

Abstract
This study investigates the potential of supersaturated self-nanoemulsifying drug delivery systems (super-SNEDDS) to improve the bioavailability of poorly water-soluble drugs compared to conventional SNEDDS. Conventional SNEDDS contained simvastatin (SIM) at 75% of the equilibrium solubility (Seq). Super-SNEDDS containing SIM at 150 and 200% of Seq were produced by subjecting the SNEDDS preconcentrates to a heating and cooling cycle. The super-SNEDDS were physically stable over 10 months. During in vitro lipolysis of SNEDDS and super-SNEDDS the SIM concentration in the aqueous phase increased for the first 30 min almost proportional to the drug loads and amounts of preconcentrate employed. The 200% drug-loaded super-SNEDDS generated an amorphous SIM precipitate at the end of in vitro lipolysis. In vivo, the relative bioavailability of SIM from super-SEDDDS increased significantly to 180 ± 53.3% (p = 0.014) compared to the dosing of two capsules of (dose equivalent) 75% drug-loaded SNEDDS. A significant increase in the terminal half-life of elimination was observed for super-SNEDDS (2.3 ± 0.6 h) compared to conventional SNEDDS (1.4 ± 0.3 h) as well as a decreased area under the curve ratio of the SIM metabolite simvastatin acid to the parent compound (0.57 ± 0.20 and 0.90 ± 0.3), possibly due to a combination of saturation effects on presystemic metabolising enzymes and prolonged absorption along the small intestine. In summary, this study demonstrated that super-SNEDDS are a viable formulation option to enhance the bioavailability of poorly water-soluble drugs such as simvastatin while reducing the pill burden by an increased drug load of SNEDDS.
Key words: bioavailability, in vitro digestion, in vitro lipolysis, simvastatin, supersaturated self-nanoemusifying drug delivery systems (super-SNEDDS) poorly soluble drugs
Introduction
The ever-increasing number of poorly water-soluble compounds emerging from modern drug discovery programs requires scientists to break new ground in the field of drug delivery. Amongst these approaches, lipid and surfactant-based drug delivery systems, in particular self-nanoemulsifying drug delivery system (SNEDDS), are promising delivery options that have attracted much attention both academically and commercially [1–4]. SNEDDS consist of a mixture of oil, surfactant, co-surfactant and co-solvent [5]. The co-administered drug is dissolved in the mixture forming an isotropic SNEDDS preconcentrate. Hence, the often rate-limiting dissolution step of crystalline compounds is avoided. Upon contact with aqueous medium and following gentle agitation, the preconcentrates spontaneously generate ultrafine oil/water nanoemulsions [6]. The interest in this delivery form was initiated by the successful commercialisation of the product Neoral® containing the drug cyclosporine in a SNEDDS preconcentrate showing improved and more reliable bioavailability independent from food intake compared to its precursor formulation Sandimmune® [7–9]. It is speculated that the improved in vivo performance of Neoral® could be attributed to the small size of the emulsion droplets, the generation of mixed micelles during the digestion of the SNEDDS facilitating drug solubilisation, and the possible inhibition of P-glycoprotein by some of the excipients [10,11].
Despite the success of Neoral®, only few other products have reached the market as SNEDDS [3, 12]. One of the reasons might be the complexity of these delivery systems when interacting with the human physiology, in particular the digestive system, which is still poorly understood. However, potential limitations can be identified at an earlier stage, e.g. when the solubility of a compound in the excipients might not be adequate to allow the drug to be dissolved and formulated as a convenient single-unit dose [13, 14]. Moreover, the full potential of the drug’s solubility in the excipients is often not fully exploited due to concerns that increased drug loads could induce potential precipitation of the co-administered drug during the dispersion and subsequent digestion of the formulation [4]. Consequently, most studies investigating SNEDDS have utilised drug loads at approximately 50–90% of the equilibrium solubility (Seq) of the compound in the SNEDDS [15, 16]. However, recent studies reported the precipitation of drug during in vitro digestion of SNEDDS in an amorphous form with improved dissolution of the precipitated drug [17, 18]. The importance of this observation for the design of SNEDDS and the in vivo performance has not been addressed adequately in the literature so far.
The poor water solubility of simvastatin (SIM; approximately 0.01 mg/l) combined with the substantial metabolism of the compound in the gut wall and in the liver results in a low systemic bioavailability of approximately 5% when administered in a conventional formulation, e.g. tablets or powder-filled capsules [19]. From a pharmacological point of view, the hepatic extraction of the prodrug SIM is desired since SIM unfolds its action in the liver inhibiting the hydroxymethylglutaryl coenzyme A (HMG-CoA) reductase after metabolic conversion to the active simvastatin acid (SIMA) [19]. The inhibition of HMG-CoA reductase by SIMA causes an early interruption of the mevalonate pathway, ultimately reducing cholesterol synthesis [20]. The uptake of SIM (log P = 4.5) into the hepatocytes is dominated by diffusion due its lipophilicity, whereas the less lipophilic SIMA depends on the less efficient transporter-mediated uptake into the hepatocyte [19, 21, 22]. Hence, it might be desirable to reduce the presystemic metabolism of SIM in the enterocyte. Previous approaches to increase the bioavailability of SIM include its formulation as lipid-based drug delivery systems (SMEDDS, solid lipid nanoparticles) and as a delayed release (DR) formulation [19, 23, 24]. The rationale behind the latter is to circumvent the intestinal metabolism of SIM. Due to the decrease in the CYP 3A expression from proximal to distal areas of the intestine, the developed DR formulation was able to increase the bioavailability of SIM while the metabolism to SIMA was reduced [19].
The current study sets out to investigate the utilisation of supersaturated SNEDDS (super-SNEDDS), i.e. SNEDDS that contain SIM as a model drug above its Seq. Specifically, SNEDDS were formulated containing SIM at elevated drug loads up to 200% of Seq in SNEDDS. SNEDDS and super-SNEDDS were investigated during in vitro lipolysis following the characterisation of the digestion phases with regard to the ability to maintain SIM in solution and the solid-state properties of precipitated drug. Finally, the in vivo performance of SNEDDS and super-SNEDDS were evaluated in a dog model.
Materials and Methods
Materials
SIM (USP grade) was purchased from Dayang Chemicals (Hangzhou, Zhejiang, China), SIMA was obtained from Molcan (Toronto, Ontario, Canada) and the internal standard lovastatin (IS) was purchased from American Radiolabeled Chemicals (St. Louis, MO, USA). Porcine pancreatic lipase and bile extract, 4-bromobenzeneboronic acid, trizma maleate, calcium chloride, and sodium hydroxide were purchased from Sigma-Aldrich (St. Louis, MO, USA). Epikuron 200 (phosphatidylcholine) was supplied by Cargill (Hamburg, Germany). Cremophor RH40 (PEG 40 hydrogenated castor oil) was received from BASF (Ludwigshafen, Germany). Captex 300 (medium chain (MC) triglycerides) and Capmul MCM (MC mono-, di-, and triglycerides, MC mixed glycerides) were kindly donated from Abitec (Columbus, OH, USA). Acetonitrile (high-performance liquid chromatography (HPLC) grade), and sodium chloride were obtained from Merck (Darmstadt, Germany). Purified water was delivered by a Millipore Milli-Q Ultrapure Water purification system (Billerica, MA, USA). All other chemicals were of analytical grade.
Determination of Saturation Solubility
The equilibrium solubilities of SIM in SNEDDS preconcentrates at 25 and 37°C were determined by the shake flask method as described previously [25] followed by the quantification of SIM on a Agilent 1200 HPLC system (Agilent Technologies, Santa Clara, CA, USA).
Preparation of Preconcentrates
The MC mixed glycerides (Capmul MCM) and the surfactant (Cremophor RH 40) were freshly molten at approximately 50°C before they were blended in dust-free, sealed glass vials on a vortex mixer with the MC triglycerides (Captex 300). After cooling to room temperature, the cosolvent (ethanol) was added to the mixture producing preconcentrates that consisted of 55% lipid (Captex: Capmul MCM ratio 1:2), 35% surfactant, and 10% cosolvent (percentages are reported as w/w).
In order to produce drug-loaded preconcentrates, the required amount of SIM was weighed accurately into dust-free glass vials and the blank preconcentrates were added. Prior to sealing, the vials were purged with nitrogen to prevent oxidation and the mixture was shaken vigorously until the entire drug had dissolved generating a clear, isotropic preconcentrate.
Since the amount of SIM needed for the preparation of super-SNEDDS exceeded the equilibrium solubility of SIM in the preconcentrates at room temperature, the procedure described above initially produced a suspension of SIM in preconcentrates. These suspensions were placed in a Sonorex super RK 106 ultrasonicator (Bandelin electronic GmbH, Berlin, Germany) for approximately 10 min followed by a heating cycle (3 h at 60°C) in a programmable heating oven (FD E2, Binder GmbH, Tuttlingen, Germany) after which they were allowed to cool down to room temperature overnight generating super-SNEDDS preconcentrates. Super-SNEDDS were produced at levels corresponding to 150 and 200% of the drug’s equilibrium solubility at 37°C. Complete dissolution of the drug in all preconcentrates was confirmed by polarising light microscopy (PLM; Motic BA 300 POL, Richmond, BC, Canada).
Stability of SNEDDS and Super-SNEDDS Preconcentrates
The physical stability of super-SNEDDS was assessed during storage at 25°C up to 10 months. Sealed glass vials containing approximately 5 g super-SNEDDS were investigated visually and by PLM for potential precipitation of crystalline SIM on a regular basis. In order to address concerns about chemical degradation of the lactone SIM during the heating cycle, the SIM content in super-SNEDDS was determined by HPLC immediately after equilibration to room temperature following the heating cycle. The determined drug content was calculated as the percentage of the nominal value.
Particle Size Characterisation of Dispersed Preconcentrates
The particle size of SNEDDS and super-SNEDDS was determined by dynamic light scattering (DLS) following the dispersion of 1 ml SNEDDS or super SNEDDS in 300 ml Milli-Q water. Dispersion was facilitated by a type 2 USP dissolution apparatus (ERWEKA, DT 600, Heusenstamm, Germany) set at 37°C and 100 rpm. After 30 min, the particle size of the resulting dispersion was measured at 37°C without further dilution using a nanosizer ZS (Malvern Instruments, Worcestershire, UK) reporting the particle size as the z average (nanometers) of three independent measurements.
In Vitro Lipolysis
The dynamic in vitro lipolysis model with the continuous addition of calcium (0.045 mM/min of a 0.5 M CaCl2 solution) to control the lipolysis rate [25–27] was employed to simulate the digestion of 1 or 2 g SNEDDS (75% drug load) and 1 g super-SNEDDS (150 and 200% drug load) under fasted state conditions at 37°C. The digestion of lipids was initiated by the addition of a freshly prepared 50 ml pancreatic extract to 250 ml digestion medium (2 mM trizma maleate buffer pH 6.5, 150 mM sodium chloride, supplemented by 5 mM bile salt and 1.25 mM phospholipids, final lipase activity of 350 U/ml).
In vitro lipolysis was carried out for 60 min during which the pH was maintained at 6.5 by a pH-stat (Metrohm Titrino 744, Tiamo Version 1.3, Switzerland) dispensing 1 N NaOH to compensate for the drop in the pH caused by the release of free fatty acids as a result of the lipolytic activity of the pancreatic lipase. During lipolysis, 9 ml digestion medium were taken in 15-min intervals and the lipase activity within the samples was immediately inhibited by the addition of 45 μL of the lipase inhibitor 4-bromobenzeneboronic acid (1 M in methanol). Of each sample, 8.0 g was subjected to ultracentrifugation for 50 min at 50,000 rpm (2.3 × 05 g at rmax) at 37°C in a Beckman L-80 Ultracentrifuge using a 70.1 Ti rotor (Beckman Coulter, Brea, CA, USA). This resulted in the separation of the digestion medium in a clear supernatant and an off-white pellet. The supernatant and pellet were quantified for their drug content by HPLC following appropriate dilution with acetonitrile.
Pellet Characterisation
X-ray Powder Diffraction
The solid state of SIM present in the isolated pellets obtained after 60 min of in vitro lipolysis of super-SNEDDS (200% drug load) was investigated by X-ray powder diffraction (XRPD). Super-SNEDDS containing 200% drug load were necessary for the pellet characterization as preliminary experiments (blank pellets spiked with the corresponding amount of crystalline drug) were not able to detect SIM by XRPD when lower drug loads were employed. The isolated pellets were placed on aluminium holders and were scanned at from 5° to 35° (2θ) using a scanning speed of 0.1285°/min and a step size of 0.0084°. The X-ray diffractometer (PANalytical X’Pert Pro MPD PW 3040/60, PANalytical B.V., Almelo, The Netherlands) used CuKα as radiation source (1.542 Å) and was operated at a voltage of 40 kV and a current of 30 mA. The diffractograms were obtained within 2 h after sampling and were compared with those obtained from the in vitro digestion of drug-free formulations spiked with the amount of crystalline SIM present in the drug-loaded formulations.
Dissolution Studies
The dissolution of precipitated SIM present in the pellets obtained from digested super-SNEDDS (200% drug load) was investigated in lipolysis medium during 3 h at pH 6.5 and 37°C under sink conditions (i.e. the concentration of SIM was below 10% of SIM solubility in the dissolution medium as determined previously [25]). In order to achieve detectable amounts of SIM during the dissolution studies, three pellets were isolated and combined after ultracentrifugation of the lipolysis medium withdrawn after 60 min in vitro lipolysis. The pellets were dispersed in 10 ml Milli-Q water and added to 890 ml lipolysis medium (of the same composition as described above but without lipase). Dissolution was carried out in a standard USP dissolution 2 apparatus (Erweka DT 600) with the paddle speed set to 75 rpm. At specified time points (1, 2, 5, 10, 20, 40, and 60 min), 3 ml of the dissolution media were withdrawn and the volume was replaced by fresh lipolysis medium. The samples were filtered through a 0.2 μm syringe driven filter unit (Millex GV, Millipore, Tullagreen, Ireland), discarding the first millilitre of the filtrate. The rest of the filtrate was diluted appropriately with acetonitrile and was subsequently analysed for SIM by HPLC. An analogous procedure was followed for control experiments using pellets from drug-free formulations that were spiked with the amount of crystalline SIM corresponding to the amount of SIM present in pellets obtained from super-SNEDDS.
HPLC Analysis of In Vitro Simvastatin Samples
The quantification of SIM in samples obtained from in vitro lipolysis, stability, solubility and dissolution studies was performed on an Agilent 1200 Series chromatographic system (Agilent Technologies, Santa Clara, CA, USA) and was based on a validated method previously described [23]. Briefly, samples (20 μl) were separated on a Gemini NX C18 column (4.6 × 250 mm, 5 μm) maintained at 40°C. As mobile phase, a mixture of acetonitrile/water (70: 30%, V/V) was used at a flow rate of 1.2 ml/min and the effluent was monitored at 238 nm. From the slope of a standard curve of SIM in acetonitrile (1–100 μg/ml), the concentration of SIM in the samples was determined.
In Vivo Study
All animal care and experimental procedures were approved by the Animal Welfare Committee, appointed by the Danish Ministry of Justice and were carried out in compliance with EC Directive 2010/63/EU, the Danish law regulating experiments on animals and NIH Guidelines for the Care and Use of Laboratory Animals. Six male beagle dogs (11.8–13.8 kg) received SNEDDS and super-SNEDDS orally in a non-randomised cross-over study allowing a washout period of 1 week between each treatment. The SNEDDS and super-SNEDDS were prepared freshly the day before the treatments and were accurately weighed into hard gelatin capsules (0.8 g/capsule). The treatments included one and two capsules of SNEDDS (75% drug load) and one capsule super-SNEDDS (150% drug load). The dogs were fasted for 20–24 h prior to the administration of the capsules and were fed again 8 h after administration.
Blood samples (0.5 ml) were obtained by individual vein puncture from the cephalic vein at designated time points (15 min pre-dose and 0.5, 1, 2, 3, 4, 6, 8, 24, and 28 h after administration) and were transferred into EDTA-coated tubes. The plasma was separated by centrifugation at 3,200×g (15 min at 4–8°C) and stored at −20°C until further analysis.
Quantification of Simvastatin and Simvastatin Acid in Plasma Samples
Prior to the quantification of SIM and its metabolite SIMA by liquid chromatography-tandem mass spectrometry, the frozen plasma samples were thawed in the dark at ambient temperature. Fifty microlitres of the plasma samples, calibration standards, quality control (QC) and blank samples were manually transferred to a 96-well plate following precipitation of the proteins with 200 μl acetonitrile, containing 0.5 ng/ml lovastatin as IS using a Biomek NX liquid handling robotic system (Beckman Coulter, Indianapolis, IN, USA). The plate was centrifuged for 20 min at 4°C and 6,200×g (Sigma 4 K15, Sigma, Osterode am Harz, Germany) after which the supernatants were transferred and injected (10 μl) into a Waters Acquity UPLC system (Waters, Milford, MA, USA). The analytes were separated on a Waters Acquity BEH C8 column (1.7 μm, 2.1 × 50 mm) maintained at 40°C. A constant flow rate of 0.6 ml/min was employed using the gradient shown in Table I. The mass spectrometer (Sciex API4000, AB Sciex, Foster City, CA, USA) was operated in multiple reaction monitoring mode using positive electrospray ionization for the ionisation of the analytes. The calibration curve consisted of eight points ranging from 1 to 1,000 ng/ml and was prepared by a Tecan Genesis RSP200 robotic system (Tecan Group Ltd., Männedorf, Switzerland). QC samples were prepared at 10 and 200 ng/ml. For both simvastatin and simvastatin acid, the lower limit of quantification by this procedure was 10 ng/ml.
Table I.
Gradient employed for the quantification of simvastatin and simvastatin acid by liquid chromatography-tandem mass spectrometry
| Time (min) | A (%) | B (%) |
|---|---|---|
| 0.00 | 98 | 2 |
| 1.50 | 5 | 95 |
| 2.00 | 5 | 95 |
| 2.20 | 98 | 2 |
| 3.00 | 98 | 2 |
A Milli-Q water + 0.1% formic acid; B acetonitrile + 0.1% formic acid
Pharmacokinetic Analyses
The pharmacokinetic parameters of SIM and SIMA were obtained by non-compartmental analysis using WinNonlin Professional software (Version 5.2, Pharsight Corporation, Mountain View, CA, USA). The linear trapezoidal method was used to determine the area under the curve (AUC) extrapolated to infinity (AUC0-inf). The maximum plasma concentration (Cmax) and the time (tmax) required to reach Cmax were obtained directly from the individual plasma concentration versus time curves. The half-life of elimination (t1/2) was calculated from the terminal slope of the elimination phase obtained from the semi-logarithmic plots of the plasma concentration versus time plots. The bioavailability (Frel) of SIM from the treatments with two capsules SNEDDS (75% drug load) and one capsule super-SNEDDS (150% drug load) relative to the treatment with one capsule SNEDDS (75% drug load, reference) were determined as
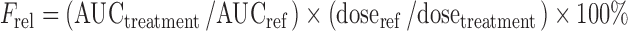 |
Statistical Analysis
Statistical analyses were carried out using GraphPad Prism (Version 5.04, GraphPad Software, San Diego, CA, USA). Unpaired Student’s t tests were applied to determine statistically significant differences (p = 0.05) between two groups, whereas analyses of variance (ANOVA) following Tukey’s post-test was utilised for differences between more than two groups (p = 0.05).
Results and Discussion
Solubility Studies in SNEDDS
Since the Seq of a compound in the preconcentrates is affected by the temperature, Seq for SIM was determined both for conditions representing storage temperature (25°C) and physiological levels (37°C). At 37°C, the Seq of SIM was approximately 10% higher (112.9 ± 2.7 mg/g) [25] compared to Seq of SIM determined at 25°C (101.9 ± 2.3 mg/g). All drug loads in this work refer to the Seq of SIM in SNEDDS at 37°C.
Stability of Super-SNEDDS
The potential crystallisation of SIM in the super-SNEDDS during storage was a major concern in this study. However, when the super-SNEDDS were inspected periodically, no crystalline drug was observed by PLM during the storage time of 10 months. After the heating cycle, 96.6 ± 1.4% (mean ± SD) of the declared SIM amount was found in super-SNEDDS indicating a reasonable stability of the compound during the heating cycle.
Dispersion Study
The particle size of dispersions generated by drug-free SNEDDS and SNEDDS containing a range of different drug loads up to the saturation concentration of SIM (100% Seq) has been previously reported [25]. In agreement with the earlier study, the particle size of dispersed SNEDDS (1:300) containing 75% drug load (34.9 ± 0.5 nm) did not vary from that obtained from drug-free SNEDDS (34.2 ± 1.2 nm) and both had a clear to slightly bluish appearance. No crystals were visible under the PLM upon dispersion of SNEDDS containing 75% drug load. In contrast, super-SNEDDS (150 and 200% drug load) had a slightly milky appearance immediately after dispersion which developed into a white, fluffy suspension within approximately 15–20 min. Due to the macroscopic scale of the particles, they could not be measured by DLS. The suspended particles appeared as fine, needle-shaped crystals under PLM (data not shown) indicating precipitation of SIM from super-SNEDDS. This can be attributed to the loss of the solubilisation capacity upon dispersion of the preconcentrates. In particular, the cosolvent can be expected to be diluted into the bulk. Since the cosolvent contributed to approximately 30% of the overall SIM solubility in the formulation [25], the dispersion triggered a dramatic precipitation of the drug when the drug load was increased to 150 and 200%.
In Vitro Lipolysis
Figure 1 shows the concentration of SIM in the aqueous phase obtained from the centrifuged lipolysis medium during the in vitro lipolysis of SNEDDS and super-SNEDDS. No SIMA was detected in the lipolysis medium. Compared to the lipolysis of 1 g conventional SNEDDS (75% drug load) the concentration of SIM in the aqueous phase increased almost proportionally with the amount of preconcentrate and drug loads employed. For example, the SIM concentration increased approximately 2.2-fold for 2 g SNEDDS (75% SIM load), and 1.8-fold for 1 g super-SNEDDS (150% SIM load) during the first 30 min of in vitro lipolysis. After 30 min in vitro lipolysis, the concentration of SIM declined to its equilibrium solubility Seq in the lipolysis medium (as indicated by asterisks in Fig. 1).
Fig. 1.
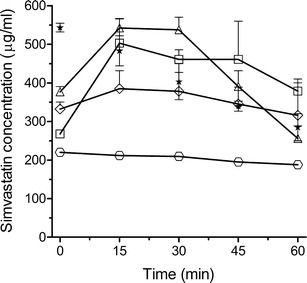
The concentration of SIM in the lipolysis medium during in vitro lipolysis of SNEDDS and super-SNEDDS. The investigated formulations consisted of 1 g SNEDDS (empty circle; 75%), 2 g SNEDDS (white square; 75%), 1 g super-SNEDDS (white diamond; 150%), and 1 g super-SNEDDS (empty triangle; 200%). The percentages refer to the equilibrium solubility (S eq) of SIM in the preconcentrates. The asterisk represents the corresponding S eq of SIM in the lipolysis medium containing the digestion products of 1 g blank SNEDDS. Data represents mean ± SD, n = 3
The SIM concentration in the aqueous phase during the in vitro lipolysis of 1 g super-SNEDDS (200% SIM load) increased approximately 2.7-fold compared to the concentration generated during the in vitro lipolysis of 1 g SNEDDS (75% SIM load). Moreover, the in vitro lipolysis of 1 g of 200% drug-loaded super-SNEDDS generated SIM concentration above the Seq of SIM in the lipolysis between the initial 15–45 min of in vitro lipolysis, indicating the generation of a temporarily supersaturated solution of SIM in the lipolysis medium. However, the supersaturation of the lipolysis medium with SIM could not be maintained longer than 30 min. As expected from supersaturated aqueous solutions, rapid precipitation was observed after 30 min in vitro lipolysis.
The solubilisation of SIM in the lipolysis medium observed during in vitro lipolysis is in contrast to the results from a previous study which employed the same composition of SNEDDS and super-SNEDDS containing the model drug halofantrine (clogP 8.5) [18]. In the previous study, the concentration of halofantrine solubilised in the lipolysis medium was almost identical during in vitro lipolysis of SNEDDS (75% halofantrine load) and super-SNEDDS (150% halofantrine load). The concentration of halofantrine in the lipolysis medium could only be elevated with increasing amounts of preconcentrates subjected to in vitro lipolysis. This might be explained by the different equilibrium solubilities of halofantrine and SIM in the lipolysis medium containing the digestion products of blank SNEDDS. In the present study, the equilibrium solubility of SIM at the start of in vitro lipolysis was approximately 540 μg/ml and showed a linear (R2 = 0.996) decrease to 290 μg/ml after 60 min in vitro lipolysis. In the previous study, using identical experimental conditions, the solubility of halofantrine was considerably lower (as characterised by a decline from approximately 110–35 μg /ml). It is likely that the different lipophilicities of the compounds contribute to the difference in the observed solubilities favouring greater solubility of the less lipophilic SIM in the digestion medium containing the digestion products of medium-chain lipids.
Characterisation of the Pellet
X-ray Powder Diffraction
The isolated pellet obtained after 60 min in vitro lipolysis of super-SNEDDS (200% drug load) was characterised by XRPD and compared with a spiked blank pellet containing amounts of crystalline SIM equivalent to those previously determined in super-SNEDDS pellets (Fig. 2).
Fig. 2.
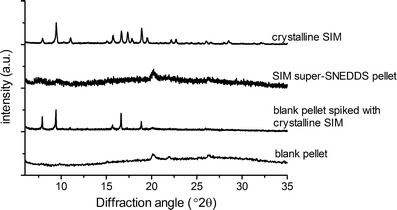
XRPD patterns of crystalline SIM, pellets obtained from super SNEDDS (200% drug load), spiked blank pellets and blank pellets obtained after in vitro lipolysis. Data are exemplary for replicate runs, n = 2 (crystalline SIM), n = 3 (pellets)
The characteristic peaks of crystalline SIM were present in the diffractograms obtained from spiked blank pellets but were absent in the super-SNEDDS pellets. This suggested that the super-SNEDDS generated an amorphous SIM precipitate during in vitro lipolysis. Precipitation of drug in an amorphous form during in vitro lipolysis has been found previously for cinnarizine and halofantrine formulated as SNEDDS and super-SNEDDS, respectively [17, 18].
Dissolution Study
Since the amorphous form of a drug is known to show a faster dissolution rate compared to the crystalline form, dissolution studies of SIM from the pellet generated by super-SNEDDS were carried out to confirm the XRPD results. The dissolution rate of SIM originating from super-SNEDDS pellets was compared with the dissolution of blank pellets spiked with crystalline SIM (Fig. 3). According to the dissolution profiles, approximately 90% of the SIM originating from the super-SNEDDS pellet dissolved within the first 3 min after initiation of the dissolution study, whereas approximately 20 min were required for crystalline SIM to obtain comparable dissolution. Thus, the fast dissolution rate of SIM from the super-SNEDD pellets confirmed the amorphous nature of the SIM precipitate.
Fig. 3.

Dissolution profiles of simvastatin from isolated pellets precipitated after 60 min in vitro lipolysis of super-SNEDDS (white square; 200% drug load) and spiked blank pellets (black square). Data represents mean ± SD, n = 3
In Vivo Dog Study
The mean SIM plasma concentration after oral administration of a single dose of one and two capsules SNEDDS (75% SIM load) and one capsule super-SNEDDS (150% SIM load) is presented in Fig. 4 and the corresponding pharmacokinetic parameters for SIM are shown in Table II.
Fig. 4.
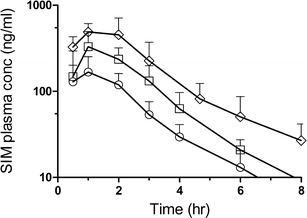
Semi-logarithmic plots of the mean plasma concentration of SIM after oral administration of one capsule SNEDDS (white circle; 75% SIM load), two capsules SNEDDS (white square; 75% SIM load), and one capsule super-SNEDDS (white diamond; 150% SIM load) to beagle dogs. The plots have been truncated to 8 h for illustration purposes. Data represents mean ± SD, n = 6
Table II.
Pharmacokinetic parameters of SIM after oral administration of SNEDDS and super-SNEDDS to beagle dogs
| Pharmacokinetic parameter | 1 Caps SNEDDS (67.7 mg SIM) | 2 Caps SNEDDS (135.5 mg SIM) | 1 Caps super-SNEDDS (135.5 mg SIM) |
|---|---|---|---|
| C max (ng/ml) | 181 ± 91.7 | 345 ± 147 | 532 ± 209 |
| t max (h) | 0.8 ± 0.3 | 1.2 ± 0.4 | 1.2 ± 0.4 |
| AUC0-inf (ng∙h/ml) | 448 ± 167 | 859 ± 384 | 1549 ± 660* |
| t 1/2 (h) | 1.4 ± 0.3 | 1.4 ± 0.5 | 2.3 ± 0.6* |
| Relative bioavailability (%)a | 100 | 101 ± 36.8 | 180 ± 53.3* |
Data represents mean ± SD, n = 6
aRelative bioavailability compared to one capsule SNEDDS
*p < 0.05, statistically significant difference when compared with data obtained after administration of dose equivalent two capsules SNEDDS
SIM was rapidly absorbed from all formulations with maximum plasma concentrations observed between 0.8 ± 0.3 h (one capsule SNEDDS) and 1.2 ± 0.4 h (for two capsules SNEDDS and one capsule super-SNEDDS). This was followed by a rapid decline resulting in plasma concentrations below the quantification limit of SIM (and its metabolite) in the plasma after 8 h. Therefore, all (semi-logarithmic) plasma concentrations versus time curves were truncated to 8 h. The data suggests that the dosing of two capsules SNEDDS did not impede the gastric emptying despite the concomitant intake of a slightly increased lipid amount when compared to one capsule. Both the Cmax and the AUC0-inf reached after the dosing of two capsules SNEDDS (135.5 mg SIM) were proportional to the dosing of one capsule SNEDDS (67.7 mg SIM; Cmax ratio 2 caps/1 caps = 2.1 ± 0.8; AUC0-inf ratio 2 caps/1 caps = 2.2 ± 1.2). Following the administration of one capsule super-SNEDDS, the ratio of “AUC0-inf super-SNEDDS/AUC0-inf one capsule SNEDDS” increased significantly (p = 0.045) to 3.6 ± 1.1 while the corresponding Cmax ratio increased to 3.3 ± 1.3, although this was not significantly different from the Cmax ratio observed for the dose-equivalent two capsules SNEDDS.
The bioavailability of SIM from two capsules SNEDDS relative to one capsule SNEDDS remained unchanged. In contrast, the bioavailability of SIM dosed as super-SNEDDS (150% SIM load) increased significantly (p = 0.014) compared to (dose equivalent) two capsules of conventional SNEDDS (75% drug load). At the current stage, the exact mechanism for this observation cannot be elucidated. However, two possible interpretations might account for the increased bioavailability of SIM.
For the first interpretation, super-SNEDDS are regarded as special types of immediate release (IR) formulations. It is known that IR formulations can potentially lead to the saturation of intestinal metabolic enzymes, subsequently leading to an enhanced bioavailability of the parent compound [28, 29]. SIM is subject to pre-systemic metabolism by enzymes of cytochrome P450 (mainly CYP 3A4) located in the enterocytes [19, 28]. It is possible that the intestinal metabolic enzymes become saturated by the instantaneously released high dose of SIM, facilitating the rapid absorption of unchanged SIM lactone. This interpretation could explain the data obtained for the SIM metabolite, SIMA, shown in Fig. 5 and Table III, respectively, in which no difference was found between the AUC of two capsules SNEDDS and one capsule super-SNEDDS. Both the AUC0-inf and Cmax of SIMA obtained after the dosing of super-SNEDDS were identical to those observed for the dose-equivalent two capsules SNEDDS. This might point to a maximal enzymatic turnover leading to the same SIMA levels in dose-equivalent SNEDDS and super-SNEDDS.
Fig. 5.
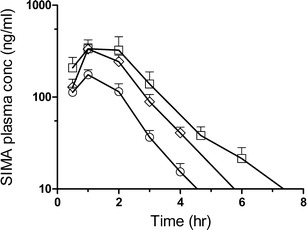
Semi-logarithmic plots of the mean plasma concentration of SIMA after oral administration of one capsule SNEDDS (white circle; 75% SIM load), two capsules SNEDDS (white square; 75% SIM load), and one capsule super-SNEDDS (white diamond; 150% SIM load) to beagle dogs. The plots have been truncated to 8 h for illustration purposes. Data represents mean ± SD, n = 6
Table III.
Pharmacokinetic parameters of SIMA after oral administration of SNEDDS and super-SNEDDS to beagle dogs
| Pharmacokinetic parameter | v1 Cap SNEDDS (67.7 mg SIM) | 2 Caps SNEDDS (135.5 mg SIM) | 1 Cap super-SNEDDS (135.5 mg SIM) |
|---|---|---|---|
| C max (ng/ml) | 175 ± 56.2 | 355 ± 88.9 | 380 ± 297 (264 ± 91.2) |
| t max (h) | 1.0 ± 0.0 | 1.3 ± 0.5 | 1.3 ± 0.6 (1.1 ± 0.5) |
| AUC0-inf (ng∙h/ml) | 373 ± 140 | 732 ± 210 | 958 ± 768 (656 ± 236) |
| t 1/2 (h) | 1.0 ± 0.4 | 1.0 ± 0.4 | 1.5 ± 0.9 (1.3 ± 0.6) |
| Relative bioavailability (%)a | 100 | 105 ± 31.0 | 116 ± 43.6 (99.6 ± 16.6) |
Data represents mean ± SD, n = 6
Numbers given in parenthesis reflect the values obtained after exclusion of one animal showing a large deviation in plasma SIMA concentrations compared to the other subjects
aRelative bioavailability compared to one capsule SNEDDS
For the second interpretation and under the assumption that the observed precipitation of amorphous SIM in vitro also occurs in vivo, super-SNEDDS could be regarded as a DR formulation, the amorphous precipitate resembling a drug reservoir from which drug is released after precipitation. The metabolic enzymes are not distributed equally in the intestine but show regional differences in their expression. For example, the expression of CYP 3A declines from proximal to distal areas of the intestine [30]. It has, therefore, been suggested that the use of formulations targeting those areas with a reduced expression of metabolic enzymes might be beneficial for the bioavailability of compounds that are susceptible to pre-systemic metabolism [19, 29]. In fact, Tubic-Grozdanis et al. [19] have recently shown by virtue of a DR formulation of SIM that the compound’s pre-systemic metabolism by CYP 3A enzymes in the gut wall could be reduced considerably. The authors reported an improved bioavailability of the parent drug SIM, a reduced SIMA/SIM-AUC ratio (0.94 ± 0.56 and 0.35 ± 0.21 for IR and DR formulations, respectively) and an increased t1/2 of SIM by designing a formulation that released SIM in the distal areas of the intestine [19]. These results are in line with the present study where a significantly (p = 0.007) increased t1/2 was found for SIM formulated as super-SNEDDS (2.3 ± 0.6 h) compared to conventional SNEDDS (1.4 ± 0.3 h). Moreover, the SIMA/SIM–AUC ratio of SNEDDS (0.90 ± 0.3) corresponded well to that previously reported for IR formulations while the SIMA/SIM–AUC ratio decreased to 0.57 ± 0.20 for super-SNEDDS. It might be possible that the fraction of SIM that has not been absorbed in the proximal regions of the small intestine could be transported as a potentially amorphous precipitate to distal areas of the intestine, where it can dissolve. Assuming the presence of a rapidly dissolving amorphous SIM in vivo, the precipitate could serve as a reservoir which still contributes to the SIM bioavailability.
In summary, it appears that super-SNEDDS facilitate SIM absorption by a complex mechanism that requires further investigation, e.g. by investigating if the precipitation observed in vitro also occurs in vivo. The major objective of in vitro lipolysis is to predict the in vivo performance of lipid-based drug delivery systems. Based on the concentration of SIM in the aqueous phase during in vitro lipolysis, the dynamic in vitro lipolysis model was able to predict the rank order of the in vivo performance of one and two capsules SNEDDS. However, the model underestimated the performance of super-SNEDDS in vivo. Similarly, in a previous study using the identical composition of SNEDDS and the model drug halofantrine, the in vivo performance of super-SNEDDS was comparable to the dosing of multiple units of conventional SNEDDS while during in vitro lipolysis the same halofantrine concentration in the aqueous phase was measured both for super-SNEDDS and one capsule conventional SNEDDS [18]. As with SIM, halofantrine precipitated in an amorphous form during in vitro lipolysis. Although the fate of the drug in vivo has not been investigated in both studies, it appears likely that the solid state properties of potentially precipitated drug have an impact on the in vivo performance. Therefore, the solid-state characterisation of the drug during in vitro lipolysis adds valuable information to the in vitro model.
This study is the second in vivo study that employed supersaturated SNEDDS as a novel drug delivery system for poorly water-soluble drugs. The first study showed that the bioavailability of halofantrine from super-SNEDDS was equivalent to conventional SNEDDS. The present study, however, provides evidence that super-SNEDDS are indeed superior to conventional SNEDDS, at least for the delivery of the poorly water-soluble drug simvastatin.
When correlating the SIM amount in the aqueous phase during in vitro lipolysis, a correlation with the in vivo data could not be found since the model does not accommodate processes beyond solubilisation, such as the first pass effect. From a biopharmaceutical point of view, super-SNEDDS are a feasible approach that could be evaluated for new compounds. There is still work to be conducted on the technical risk of precipitation during storage, though this was not observed in the current study nor in our previous work on super-SNEDDS with halofantrine.
Conclusion
The broader application of SNEDDS has been often restricted due to solubility limitations of lipophilic compounds in common excipients used in lipid and surfactant-based drug delivery systems ultimately requiring the administration of multiple units of SNEDDS. The current study has shown that supersaturated SNEDDS are a feasible approach to increase the drug load in SNEDDS and to increase the bioavailability of simvastatin. Super-SNEDDS contained drug loads up to twice of the drug’s equilibrium solubility and were physically stable for more than 10 months. The in vitro lipolysis of 2 g of SNEDDS (75% drug load) generated concentrations of solubilised SIM in the lipolysis medium comparable to 1 g of dose-equivalent super-SNEDDS (150% drug load). The digestion of super-SNEDDS containing 200% drug load produced temporary supersaturated solutions of SIM in the in vitro lipolysis medium followed by rapid precipitation of amorphous SIM. In a pharmacokinetic study in beagle dogs, the super-SNEDDS showed considerably higher bioavailability than multiple units of conventional SNEDDS (with the same dose).
Acknowledgments
The authors would like to thank the personnel in the animal facilities at H. Lundbeck A/S for their skilful handling of the animals.
References
- 1.Chakraborty S, Shukla D, Mishra B, Singh S. Lipid - an emerging platform for oral delivery of drugs with poor bioavailability. Eur J Pharm Biopharm. 2009;73(1):1–70. doi: 10.1016/j.ejpb.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 2.Fahr A, Liu X. Drug delivery strategies for poorly water-soluble drugs. Expert Opin Drug Deliv. 2007;4(4):403–416. doi: 10.1517/17425247.4.4.403. [DOI] [PubMed] [Google Scholar]
- 3.Hauss DH. Oral lipid-based formulations. Adv Drug Deliv Rev. 2007;59(7):667–676. doi: 10.1016/j.addr.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 4.Pouton CW, Porter CJH. Formulation of lipid-based delivery systems for oral administration: materials, methods and strategies. Adv Drug Deliv Rev. 2008;60(6):625–637. doi: 10.1016/j.addr.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 5.Humberstone AJ, Charman WN. Lipid-based vehicles for the oral delivery of poorly water soluble drugs. Adv Drug Deliv Rev. 1997;25(1):103–128. doi: 10.1016/S0169-409X(96)00494-2. [DOI] [Google Scholar]
- 6.Anton N, Vandamme T. Nano-emulsions and micro-emulsions: clarifications of the critical differences. Pharm Res. 2011;28(5):978–985. doi: 10.1007/s11095-010-0309-1. [DOI] [PubMed] [Google Scholar]
- 7.Kovarik JM, Mueller EA, Van Bree JB, Tetzloff W, Kutz K. Reduced inter- and intraindividual variability in cyclosporine pharmacokinetics from a microemulsion formulation. J Pharm Sci. 1994;83(3):444–446. doi: 10.1002/jps.2600830336. [DOI] [PubMed] [Google Scholar]
- 8.Mueller EA, Kovarik JM, van Bree JB, Grevel J, Lücker PW, Kutz K. Influence of a fat-rich meal on the pharmacokinetics of a new oral formulation of cyclosporine in a crossover comparison with the market formulation. Pharm Res. 1994;11(1):151–155. doi: 10.1023/A:1018922517162. [DOI] [PubMed] [Google Scholar]
- 9.Mueller EA, Kovarik JM, van Bree JB, Tetzloff W, Grevel J, Kutz K. Improved dose linearity of cyclosporine pharmacokinetics from a microemulsion formulation. Pharm Res. 1994;11(2):301–304. doi: 10.1023/A:1018923912135. [DOI] [PubMed] [Google Scholar]
- 10.Fatouros DG, Karpf DM, Nielsen FS, Müllertz A. Clinical studies with oral lipid based formulations of poorly soluble compounds. Ther Clin Risk Manag. 2007;3(4):591–604. [PMC free article] [PubMed] [Google Scholar]
- 11.Porter CJH, Trevaskis NL, Charman WN. Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat Rev Drug Discov. 2007;6(March):231–248. doi: 10.1038/nrd2197. [DOI] [PubMed] [Google Scholar]
- 12.Strickley RG. Solubilizing excipients in oral and injectable formulations. Pharm Res. 2004;21(2):201–230. doi: 10.1023/B:PHAM.0000016235.32639.23. [DOI] [PubMed] [Google Scholar]
- 13.Rane SS, Anderson BD. What determines drug solubility in lipid vehicles: is it predictable? Adv Drug Deliv Rev. 2008;60(6):638–656. doi: 10.1016/j.addr.2007.10.015. [DOI] [PubMed] [Google Scholar]
- 14.Thi TD, Van Speybroeck M, Barillaro V, Martens J, Annaert P, Augustijns P, et al. Formulate-ability of ten compounds with different physicochemical profiles in SMEDDS. Eur J Pharm Sci. 2009;38(5):479–488. doi: 10.1016/j.ejps.2009.09.012. [DOI] [PubMed] [Google Scholar]
- 15.Christensen J, Schultz K, Mollgard B, Kristensen HG, Müllertz A. Solubilisation of poorly water-soluble drugs during in vitro lipolysis of medium- and long-chain triacylglycerols. Eur J Pharm Sci. 2004;23(3):287–296. doi: 10.1016/j.ejps.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 16.Kaukonen AM, Boyd BJ, Porter CJH, Charman WN. Drug solubilization behavior during in vitro digestion of simple triglyceride lipid solution formulations. Pharm Res. 2004;21(2):245–253. doi: 10.1023/B:PHAM.0000016282.77887.1f. [DOI] [PubMed] [Google Scholar]
- 17.Sassene PJ, Knopp MM, Hesselkilde JZ, Koradia V, Larsen A, Rades T, et al. Precipitation of a poorly soluble model drug during in vitro lipolysis: characterization and dissolution of the precipitate. J Pharm Sci. 2010;99(12):4982–4991. doi: 10.1002/jps.22226. [DOI] [PubMed] [Google Scholar]
- 18.Thomas N, Holm R, Müllertz A, Rades T. In vitro and in vivo performance of novel supersaturated self-nanoemulsifying drug delivery systems (super-SNEDDS) J Control Release. 2012;160(1):25–32. doi: 10.1016/j.jconrel.2012.02.027. [DOI] [PubMed] [Google Scholar]
- 19.Tubic-Grozdanis M, Hilfinger J, Amidon G, Kim J, Kijek P, Staubach P, et al. Pharmacokinetics of the CYP 3A substrate simvastatin following administration of delayed versus immediate release oral dosage forms. Pharm Res. 2008;25(7):1591–1600. doi: 10.1007/s11095-007-9519-6. [DOI] [PubMed] [Google Scholar]
- 20.Pedersen TR, Tobert JA. Simvastatin: a review. Expert Opin Pharmacother. 2004;5(12):2583–2596. doi: 10.1517/14656566.5.12.2583. [DOI] [PubMed] [Google Scholar]
- 21.Hamelin BA, Turgeon J. Hydrophilicity/lipophilicity: relevance for the pharmacology and clinical effects of HMG-CoA reductase inhibitors. Trends Pharmacol Sci. 1998;19(1):26–37. doi: 10.1016/S0165-6147(97)01147-4. [DOI] [PubMed] [Google Scholar]
- 22.Serajuddin ATM, Ranadive SA, Mahoney EM. Relative lipophilicities, solubilities, and structure-pharmacological considerations of 3-hydroxy-3-methylglutaryl-coenzyme a (HMG-COA) reductase inhibitors pravastatin, lovastatin, mevastatin, and simvastatin. J Pharm Sci. 1991;80(9):830–834. doi: 10.1002/jps.2600800905. [DOI] [PubMed] [Google Scholar]
- 23.Kang BK, Lee JS, Chon SK, Jeong SY, Yuk SH, Khang G, et al. Development of self-microemulsifying drug delivery systems (SMEDDS) for oral bioavailability enhancement of simvastatin in beagle dogs. Int J Pharm. 2004;274(1–2):65–73. doi: 10.1016/j.ijpharm.2003.12.028. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Z, Bu H, Gao Z, Huang Y, Gao F, Li Y. The characteristics and mechanism of simvastatin loaded lipid nanoparticles to increase oral bioavailability in rats. Int J Pharm. 2010;394(1–2):147–153. doi: 10.1016/j.ijpharm.2010.04.039. [DOI] [PubMed] [Google Scholar]
- 25.Thomas N, Müllertz A, Graf A, Rades T. Influence of lipid composition and drug load on the in vitro performance of self-nanoemulsifying drug delivery systems. J Pharm Sci. 2012;101(5):1721–1731. doi: 10.1002/jps.23054. [DOI] [PubMed] [Google Scholar]
- 26.Zangenberg NH, Müllertz A, Kristensen HG, Hovgaard L. A dynamic in vitro lipolysis model: I. Controlling the rate of lipolysis by continuous addition of calcium. Eur J Pharm Sci. 2001;14(2):115–122. doi: 10.1016/S0928-0987(01)00169-5. [DOI] [PubMed] [Google Scholar]
- 27.Zangenberg NH, Müllertz A, Kristensen HG, Hovgaard L. A dynamic in vitro lipolysis model. II: Evaluation of the model. Eur J Pharm Sci. 2001;14(3):237–244. doi: 10.1016/S0928-0987(01)00182-8. [DOI] [PubMed] [Google Scholar]
- 28.Shitara Y, Sugiyama Y. Pharmacokinetic and pharmacodynamic alterations of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors: drug–drug interactions and interindividual differences in transporter and metabolic enzyme functions. Pharmacol Ther. 2006;112(1):71–105. doi: 10.1016/j.pharmthera.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 29.Sawada T, Sako K, Yoshihara K, Nakamura K, Yokohama S, Hayashi M. Timed-release formulation to avoid drug–drug interaction between diltiazem and midazolam. J Pharm Sci. 2003;92(4):790–797. doi: 10.1002/jps.10336. [DOI] [PubMed] [Google Scholar]
- 30.McKinnon RA, McManus ME. Localization of cytochromes P450 in human tissues: implications for chemical toxicity. Pathol (Phila). 1996;28(2):148–155. doi: 10.1080/00313029600169783. [DOI] [PubMed] [Google Scholar]