 5 mL sample of the solution was taken out periodically from 0 to 180 min, and the same amount of the medium at the same temperature was replaced. To quantitatively compare the precipitation kinetics of both formulations, the area under the curve (AUCP, in vitro) and the area under the first moment-versus-time curve (AUMCP, in vitro) of the remaining soluble compound (%) versus time in precipitation profile were calculated from 60 min to 180 min of the incubation time. A higher AUC for a precipitation profile represented less precipitation after medium changes. The in vitro mean precipitation time (MPT) was defined as the average time for the drug to solubilize or supersaturate in the solution, and was calculated by the Eq. 2.
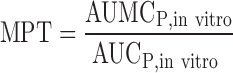 |
2 |
NVS562 human food effect was studied after a 50 mg single dose given as either SEDDS capsule or tablet under fasted and fed conditions. The calculated values of in vitro MPT were used as initial estimates of the in vivo precipitation time in the GastroPlus ACAT model.
Case Study 4: Food Effect Predictions Using the Dog Model for Weak Bases
NVS701 is a weak base with moderate to high permeability based on Caco-2 permeability and calculated human effective permeability (Peff) as listed in Tables I and II. NVS701 has low and pH-dependent solubility (Tables I and II). A significant positive food effect was observed with a capsule formulation (capsule F1) of 200 mg dose as Cmax and AUC increased by about twofold when administered 30 min after a high fat meal. As a positive food effect was undesirable, formulation approaches were proposed to slow drug precipitation in the intestine to maximize the bioavailability under the fasted state. A microemulsion formulation (Capsule F2) was developed with the expectation to provide higher solubility and to prevent precipitation. Both F1 and F2 capsules (50 mg) were tested in dogs under fasted and high fat meal conditions. GastroPlus ACAT models were developed by fitting the corresponding concentration-time profiles for each dog cohort with an optimized MPT. The obtained MPT values from the dog models were directly used in the human model to simulate the absorption under corresponding dosing scenarios. The human food effect was studied in healthy volunteers in a two-period, open label, single dose, cross-over clinical trial for both formulations. In two study periods, subjects were randomized to receive a single 200 mg dose under fasted and fed condition. Absorption modeling was performed for each formulation under fasted or fed conditions.
Case Study 5: Negative Food Effect for a BCS/BDDCS Class 3 Drug
NVS001 is a moderate to strong base (pKa = 8.4) with a relatively low lipophilicity at physiological pH (log DpH=7.4 = 1.01) (Table II). NVS001 has a high water solubility and low Caco-2 permeability (Tables I and II). NVS001 is a high affinity (Km: 3 μM) and moderate-capacity (Jmax: 29 × 10−5 nmol/min∙cm2) substrate for the p-glycoprotein and is transported by organic anion-transporting peptide OATP2B1 in HEK293 cells with an estimated Km of 72 μM and a maximum uptake clearance of 0.5 μL/min/mg protein. The majority of the absorbed oral dose was eliminated unchanged in the feces (77.7%) (Table I). NVS001 was orally administrated to fasted and non-fasted primates and food reduced Cmax and AUC(0−∞) by 92% and 85%, respectively (Table III, Fig. s2). Since NVS001 had a low permeability, both via in silico calculated Peff (Table I) and Peff derived from in vitro Caco-2 data (Table II), the intestinal uptake transporter OATP2B1 likely played a major role in the absorption. However, the organic anion transporting peptide was likely inhibited by food, as there was an over 85% reduction of the systemic exposure in non-fasted primates. Therefore, the kinetic data of uptake transporters were incorporated in the human model. The passive permeability data and p-glycoprotein kinetic data were included in both fasted and fed model. A single oral dose (300 mg) under fasted or fed state was given to healthy volunteers in a two-way and cross-over study. The human oral PK profile for the fasted state was first simulated using the GastroPlus ACAT model. The measured values of Jmax and Km of P-gp and OATP2B1 were used as inputs. The relative physiological distribution of P-gp and OATP2B1 (Table s1) have been reported previously (38) and were used in the ACAT model. However, the relative expression levels of OATP-2B1 between in vitro HEK293 cells and in vivo enterocytes remain unknown. The in vitro-to-in vivo scaling factor of Km was set as 1.0, assuming that the substrate’s affinity to the transporter are equivalent between in vitro expressed cells and in vivo enterocytes. The scaling factor for Jmax of influx transporter of OATP2B1 was then fitted against the observed plasma concentration-time curves to account for the unknown ratio of the expression level. Further, the mechanism-based analysis of human oral PK profile under fed state was performed, assuming that the inhibitory interactions on OATP2B1 between food and NVS001 are competitive. Therefore, the apparent Km of OATP2B1-mediated uptake of NVS001 in the gut tended to increase under fed state. A parameter optimization was performed to identify the best scaling factor for Km to describe the PK profile and match the AUC under fed state conditions.
Case Study 6: A priori Prediction of Food Effects for BCS/BDDCS Class 4 Drugs
NVS169 and NVS113 are both ampholytic compounds. Their aqueous solubility was low over the pH range of 1–8 for unformulated drug substance. Both compounds are substrates of P-gp and exhibited a low permeability (Table II). This suggested a risk for limited oral absorption and an uncertain direction for the food effect. To mitigate these dual challenges caused by limited solubility and permeability, a microemulsion formulation (ME) was designed for NVS169. An optimized solid formulation (SF) dosage form was designed for NVS113.
For NVS169, a ME formulation was initially tested in rats after oral dosing (1–200 mg/kg) under fasted conditions. For NVS113, a SF formulation was evaluated in the canine food effect model similar to case study 4. Simcyp ADAM or GastroPlus ACAT models were developed for rat or dog profiles to identify the in vivo solubility and/or permeability that best captured the concentration-time profiles. The optimized values were incorporated in a human model to simulate PK parameters and profiles after a single oral dose of 100 mg NVS169 or 30 mg NVS113 under fasted and fed condition a priori (Table III). Once clinical food effect profile data were available, they were used for assess the accuracy of the simulations.
RESULTS
Prospective predictions were performed to estimate the magnitude of food effects for most of compounds (Table III). For NVS001, retrospective modeling had to be done due to the unknown relative expression of uptake transporters. For NVS562, the model was further refined by fitting the parameter of MPT to observed clinical data to drive future clinical studies. The magnitudes of predicted food effects (represented as fold change of AUC and Cmax) were compared with observed clinical results. (Table III). Overall, the fold changes of AUC and Cmax (fed vs. fasted) estimated by current models for all of case studies fell within 1.6-fold of the observed values, indicating that the current modeling approach can accurately analyze food effects across BCS/BDDCS Class Compounds.
Case Example 1: NVS732
Initially, based on in silico data, NVS732 was classified as a pBCS/pBDDCS class 3 drug (Table I) due to its low calculated human Peff (Table I), its low to moderate Caco-2 permeability and its high aqueous solubility (Table I and II). In the rat in vivo, however, Fa was high, indicating near complete absorption (87%) (Table I). Thus, NVS732 is an example where low in vitro Caco-2 permeability does not always result in low absorption in vivo (Table I) (39). Rat ADME data confirmed NVS732 as an rBCS/rBDDCS class 1 drug since absorption was high, and metabolism was extensive (64.3%). In human, NVS732 demonstrated hBCS/hBDDCS class 1 drug behavior (Tables I and III). A high-fat meal did not affect the extent of absorption or AUC (Fig. 3). While Cmax was reduced by 19% and Tmax was delayed by approximately 1 h, this decrease was not considered clinically relevant. The predicted plasma concentration versus time curve and PK parameters were in agreement with observed data (Fig. 3). The GastroPlus model successfully predicted bioequivalent exposure under fasted and fed dosage regimen (Table III). Predicted and observed results were consistent with the BCS/BDDCS, which stipulates that Class 1 drugs generally do not show any food effects (1, 7).
Fig. 3.
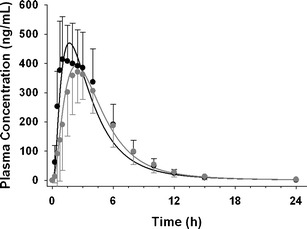
Observed plasma concentrations are shown in solid circles (black fasted, gray fed). Simulated PK profile are presented as solid curves (black fasted, gray fed) after a single oral dose of 100 mg NVS732 in healthy volunteers
Case Example 2: NVS406
Based on in silico calculations, NVS406 was classified as a pBCS/pBDDCS Class 2 drug (Table I). NVS406 solubility was low in fasted state (0.004 mg/mL in FaSSIF) and fed state (0.1 mg/mL in FeSSIF). Rat ADME data confirmed NVS406 as a rBCS/rBDDCS Class 2 drug, as metabolism was >90% (Table I), with moderate absorption (45%) likely limited by low GI solubility. In human, NVS406 showed a significant positive food effect; which is consistent with hBCS/BDDCS Class 2 drugs (Table I and III). The mean of AUC and Cmax increased by ∼4.4 or ∼4.3-fold and 6.1 or 6.1-fold under fed state conditions after a single dose of 150 mg or 450 mg of an oral coarse suspension, respectively (Table III). The magnitude of this food effect and plasma concentration profiles were well predicted when compared with observed data (Fig. 4, Table III). For the cohort receiving a 450 mg dose without food, the initial absorption and distribution phase from 0 to 24 h was well captured by the model, but the concentrations after 24 h of dose were underestimated (Fig. 4b). Overall, the predicted magnitude of food effect was still in good agreement with the observed results. Prediction fold-errors were less than 1.6 for AUC and 1.3 for Cmax, respectively (Table III). In the fasted state, a flattened terminal concentration-time curve was observed, and the concentration after 12 h of the dose appeared to remain constant for up to 48 h and then declined more rapidly. One explanation may be a prolonged and slow absorption in the colon. According to the regional absorption plot (Fig. 4c), colonic absorption from the caecum and ascending colon accounted for ∼45% of the total dose absorbed in the fasted state. However, the significance of the colonic absorption was attenuated in the fed state and only accounted for less than 13% of the total absorbed dose. The main absorption area under fed state located in the upper intestine where sufficient drug is available as a result of improved in vivo solubility, whereas the extent of colonic absorption was not pronounced due to the poor aqueous solubility. To design a formulation that could mitigate the positive food effect, it was important to explore the interaction between solubility and dose and their impacts on the extent of absorption under both fasted and fed conditions. A parameters analysis (PSA) was conducted under fasted and fed condition and three-dimension surface response plots (Fig. 4d, e) elucidated that Fa has a steep drop from ∼80% to ∼30% when duodenal solubility decreased from 1 mg/mL to 0.1 mg/mL under both fasted and fed states. Fa gradually decreased when doses were increased from 200 to 1,000 mg. This information was provided to guide formulation development of a new dosage form that could significantly enhance solubility along the gastrointestinal tract. To this end, a new liquid formulation and a coarse suspension were tested in the dog food effect model using a single oral dose of 5 mg/kg (equivalent to a ∼190 mg dose in 70 kg adult). Significant differences for mean AUC and Cmax values were not observed for the liquid formulation between fasted and fed condition whereas a strong positive food effect (> 4.5 fold increase on AUC and Cmax) was observed for the coarse suspensions (Table III). This suggested that the utility of a lipid-based formulation appeared to be an effective strategy to boost dissolution/solubility and enhance the oral bioavailability of a poorly water soluble compound under the fasted state.
Fig. 4.
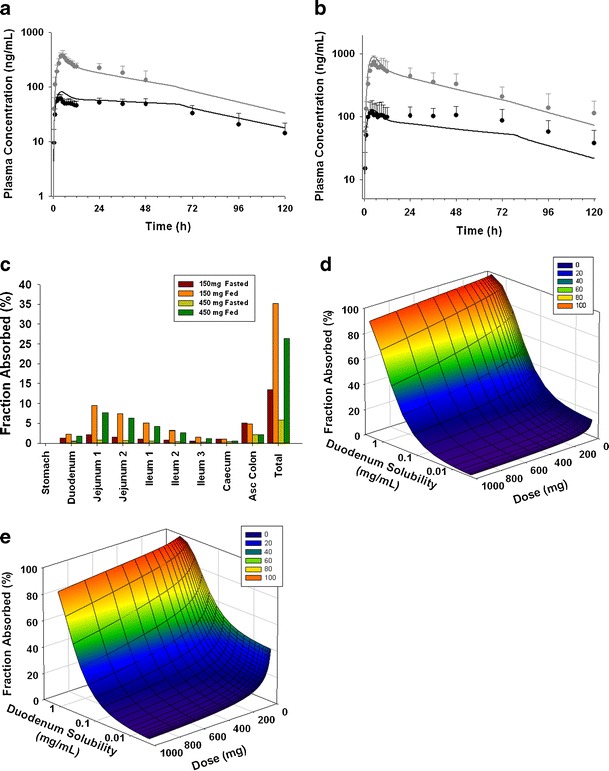
Observed plasma concentrations of NVS406 are shown in solid circles (black fasted, gray fed). Simulated PK profile are presented as solid curves (black fasted, gray fed) after a single administration of a 150 mg of NVS406 and b 450 mg of NVS406 given as an oral suspension in healthy volunteers. c Fraction of regional absorption for NVS406 after a 150 or 450 mg dose in fasted and fed healthy volunteers. Surface response plot of the change of fraction absorbed (F a) with respect to duodenum solubility (representing in vivo solubility) and dose under d fasted or e fed condition
Case Example 3: NVS562
NVS562 was classified between pBCS/rBDDCS classes 2–4 due to a low aqueous solubility and a moderate permeability (Table I). For NVS562, the in silico human Peff was high (Table I), while the measured Caco-2 permeability was low leading to a low derived human Peff permeability of 0.95 × 10−4 cm/s which less than that for metoprolol (1.8 × 10−4 cm/s) (Table II). As rat in vivo metabolism data were not available, NVS562 was still classified as rBCS/rBDDCS class 2 drug as rat in vivo absorption was high (> 80%) (Table I, Fig. 1).
The biorelevant solubility values for SEDDS of the free base and tablet formulations with the salt were listed in Table II. For NVS562, human food effects were formulation dependent (Table III). With SEEDS capsules, a high-fat meal did not affect the extent of absorption and the AUC values were unchanged. The geometric mean of Cmax was reduced by 15% and Tmax was delayed by approximately 3 h in the fed state. However, with a tablet formulation, the geometric mean of AUC and Cmax increased by 1.6 and 1.6 fold and Tmax was delayed by approximately 2 h (Table III). The in vitro two-step dissolution results are shown in Fig. 5a. Apparently supersaturation was maintained for over 2 h with the SEDDS formulation in FaSSIF and FeSSIF media, thus a high fat meal caused only negligible effects on drug dissolution and precipitation. The SEDDS formulation likely enhanced NVS562 solubility in the intestine, thus imparting desirable hBCS/BDDCS class 1 characteristics with minimal food effects on drug exposure. On the contrary, the tablet formulation did not mitigate positive food effects (Table III) likely due to precipitation under fasted and fed state, which was identified by the two-step dissolution test. The MPT values obtained from the in vitro and in vivo method as well as the AUC values predicted were summarized in Table s2. The plasma concentration time curves simulated by the model using in vivo optimized MPT were represented in Fig. 5b and c. For the SEDDS formulations under fasted and fed condition as well as a tablet formulation under fed condition, a model with in vitro MPT values (3,200–3,500 s) resulted in acceptable prediction PK profiles (data not shown) for AUC. A minor improvement of the simulation accuracy was achieved when MPT values were approximately doubled (e.g. 6,000 s; Fig. 5b). For the tablet formulation in the fasted state, the in vitro MPT (2,210 s, Table s2) slightly underestimated the extent of absorption. An MPT of 3,500 s best described the observed PK profile (Fig. 5c). Although the AUC value was slightly under predicted with the tablet formulation in the fasted state, the food effect trend was well predicted for both SEDDS and tablet formulations using in vitro MPT values (Table s2). A parameter analysis (PSA) for Fa change with respective to MPT is shown in Fig. 5d. MPT has less influence on Fa for SEDDS as solubility of NVS562 is sufficient, irrespective of pH and meal conditions. In such scenarios, the extent of absorption was always nearly complete (> 80%) and thereby bias of food effects prediction was generally minor using the PBPK model. On the other hand, for suspensions where solubility of NVS562 is limited, slower precipitation rate (or long MPT) prolonged the “supersaturation” state of compounds and likely allowed higher extent of absorption, suggesting that MPT is potentially a key driver for Fa in suspensions. Thus, inaccurate estimation of MPT can result in a large bias towards the prediction of food effects.
Fig. 5.
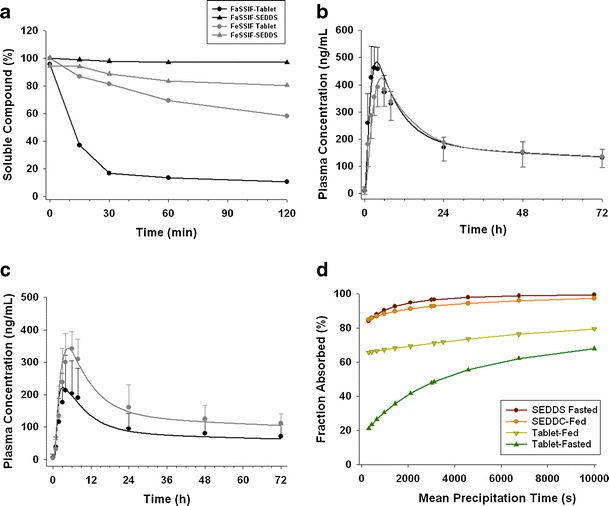
a In vitro precipitation profiles of two formulations containing NVS562 in FaSSIF or FeSSIF media. Observed plasma concentrations are shown in solid circles (black fasted, gray fed) and simulated PK profile are presented as solid curves (black fasted, gray fed) after a single administration of 150 mg of NVS562 given as a b SEDDS or c tablet in healthy volunteers. d Parameter sensitivity of precipitation time to the change of fraction absorbed
Case Example 4: NVS701
NVS701 was identified as a pBCS/pBDDCS class 2 to 4 due to a low aqueous solubility, and a moderate permeability (Table I). Rat ADME data confirmed NVS701 as an rBCS/rBDDCS 2 drug due to extensive metabolism (84.7%) and low absorption. In human, NVS701 demonstrated hBCS/hBDDCS class 2 drug behavior (Table I) and a positive food effect (Table III). A high-fat meal increased the extent of absorption following the administration of Capsules F1 and Capsule F2 similarly in dogs and humans (Table III). For both formulations, the values of MPT optimized by fitting plasma concentration-time curves under fed state were higher than the values under fasted conditions, suggesting that solubilized NVS701 can remain in a supersaturated state in the intestine for a longer time after high fat meals. The simulated results were also in agreement with the fact that meals can stimulate bile flow and enhance solubility of poorly water soluble compound in small intestine (7, 40–42). The resulting human model captured the mean observed data reasonably well in fasted and fed conditions for both formulations using the MPT obtained from the optimized dog model (Fig. 6). The predicted magnitude of food effects in human in terms of changes on AUC and Cmax was less than 1.1 fold of the observed effects (Table III).
Fig. 6.
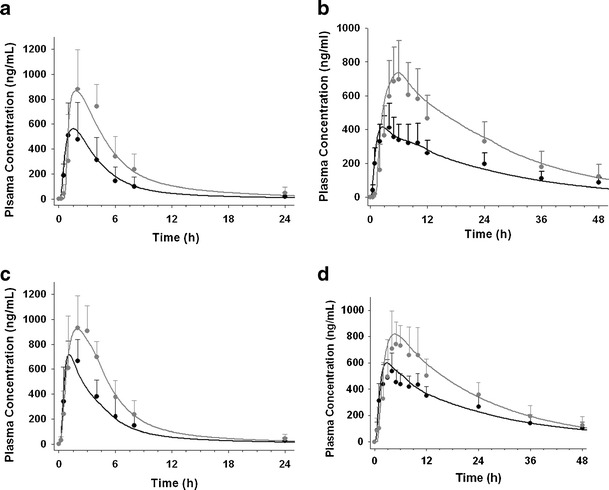
Observed plasma concentrations are shown in solid circles (black fasted, gray fed) and simulated PK profile are presented as solid curves (black fasted, gray fed) after a single administration of 50 mg of NVS701 given as a a marketed capsule (capsule F1) or c solid suspended microemulsion (capsule F2) in dogs or 200 mg of NVS701 given as a b capsule F1 or d capsule F2 in healthy volunteers
Case Example 5: NVS001
Based on in silico and early discovery data, NVS001 was classified as a pBCS/pBDDCS Class 3 drug due to its high solubility and low permeability (Table I). In the rat, metabolism was low (17.3%), thus NVS001 was classified as a pBCS/pBDDCS class 3 drug. In human, NVS001 also demonstrated hBCS/hBDDCS class 3 behaviors. Food greatly reduced NVS001 Cmax by 72% and AUC0−∞ by 69% (Table III). To best describe the concentration-time curve of NVS001 in the fasted state, an in vitro-in vivo scaling factor for Jmax of 0.055 was fitted against the data, suggesting that the expression level of OATP2B1 in the gut are lower than those in the in vitro expressed HEK293 cells. For oral absorption modeling, the same scaling factor for Jmax was used in the fasted and fed state since transporter expression level was constant between fasted and fed conditions. On the other hand, a scaling factor for Km was estimated to be 50 to fit the concentration-time curve (Fig. 7). The fitted Km scaling factor in the fed state implied that the food components competitively inhibited the uptake transporter of OATP2B1 and resulted in reduced NVS001 absorption. Overall, the observed negative food effect was consistent with the predicted results obtained with the ACAT model (Table III).
Fig. 7.
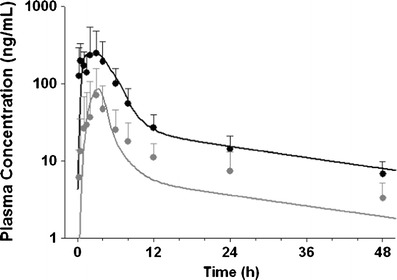
Observed plasma concentrations are shown in solid circles (black fasted, gray fed) and simulated PK profile are presented as solid curves (black fasted, gray fed) after a single administration of 300 mg of compound NVS001 given as a tablet in healthy volunteers
Case Example 6: NVS169 and NVS113
NVS169 and NVS113 were identified as borderline pBCS/pBDDCS class 4 and 2 compounds based on in silico prediction, respectively (Table I) with low solubility and low permeability, but high clogP. The in vitro measured solubility for NVS113 was even lower than the in silico predicted solubility (Tables I and II). The rat ADME study showed metabolism higher than 60% for both NVS113 (65%) and NVS169 (81.3%) (Table I), thus resulting in an rBCS/rBDDCS class 2. During clinical development, formulation efforts greatly increased the solubility/dissolution for both drug substances (Table II). For NVS169, the fraction absorbed in rats exceeded 80% with an ME formulation, suggesting that absorption was not limited by solubility. The values of Peff and solubility that can best describe the rat PK profiles were 1.78 × 10−4cm/s and 0.78 mg/mL. Using these values in the human model, no changes in AUC and Cmax with or without meals were predicted. The predictions were partly in agreement with the clinical observations, where AUC was not significantly altered, but Cmax decreased by 40% (Table III). For NVS113, no food effect was observed in canine model. Similarly, in vivo solubility values were obtained and used in human PBPK model. The human model predicted an insignificant food effect with less than 10% decrease of AUC and Cmax when comparing PK data under fed and fasted state. However, the observed clinical data showed a more significant reduction of AUC (∼30%) in the fed state (Table III). The reasons are not entirely understood, but may be due to an unknown drug complexation with food components or possible transporter interactions.
DISCUSSIONS
Food can induce changes in physiological conditions, such as delayed in gastric emptying, change of gastrointestinal pH, stimulation of bile flow, and interaction of intestinal influx or efflux transporters (1, 2, 8, 9). A positive food effect is manifested by a higher systemic exposure when the drug is given with food compared to the fasted state and mainly seen for BCS/BDDCS 2 drugs. A negative food effect is manifested by a reduced exposure when the drug is given with food and is mainly seen for BCS/BDDCS 3 drugs. BCS/BDDCS Class 1 drugs often show no food effect. For BCS/BDDCS 4 drugs food effects is often difficult to predict, as they can be dose and formulation dependent. Presence of a food effect can impact drug labeling (2) and convenience to the patient. Briefly, a food effect is observed if the 90% confidence interval for the ratio of population geometric means between fed and fasted treatments fails to meet the limits of 80–125% for either AUC or Cmax (2).
Potential human food effects can be predicted to aid product or formulation development (43) at all drug development stages using in silico, in vitro, and in vivo methods as shown in Fig. 1. Here, both BCS and BDDCS classifications (1, 7) aided in identifying likely food effects for seven proprietary compounds from early discovery through early development using readily available data (Fig. 1, Table I).
For early discovery, a preliminary pBCS/pBDDCS class is established for a NCE when little or no in vivo ADME properties are available. To establish a pBCS, key in silico parameters (e.g. human effective permeability, and intrinsic solubility) can be calculated using e.g. the GastroPlus ADMET predictor. Alternative in silico tools are available (e.g. ALOGPS, VolSurf, etc.) to calculate these and related physicochemical parameters. As shown in Fig. 1, we assigned a preliminary BCS class of NCEs based on a calculated intrinsic solubility and a human effective permeability (Peff). A differentiating solubility value of 0.5 mg/mL was arbitrarily chosen; as this concentration is achieved for a 125 mg efficacious dose given with 250 mL water. For permeability assessment, metoprolol was chosen as a high permeability reference compound (44, 45) (Tables I and II). Calculated LogP (cLogP) values were used to describe the probability of extensive metabolism. This was done retrospectively, as this approach was only recently published (10). Compounds with a cLogP value greater than 2, are likely to be eliminated mainly via metabolism and can be assigned to Class 1 or Class 2 in the pBDDCS (10). Our practical preliminary pBCS/pBDDCS classes could thus identify food effect risks early and guide a formulation strategy of the NCE during later development stages.
During the candidate selection stage, rat in vivo mass balance/disposition data from radiolabeled drug substances allowed estimation of Fa and extent of metabolism (46) leading to a rat based BCS/BDDCS or rBCS/rBDDCS (Fig. 1). High metabolism or extensive metabolism were defined if >60% of the drug was metabolized after intravenous dosing and resulted in an rBDDCS class 1 or 2, depending on solubility, F, or Fa. The rBCS/rBDDCS was used to confirm or complement the pBCS/pBDDCS, derived via the earlier in silico approach. For the seven Novartis compounds, there was a general agreement with the food effects that could be expected from the rBCS/rBDDCS and pBCS/pBDDCS when compared to observed food effects in the clinic (Table I). Thus, overall, the pBCS/pBDDCS and rBCS/rBDDCS classes predicted the likely direction clinical food effects.
In early clinical development, quantitative food effect predictions along with exposure profiles are typically desired by project teams, and often more than one formulation is under consideration for human trials. Advanced PBPK modeling tools readily allow the mechanism-based simulation of concentration-time profiles for liquid or solid dosage forms both in the fasted and fed states using physiological based absorption model and input parameters such as physicochemical properties and biorelevant solubility (33). As shown in Fig. 2, human systemic PK parameter (e.g., Vss, CL), could be scaled from preclinical data using, e.g., the latest PhRMA scaling methods (47, 48) when human IV data were not available. In vivo relevant biopharmaceutical properties, some of which have not been extensively discussed in the literature, i.e., drug precipitation, transporter interactions, and formulation effects were included our absorption models. Here, we provide case examples and potential solutions on how to generate in vivo precipitation and solubility data toward human food effect profile predictions. We used in vivo observed dog data to describe an ACAT model and in vivo solubility/precipitation data can be obtained by fitting the observed dog exposure profiles (cases NVS701, NVS562). These data were then used for human profile predictions (shown schematically in Fig. 2). Food effects can be formulation dependent and can sometimes be mitigated with optimized formulations (case NVS562). We also analyzed transporter involvement in food effects for a BCS/BDDCS class 3 compound (case NVS001). The impacts of formulation on the food effects of BCS class 4 compounds were studied with prospective predictions (NVS169, NVS113).
For weak bases, precipitation can limit oral exposure both in fasted and fed states (1). To model in vivo precipitation, an MPT parameter was used to represent the time of supersaturation under fasted and fed conditions. We proposed two methods for this parameter estimation: (1) calculating the AUMC/AUC ratio based on drug precipitation profiles obtained from a two-step biorelevant dissolution test or (2) fitting this parameter against the observed PK profiles in dogs under fasted and fed conditions. For NVS562, the simulation results showed that the parameter of MPT obtained from the two-step biorelevant dissolution test underestimated the human plasma concentration after a single suspension dose under fasted state. A donor-to-acceptor transfer model has been reported to simulate the slow transfer process for poorly soluble weak bases from stomach to intestine, which may potentially better predict MPT (34). However, this approach has not been fully validated. Here, the human plasma concentration time profiles of NVS701 were well described using the MPT obtained from fitting dog PK data under fasted or fed condition. Dog is the most studied species for predicting human food effects (12–14) as clinical service forms (e.g., capsule or tablet) can be directly administered to dogs. For NVS701, the dog model was not only used as a surrogate human food effect model but was a useful tool to generate the MPT in human models. To our knowledge, no prior attempts have been published to successfully apply optimized MPT data from canine model to quantitatively predict the human food effects. Yet, the prediction accuracy using such an approach may depend on the compound properties and formulations. It was critical that human relevant disposition parameters and physicochemical properties are used. If the MPT parameter is found to be inadequate and not translatable between dog and human, then the model may have to be refined when observed human PK data become available.
Transporter interactions can be important for BCS class 3 compounds (7). A priori predictions of negative food effects caused by food and intestinal influx transporter interactions can be difficult as inhibitory effects of dietary substances on transporter cannot be measured. A retrospective analysis demonstrated the impact of food on intestinal transporter inhibition by optimizing the value of an in vitro to in vivo scaling factor for Km for the related uptake transporters. If absorption of BCS class 3 compounds is mediated by an intestinal influx transporter, high-fat meals will decrease the extent of absorption due to inhibition or competition of uptake transporters in the intestine. Wu and Benet had noted that the overall exposure changes with class 3 compounds depend upon whether meals have more pronounced effects on the efflux or influx transporters in the absorption process. An unexpected increase or minimal meal effect in the extent of absorption can be observed (e.g., acyclovir). Due to the complexity of food and transporter interaction, a preclinical model may provide a useful insight into the direction of food effects. For NVS001, the food effects observed in primates were similar to those observed in human with over 85% reduction in AUC (Fig. s2 and Fig. 7). Such results can serve as an important translatable link for establishing mechanism-based human PBPK models.
The prediction of clinical food effects for BCS/BDDCS class 2 to 4 compounds (both of which can exhibit Class 4 behavior at higher doses or with suboptimal formulations) can be very challenging. We successfully predicted the lack of positive food effects for two BCS/BDDCS class 2 to 4 compounds (Table III). NVS169 was extensively metabolized and was likely not a substrate for absorptive transporters, similar to BCS Class 1 and 2 compounds (7). Apparently low solubility can be a rate limiting factor in NVS169 oral absorption. The calculated dose number at 100 mg for the drug substance (DS) was 3.1, which was successfully reduced to 0.51 with a microemulsion (ME) formulation (Tables I and II). Thus, solubility is currently not rate-limiting step for drug absorption. This represented a successful example where formulations can change a compound’s BCS Class and a reduced risk for food effects (as was also shown for NVS562). For NVS113, also no positive food effect had been predicted (Table III). Interestingly, a significant negative food effect was observed in the clinic—a trend which had not been identified in the dog model (Table III). Thus for NVS113, as with other Class 4 drugs, food effects are apparently mediated by factors other than solubility and which can include drug-food complexation or uptake transporter interactions. It is likely that absorptive intestinal transporters were involved in the absorption process and food may inhibit the drug uptake process, resulting in a negative food effects. Taken together, these two examples indicated the complexity in prediction and mitigation of food effects for BCS Class 4 compounds. It further illustrated that a compounds’ BCS/BDDCS class can be formulation-dependent.
CONCLUSIONS
Integration of in silico, in vitro, and preclinical in vivo data with ACAT PBPK models, allowed early identification of likely food effect risks and exposure profile predictions across BCS/BDDCS class 1 through 4 compounds. A practical preclinical BCS/BDDCS, which relies mainly on calculated parameters, could be readily implemented by DMPK or formulation scientists for all drug development stages. Rat in vivo absorption and metabolism data aided in identifying or confirming the likely human BDDCS class. Various in vitro and in vivo preclinical tools were successfully integrated with commercially available software to conduct physiologically based human absorption and exposure modeling for orally-dosed compounds with or without meals. Formulation-dependent food effects, once understood, could sometimes be solved with optimized formulations. Overall, integrated and practical approaches provided representative case examples for food effect predictions and risk identification prior to clinical studies. Yet, challenges for food effect predictions do remain, especially for BCS/BDDCS class 3 and 4 drugs due to insufficient knowledge of in vivo intestinal transporter expression levels, meal effects on intestinal transporters, and complex food-drug interactions.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
{kind=link}
JPEG 26 kb
{kind=link}
JPEG 27 kb
{kind=link}
JPEG 25 kb
DOCX 120 kb
Acknowledgement
The authors would like to thank Dr. Akash Jain and the members of the Novartis Food Effect Quality Plus team for many helpful discussions.
Conflict of Interests
None.
Footnotes
Tycho Heimbach and Binfeng Xia contributed equally to this work.
References
- 1.Fleisher D, Li C, Zhou Y, Pao LH, Karim A. Drug, meal and formulation interactions influencing drug absorption after oral administration. Clinical implications. Clin Pharmacokinet. 1999;36(3):233–254. doi: 10.2165/00003088-199936030-00004. [DOI] [PubMed] [Google Scholar]
- 2.US FDA. Food–effect bioavailability and fed bioequivalence studies. In: Guidance for industry. http://www.fda.gov/downloads/regulatoryinformation/guidances/ucm126833.pdf. 2002. Accessed 02 Jun 2012.
- 3.Zhang X, Lionberger RA, Davit BM, Yu LX. Utility of physiologically based absorption modeling in implementing quality by design in drug development. AAPS J. 2011;13(1):59–71. doi: 10.1208/s12248-010-9250-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hendeles L, Weinberger M, Milavetz G, Hill M, 3rd, Vaughan L. Food-induced “dose-dumping” from a once-a-day theophylline product as a cause of theophylline toxicity. Chest. 1985;87(6):758–765. doi: 10.1378/chest.87.6.758. [DOI] [PubMed] [Google Scholar]
- 5.Wilder BJ, Leppik I, Hietpas TJ, Cloyd JC, Randinitis EJ, Cook J. Effect of food on absorption of dilantin kapseals and mylan extended phenytoin sodium capsules. Neurology. 2001;57(4):582–589. doi: 10.1212/WNL.57.4.582. [DOI] [PubMed] [Google Scholar]
- 6.Amidon GL, Lennernas H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification—the correlation of in-vitro drug product dissolution and in-vivo bioavailability. Pharmaceut Res. 1995;12(3):413–420. doi: 10.1023/A:1016212804288. [DOI] [PubMed] [Google Scholar]
- 7.Wu CY, Benet LZ. Predicting drug disposition via application of BCS: transport/absorption/elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharm Res. 2005;22(1):11–23. doi: 10.1007/s11095-004-9004-4. [DOI] [PubMed] [Google Scholar]
- 8.Benet L. Z. WCY. Using a biopharmaceutics drug disposition classification system to predict bioavailability and elimination characteristics of new molecular entities. Somerset, NJ: NJDMDG. 2006.
- 9.Custodio JM, Wu C-Y, Benet Leslie Z. Predicting drug disposition, absorption/elimination/transporter interplay and the role of food on drug absorption. Adv Drug Deliv Rev. 2008;60(6):717–733. doi: 10.1016/j.addr.2007.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Benet LZ, Broccatelli F, Oprea TI. BDDCS applied to over 900 drugs. AAPS J. 2011;13(4):519–547. doi: 10.1208/s12248-011-9290-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singh A, Worku ZA, Van den Mooter G. Oral formulation strategies to improve solubility of poorly water-soluble drugs. Expert Opin Drug Deliv. 2011;8(10):1361–1378. doi: 10.1517/17425247.2011.606808. [DOI] [PubMed] [Google Scholar]
- 12.Lui CY, Amidon GL, Berardi RR, Fleisher D, Youngberg C, Dressman JB. Comparison of gastrointestinal Ph in dogs and humans—implications on the use of the beagle dog as a model for oral absorption in humans. J Pharm Sci. 1986;75(3):271–274. doi: 10.1002/jps.2600750313. [DOI] [PubMed] [Google Scholar]
- 13.Meyer JH, Dressman J, Fink A, Amidon G. Effect of size and density on canine gastric-emptying of nondigestible solids. Gastroenterology. 1985;89(4):805–813. doi: 10.1016/0016-5085(85)90576-1. [DOI] [PubMed] [Google Scholar]
- 14.Akimoto M, Nagahata N, Furuya A, Fukushima K, Higuchi S, Suwa T. Gastric pH profiles of beagle dogs and their use as an alternative to human testing. Eur J Pharm Biopharm. 2000;49(2):99–102. doi: 10.1016/S0939-6411(99)00070-3. [DOI] [PubMed] [Google Scholar]
- 15.Lentz KA, Quitko M, Morgan DG, Grace JE. Development and validation of a preclinical food effect model. J Pharm Sci. 2007;96(2):459–472. doi: 10.1002/jps.20767. [DOI] [PubMed] [Google Scholar]
- 16.Russell WMS, Burch RL. The principles of humane experimental technique. London: Methuen & Co. Special edition published by Universities Federation for Animal Welfare (UFAW), 1992; 1959.
- 17.Huang SM. PBPK as a tool in regulatory review. Biopharm Drug Dispos. 2012;33(2):51–52. doi: 10.1002/bdd.1777. [DOI] [PubMed] [Google Scholar]
- 18.Lukacova V, Woltosz WS, Bolger MB. Prediction of modified release pharmacokinetics and pharmacodynamics from in vitro, immediate release, and intravenous data. AAPS J. 2009;11(2):323–334. doi: 10.1208/s12248-009-9107-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parrott N, Lukacova V, Fraczkiewicz G, Bolger MB. Predicting pharmacokinetics of drugs using physiologically based modeling—application to food effects. AAPS J. 2009;11(1):45–53. doi: 10.1208/s12248-008-9079-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vieira MLT, Zhao P, Berglund EG, Reynolds KS, Zhang L, Lesko LJ, et al. Predicting drug interaction potential with a physiologically based pharmacokinetic model: a case study of telithromycin, a time-dependent CYP3A inhibitor. Clin Pharmacol Ther. 2012;91(4):700–708. doi: 10.1038/clpt.2011.305. [DOI] [PubMed] [Google Scholar]
- 21.Shaffer CL, Scialis RJ, Rong HJ, Obach RS. Using Simcyp to project human oral pharmacokinetic variability in early drug research to mitigate mechanism-based adverse events. Biopharm Drug Dispos. 2012;33(2):72–84. doi: 10.1002/bdd.1768. [DOI] [PubMed] [Google Scholar]
- 22.Shono Y, Jantratid E, Dressman JB. Precipitation in the small intestine may play a more important role in the in vivo performance of poorly soluble weak bases in the fasted state: case example nelfinavir. Eur J Pharm Biopharm. 2011;79(2):349–356. doi: 10.1016/j.ejpb.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 23.Shono Y, Jantratid E, Janssen N, Kesisoglou F, Mao Y, Vertzoni M, et al. Prediction of food effects on the absorption of celecoxib based on biorelevant dissolution testing coupled with physiologically based pharmacokinetic modeling. Eur J Pharm Biopharm. 2009;73(1):107–114. doi: 10.1016/j.ejpb.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 24.Shono Y, Jantratid E, Kesisoglou F, Reppas C, Dressman JB. Forecasting in vivo oral absorption and food effect of micronized and nanosized aprepitant formulations in humans. Eur J Pharm Biopharm. 2010;76(1):95–104. doi: 10.1016/j.ejpb.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 25.Nicolaides E, Symillides M, Dressman JB, Reppas C. Biorelevant dissolution testing to predict the plasma profile of lipophilic drugs after oral administration. Pharm Res. 2001;18(3):380–388. doi: 10.1023/A:1011071401306. [DOI] [PubMed] [Google Scholar]
- 26.Dressman JB, Reppas C. In vitro-in vivo correlations for lipophilic, poorly water-soluble drugs. Eur J Pharm Sci. 2000;11:S73–S80. doi: 10.1016/S0928-0987(00)00181-0. [DOI] [PubMed] [Google Scholar]
- 27.Parrott N, Lave T. Prediction of intestinal absorption: comparative assessment of GASTROPLUS (TM) and IDEA (TM) Eur J Pharm Sci. 2002;17(1–2):51–61. doi: 10.1016/S0928-0987(02)00132-X. [DOI] [PubMed] [Google Scholar]
- 28.Kuentz M, Nick S, Parrott N, Rothlisberger D. A strategy for preclinical formulation development using GastroPlus (TM) as pharmacokinetic simulation tool and a statistical screening design applied to a dog study. Eur J Pharm Sci. 2006;27(1):91–99. doi: 10.1016/j.ejps.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 29.Yu LX, Amidon GL. Characterization of small intestinal transit time distribution in humans. Int J Pharm. 1998;171(2):157–163. doi: 10.1016/S0378-5173(98)00174-4. [DOI] [Google Scholar]
- 30.Heimbach T, Lakshminarayana SB, Hu WY, He HD. Practical anticipation of human efficacious doses and pharmacokinetics using in vitro and preclinical in vivo Data. AAPS J. 2009;11(3):602–614. doi: 10.1208/s12248-009-9136-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xia B, Heimbach T, Lin TH, He H, Wang Y, Tan E. Novel physiologically based pharmacokinetic modeling of patupilone for human pharmacokinetic predictions. Canc Chemother Pharmacol. 2012;69(6):1567–1582. doi: 10.1007/s00280-012-1863-5. [DOI] [PubMed] [Google Scholar]
- 32.Kesisoglou F, Wu YH. Understanding the effect of API properties on bioavailability through absorption modeling. AAPS J. 2008;10(4):516–525. doi: 10.1208/s12248-008-9061-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones HM, Parrott N, Ohlenbusch G, Lave T. Predicting pharmacokinetic food effects using biorelevant solubility media and physiologically based modelling. Clin Pharmacokinet. 2006;45(12):1213–1226. doi: 10.2165/00003088-200645120-00006. [DOI] [PubMed] [Google Scholar]
- 34.Kostewicz ES, Wunderlich M, Brauns U, Becker R, Bock T, Dressman JB. Predicting the precipitation of poorly soluble weak bases upon entry in the small intestine. J Pharm Pharmacol. 2004;56(1):43–51. doi: 10.1211/0022357022511. [DOI] [PubMed] [Google Scholar]
- 35.De Buck SS, Sinha VK, Fenu LA, Nijsen MJ, Mackie CE, Gilissen RAHJ. Prediction of human pharmacokinetics using physiologically based modeling: a retrospective analysis of 26 clinically tested drugs. Drug Metab Dispos. 2007;35(10):1766–1780. doi: 10.1124/dmd.107.015644. [DOI] [PubMed] [Google Scholar]
- 36.De Buck SS, Sinha VK, Fenu LA, Gilissen RA, Mackie CE, Nijsen MJ. The prediction of drug metabolism, tissue distribution, and bioavailability of 50 structurally diverse compounds in rat using mechanism-based absorption, distribution, and metabolism prediction tools. Drug Metab Dispos. 2007;35(4):649–659. doi: 10.1124/dmd.106.014027. [DOI] [PubMed] [Google Scholar]
- 37.Poulin P, Theil FP. Prediction of pharmacokinetics prior to in vivo studies. II. Generic physiologically based pharmacokinetic models of drug disposition. J Pharm Sci. 2002;91(5):1358–1370. doi: 10.1002/jps.10128. [DOI] [PubMed] [Google Scholar]
- 38.Meier Y, Eloranta JJ, Darimont J, Ismair MG, Hiller C, Fried M, et al. Regional distribution of solute carrier mRNA expression along the human intestinal tract. Drug Metab Dispos. 2007;35(4):590–594. doi: 10.1124/dmd.106.013342. [DOI] [PubMed] [Google Scholar]
- 39.Chen ML, Yu L. The use of drug metabolism for prediction of intestinal permeability. Mol Pharmaceut. 2009;6(1):74–81. doi: 10.1021/mp8001864. [DOI] [PubMed] [Google Scholar]
- 40.Mithani SD, Bakatselou V, TenHoor CN, Dressman JB. Estimation of the increase in solubility of drugs as a function of bile salt concentration. Pharmaceut Res. 1996;13(1):163–167. doi: 10.1023/A:1016062224568. [DOI] [PubMed] [Google Scholar]
- 41.Litman T, Druley TE, Stein WD, Bates SE. From MDR to MXR: new understanding of multidrug resistance systems, their properties and clinical significance. Cell Mol Life Sci. 2001;58(7):931–959. doi: 10.1007/PL00000912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mithani SD, Bakatselou V, TenHoor CN, Dressman JB. Estimation of the increase in solubility of drugs as a function of bile salt concentration. Pharm Res. 1996;13(1):163–167. doi: 10.1023/A:1016062224568. [DOI] [PubMed] [Google Scholar]
- 43.Lentz KA. Current methods for predicting human food effect. AAPS J. 2008;10(2):282–288. doi: 10.1208/s12248-008-9025-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.US FDA. Waiver of in vivo bioavailability and bioequivalence studies for immediate-release solid oral dosage forms based on a biopharmaceutics classification system. In: Guidance for industry. 2000. http://www.fda.gov/downloads/Drugs/…/Guidances/ucm070246.pdf. Accessed 02 Jun 2012.
- 45.Fagerholm U, Johansson M, Lennernas H. Comparison between permeability coefficients in rat and human jejunum. Pharm Res. 1996;13(9):1336–1342. doi: 10.1023/A:1016065715308. [DOI] [PubMed] [Google Scholar]
- 46.Tse FLS. Pharmacokinetics in drug discovery and development: nonclinical studies. In: Welling PG, Tse FLS, editors. Pharmacokinetics: regulatory, industrial, academic perspectives. 2. New York: Dekker; 1995. pp. 300–306. [Google Scholar]
- 47.Jones RD, Jones HM, Rowland M, Gibson CR, Yates JW, Chien JY, et al. PhRMA CPCDC initiative on predictive models of human pharmacokinetics, part 2: comparative assessment of prediction methods of human volume of distribution. J Pharm Sci. 2011. doi:10.1002/jps.22553. [DOI] [PubMed]
- 48.Ring BJ, Chien JY, Adkison KK, Jones HM, Rowland M, Jones RD, et al. PhRMA CPCDC initiative on predictive models of human pharmacokinetics, part 3: Comparative assessement of prediction methods of human clearance. J Pharm Sci. 2011. doi:10.1002/jps.22552. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
JPEG 26 kb
JPEG 27 kb
JPEG 25 kb
DOCX 120 kb