

Abstract
Trichloroethylene, a chlorinated solvent widely used as a degreasing agent, is a common environmental contaminant. Emerging evidence suggests that chronic exposure to tri-chloroethylene may contribute to the development of Parkinson’s disease. The purpose of this study was to determine if selective loss of nigrostriatal dopaminergic neurons could be reproduced by systemic exposure of adult Fisher 344 rats to trichloroethylene. In our experiments, oral administration of trichloroethylene induced a significant loss of dopaminergic neurons in the substantia nigra pars compacta in a dose-dependent manner, whereas the number of both cholinergic and GABAergic neurons were not decreased in the striatum. There was a robust decline in striatal levels of 3, 4-dihydroxyphenylacetic acid without a significant depletion of striatal dopamine. Rats treated with trichloroethylene showed defects in rotarod behavior test. We also found a significantly reduced mitochondrial complex I activity with elevated oxidative stress markers and activated microglia in the nigral area. In addition, we observed intracellular α-synuclein accumulation in the dorsal motor nucleus of the vagus nerve, with some in nigral neurons, but little in neurons of cerebral cortex. Overall, our animal model exhibits some important features of Parkinsonism, and further supports that trichloroethylene may be an environmental risk factors for Parkinson’s disease.
Keywords: neurodegeneration, Parkinson’s disease, substantia nigra, trichloroethylene, tyrosine hydroxylase, α-synuclein
Parkinson’s disease (PD) is the most prevalent neurodegenerative movement disorder. The vast majority of PD cases, which account for about 90–95% of patients, are sporadic. The etiology of sporadic PD has not yet been fully elucidated. Nowadays, the prevailing view is that the causes are multifactorial and include genetic predispositions, environmental toxins and aging (Nagatsu and Sawada 2006). Recently, we reported that a group of industrial workers with PD and parkinsonism were subjected to long-term (8–33 years) chronic exposure to trichloroethylene (TCE), and we also found TCE can instigate a selective complex I mitochondrial impairment with a concomitant loss of dopaminergic neurons in the substantia nigra of Fisher 344 rats (Gash et al. 2008). The purpose of this study was to determine if selective loss of nigrostriatal dopaminergic neurons could be reproduced by systemic exposure of animals to the widely used TCE. We also investigated for the first time the neurotoxicity of TCE in different brain regions and on various neuronal cell types as well as some of the underlying mechanisms.
TCE is a chlorinated solvent widely used as a degreasing agent. It is a common environmental contaminant at a high percentage of Superfund sites at many industry and government facilities. For example, TCE is found in soil and surface water as a result of direct discharges and in groundwater as a result of leaching from disposal operations. TCE is also released into the air from degreasing operations. It was estimated that approximately 42 million pounds of TCE was released into the environment in America in 1994 (Scott and Cogliano 2000). Moreover, individuals in many communities are exposed to TCE and have associated health risks. There have been reports showing TCE exposure is involved in: nephrotoxicity and nephrocarcinogenicity such as chronic tubular damage and renal cell tumors (Vamvakas et al. 1998; Pesch et al. 2000; Bruning et al. 2003); several forms of liver disease such as hepatic necrosis, fatty liver and cirrhosis (Thiele et al. 1982; Phoon et al. 1984; Pantucharoensri et al. 2004) and neurotoxicity such as changes in human trigeminal nerve function, motor incoordination and other general symptoms (Ruijten et al. 1991; Rasmussen et al. 1993). Although studies on the human health risk of TCE have lasted for two decades, there continues to be a lack of data for understanding the effects of chronic exposure to TCE on neurotoxicity (Committee 2006). Some case reports suggest a link between chronic TCE exposure and PD. In one report, a 47-year-old woman developed PD after 7 years of professional exposure to TCE (Guehl et al. 1999). Another report described the onset of PD in three individuals chronically exposed to TCE during the post-exposure period (Kochen et al. 2003). Recently, we found that three workers with occupational exposure to TCE were diagnosed with PD; and their coworkers displayed many features of Parkinsonism as well (Gash et al. 2008).
Herein, we follow up the previous study and confirm that TCE can induce a selective dopaminergic neurodegeneration in the nigrostriatal system of Fischer 344 rats, without changing the number of either GABAergic or cholinergic neurons in the striatum. This PD phenotype may be mediated by mitochondrial dysfunction, oxidative/nitrative stress and neuroinflammation.
Materials and methods
Animal and treatments
Male Fischer 344 rats of 5-months of age were used in this study. Animals received either TCE (≥ 99.5%; Sigma, St Louis, MO, USA; dissolved in olive oil) or vehicle by oral gavage (total 0.6 mL per gavage) once a day, 5 days/week. We first performed a dose response for TCE as the rats were orally administered doses of 200, 500, or 1000 mg/kg TCE for 6 weeks (n = 6 per group). After treatment, the animals were deeply anesthetized and subjected to transcardial perfusion with phosphate-buffered saline (PBS) followed by 4% paraformaldehyde. The brains were removed, equilibrated in 30% sucrose and cryosectioned at 30 μm for tyrosine hydroxylase (TH) immunohistochemistry analysis. Next, the dose of 1000 mg/kg TCE was used to further explore the mechanisms of selective cell death and the changes in monoamine levels and behavior. At 2 and 6 weeks after 1000 mg/kg TCE or vehicle treatment, 18 rats (n = 9 for vehicle- or TCE-treated group) at each time point were anesthetized with CO2 and decapitated quickly for mitochondria enzyme activity and oxidative damage measurement in the substantia nigra; 16 rats (n = 8 for vehicle- or TCE-treated group) at each time point were anesthetized and decapitated for HPLC analysis of monoamines and their metabolites in the striatum; and 12 rats (n = 6 for vehicle- or TCE-treated group) at each time point were anesthetized and perfused as described above for histological analysis of silver staining and immunohistochemistry for detecting choline acetyltransferase (ChAT), dopamine and cAMP-regulated phosphoprotein 32 (DARPP-32), cleaved caspase 3, nitrotyrosine (3-NT), OX-42 and α-synuclein, respectively. All animal procedures were performed in accordance with the National Institute of Health guidelines and were approved by the University of Kentucky Institutional Animal Care and Use Committee.
Histological analysis
For immunohistochemistry, sections containing regions of interest were incubated overnight at 4°C with a primary antibody. The primary antibodies used were TH (polyclonal, 1 : 2000; Pel Freez, Rogers, AR, USA) to detect dopaminergic neurons, ChAT (monoclonal, 1 : 500; Chemicon, Temecula, CA, USA) to detect cholinergic neurons, DARPP-32 (polyclonal, 1 : 2000; Chemicon) to detect GABAergic neurons, 3-NT (polyclonal, 1 : 500; Chemicon) to detect protein tyrosine nitration, cleaved caspase 3 (polyclonal, 1 : 500; Cell Technology, Minneapolis, MN, USA) to detect apoptosis, OX-42 (monoclonal, 1 : 2000; PharMingen, San Diego, CA, USA) to detect microglia and α-synuclein (monoclonal, 1 : 1000; BD Pharmingen, Franklin Lakes, NJ, USA). After washes and incubation with an appropriate secondary antibody (Vector Laboratories, Burlingame, CA, USA), immunoreactive cells were visualized by the avidin-biotin immunoperoxidase method (ABC kits; Vector Laboratories) with chromogen 3, 3′-diaminobenzidine tetrahydrochloride (Sigma). In addition, sections immunostained with 3-NT and α-synuclein were counter-stained with Harris hematoxylin to reveal the location of positive signal in the cell body.
The total number of TH-positive cells was counted in a series composed of every sixth section of substantia nigra (total 8 sections per brain), ChAT and DARPP-32 positive cells were counted in a series composed of every 12th section of striatum (total 4 sections per brain), and noradrenergic neurons stained with TH in the locus coeruleus (LC) were counted every sixth section (total 4 sections per brain). In addition, adjacent sections to TH immunostaining in the substantia nigra pars compacta were stained using conventional Nissl staining and the total number of Nissl-stained neurons was counted. Bioquant Image Analysis software (Bioquant, Nashville, TN, USA) was used to estimate total cell number in regions of interest using the optical fractionator method. This method represents an unbiased quantitative technique that is independent of size and shape or any conformational changes of cells. Detailed procedures regarding stereological counting are similar to our previously published studies (Liu et al. 2008).
For silver stain, sections of the substantia nigra were processed using the FD NeuroSilver kit (FD Neuro Technologies, Inc., Baltimore, MD, USA) according to the manufacturer’s instructions. For Giemsa stain, sections of the cerebellum were mounted on the subbed slides. Giemsa’s solution (EM Science, Lawrence, KS, USA) was diluted 1/10 in 0.15 M KH2PO4 buffer and heated to 60°C. Then slides were stained in warmed dilution solution for 10 min. For Luxol fast blue stain, sections after de-fatting step were incubated in 0.1% luxol fast blue solution overnight, then differentiated in the 0.05% lithium carbonate solution and counter-stained in 1% neutral red.
HPLC assay
Striatal levels of monoamines and their metabolites were determined by HPLC with electrochemical detection as previously described (Cass et al. 2003). Results were expressed as ng/g wet weight of tissue from vehicle- and TCE-treated rats.
Rotarod testing
The Rotarod treadmill ENV-575 (MED Associates Inc., Albans, VT, USA), designed to measure motor performance and coordination, consists of a 7.0-cm diameter cylindrical treadmill connected to a computer-controlled stepper motor-driven drum, that can be programmed to operate at a constant speed or in a defined acceleration mode. When the animal falls off the rotating drum, individual sensors at the bottom of each separate compartment automatically record the amount of time (in seconds) spent on the treadmill. Rats were trained on three consecutive days at a fixed speed. Then, at each time point, rats were tested while the treadmill accelerated from 3 to 30 rpm, and time spent on the treadmill was recorded for two successive trials, so a mean score could be calculated and analyzed.
Mitochondria isolation and enzyme activity
The method has been previously described with some minor modifications (Sullivan et al. 2003; Brown et al. 2004; Patel et al. 2009). Briefly, following CO2 anesthesia, the nigral regions were isolated quickly using a rat brain matrix. Three rat substantia nigras were pooled to make one nigral mitochondrial sample. The mitochondria were isolated from synaptoneurosomes using ficoll density gradient ultracentrifugation and nitrogen decompression. Mitochondrial complex I activity in rotenone-sensitive condition was assayed by measuring the decrease in fluorescence of NADH as substrate (340 nm excitation, > 450 nm emission) using BioTek Synergy HT plate reader (Bio-Tek Instruments, Winooski, VT, USA).
Immunoblotting analysis
Oxidative damage was assessed by slot immunoblotting as previously described (Hunter et al. 2007). The protein samples were loaded into each well of the slot-blot apparatus. They were sucked through onto the nitrocellulose followed by PBS. The membrane was incubated overnight at 4°C in primary antibody solution containing antibody against 3-NT (1 : 2000; Upstate Biotechnology, Lake Placid, NY, USA) or 4HNE (1 : 10 000; Calbiochem) or Oxyblot Kit (1 : 100; Chemicon). Then, the blot was incubated in the appropriate secondary antibody. Next, the blot was developed in Super Signal 1 : 1 and a picture was taken using the BioRad set-up.
Statistical analysis
All data were analyzed using a computer statistical package (SYSTAT 10; SPSS Inc, Chicago, IL, USA). The values are expressed as the mean ± SEM, and differences among means were analyzed by using one-way analysis of variance (ANOVA) with treatment as the independent factor. Fisher’s least-significant difference post hoc analysis was employed when differences were observed in ANOVA testing (p < 0.05).
Results
TCE selectively induced dopaminergic neurodegeneration in the central nervous system
We first conducted a dose response for TCE as rats were orally administered doses of 200, 500 or 1000 mg/kg/day TCE for 6 weeks. TH immunohistochemistry was performed in the substantia nigra and the total number of TH-positive cells was counted using the optical fractionator method (Bioquant). There was a dose-dependent loss of dopaminergic neurons: 20.1% in TCE 200, 25.6% in TCE 500 and 40.6% in TCE 1000 mg/kg treatment in comparison with the vehicle treatment (Fig. 1a and b). One-way ANOVA indicates that there was a significant cell loss in the 500 and 1000 mg/kg TCE-treated groups, but not in the 200 mg/kg TCE-treated group, when compared with vehicle-treated group (Fig. 1b, p < 0.05 500 mg/kg TCE relative to vehicle, p < 0.01 1000 mg/kg TCE relative to vehicle). On adjacent sections (see Fig. S1a) and the total number of Nissl-stained neurons in the substantia nigra pars compact was 57 260 ± 3211 in vehicle group and 37 609 ± 1933 in 1000 mg/kg TCE group. Statistical analysis also confirmed there was a significant 34.3% loss of Nissl-stained neurons in the 1000 mg/kg TCE group as compared with vehicle control (p < 0.01). Thus, TCE-induced dopaminergic neuron loss is not because of TCE-induced down-regulation of the TH marker but to cell destruction or degeneration. In addition, the total number of TH-positive cells in the ventral tegmental area was counted and there were 36 909 ± 1371 in vehicle group and 36 580 ± 1727 in 1000 mg/kg TCE group. The difference was not statistically significant between the two groups (p > 0.05) indicating that dopaminergic neurons in the ventral tegmental area are not affected by TCE administration. Furthermore, silver staining indicated neurons with metallic silver deposits in the nigral area of 1000 mg/kg TCE-treated animals, but no obviously positive silver staining in corresponding vehicle subjects. Interestingly, the silver-stained neurons were surrounded by glial-like cells and glial cells can serve diverse functions including cellular maintenance through phagocytosis (Fig. 1c). Taken together, these results support that TCE can induce dopaminergic neuron degeneration in the substantia nigra pars compacta. The TCE dose of 1000 mg/kg was used for further experiments.
Fig. 1.

TCE-induced dopaminergic neuron degeneration in a dose-dependent manner. (a) Representative TH immunostaining in the substantia nigra of 6-week TCE (200, 500 and 1000 mg/kg) and vehicle-treated rats. Scale bar = 500 μm. (b) Unbiased stereological cell counting showed a significant reduction in the total number of TH-positive neurons in the substantia nigra of 500 and 1000 mg/kg TCE-treated rats as compared with vehicle treatment. *p < 0.05, **p < 0.01 vs. vehicle, n = 6 per group. (c) Silver staining revealed degenerating neurons with metallic silver deposits (arrow) and the surrounding of glia-like cells (arrowheads) in the substantia nigra after 6-week 1000 mg/kg TCE treatment. Scale bar = 50 μm.
Damage to specific subnuclei of the substantia nigra pars compacta is frequently considered to be the most important hallmark of PD; however, pathological studies have demonstrated it is also accompanied by extensive extranigral pathology, including that in the dorsal motor nucleus of the glossopharyngeal and vagus nerves, the coeruleus–subcoeruleus complex and so on (Braak et al. 2003). TH immunohistochemistry in the locus coeruleus was performed to visualize for noradrenergic neurons; and the cell counting showed there was no significant difference in the number of TH-positive cells in 1000 mg/kg TCE-treated animals as compared with control subjects (p > 0.05, see Fig. S2). Hematoxylin and eosin stain also did not reveal obvious loss of neurons in the dorsal motor nucleus of the vagus nerve in TCE treatment.
To investigate whether systemic treatment of TCE to animals can cause non-dopaminergic neuronal or myelin damage in the central nervous system, we did immunostaining of ChAT for cholinergic neurons and DARPP-32 for GABAergic neurons in the striatum, Giemsa staining for Purkinje cells in the cerebellum, and Luxol fast blue staining for myelin distribution in the brainstem of rats at 6 weeks after 1000 mg/kg TCE treatment. Our data showed both ChAT and DARPP-32 immunostaining appeared normal with undamaged neurons and their processes in the striatum (Fig. 2a). The total number of cholinergic neurons was 55 067 ± 8414 in vehicle-treated group and 51 467 ± 6382 in TCE-treated group, and GABAergic neurons was 3 211 200 ± 255 513 in vehicle-treated group and 3 082 133 ± 147 588 in TCE-treated group. There was no statistically significant difference in the number of cholinergic or GABAergic neurons in TCE-treated animals when compared with vehicle treatment (p > 0.05). Thus, TCE selectively induced dopaminergic neuronal loss in the nigrostriatal system. In addition, Purkinje cells, a class of GABAergic neurons, are located in cerebellar cortex. They send inhibitory projections to the deep cerebellar nuclei, and constitute the sole output of all motor integration in the cerebellar cortex. As our behavioral study indicated there was impairment of coordination and balance in TCE-treated animals, we performed Giemsa staining to determine whether Purkinje cells were injured in our experiment. Giemsa staining showed an intact monolayer of Purkinje cells aligned in the cerebellum of both TCE and vehicle-treated animals (Fig. 2b), suggesting that Purkinje cells were not injured by the TCE treatment. Luxol fast blue staining also indicated no difference in myelin distribution in brain between TCE-and vehicle-treated animals (Fig. 2c). Therefore, TCE oral administration in our rat model appears to specifically induce nigral dopaminergic neurodegeneration.
Fig. 2.

There was no obvious damage for non-dopaminergic neurons in central nervous system after 6-week TCE (1000 mg/kg) treatment. (a) ChAT and DARPP-32 immunostaining were respectively visualized for cholinergic and GABAergic neurons in the striatum; and no significant cell loss in TCE compared with vehicle-treated rats. Scale bar = 500 μm. (b) Giemsa staining showed intact Purkinje cell monolayer (arrow) in the cerebellum of both TCE and vehicle-treated group. Scale bar = 1 mm. (c) Luxol fast blue staining revealed no myelin disruption in the brain of both TCE and vehicle-treated animals. Scale bar = 1 mm.
TCE impaired striatal dopaminergic transmitter system and caused movement dysfunction
To evaluate the change of monoamines and their metabolites in the nigrostriatal system, striatal tissues of animals were dissected out quickly at 6 weeks after 1000 mg/kg TCE treatment. The levels of dopamine (DA) and its metabolites 3, 4-dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA), and serotonin and its metabolite 5-hydroxyindoleacetic acid, were determined by HPLC. The striatal level of DOPAC had a significant decrease of 33.1%, HVA had a decrease of 11.6%, whereas DA was not altered in TCE-treated rats when compared with vehicle-treated animal (Fig. 3a). In addition, the DOPAC level was also significantly decreased by 30.2% at 2 weeks after TCE administration (data not shown). No significant changes were found in levels of serotonin and 5-hydroxyindoleacetic acid in the striatum after treatment (Fig. 3a). Therefore, the data suggest that TCE selectively damage DA neurons rather than serotonin neurons.
Fig. 3.

TCE (1000 mg/kg) treatment affected dopamine’s metabolites in the striatum and led to movement dysfunction. (a) The levels of DOPAC and HVA were significantly decreased in TCE compared with vehicle-treated rats. (b) Rotarod testing analysis showed the duration on the rotating drum was significantly decreased on the 5th and 6th week of TCE treatment when compared with vehicle-treated animal. *p < 0.05, **p < 0.01 vs. vehicle, n = 8 per group.
To test the effect of TCE on locomotor function, we performed behavioral studies including both rotarod performance and spontaneous locomotor activity once a week in rats during 1000 mg/kg TCE treatment. The rotarod test was completed on the Rotarod treadmill. Rats treated with TCE had a significant decrease in duration on the treadmill at both 5 and 6 weeks of treatment when compared with vehicle-treated rats (Fig. 3b). This rotarod behavioral test is an established test used for the assessment of neurological deficits in rodents, and has been reported to be useful for evaluation of overall motor deficits in rat models of Parkinsonism to assess akinetic symptoms (Rozas et al. 1997). Thus, TCE may produce PD-like motor deficits in rats. However, spontaneous locomotor activity was measured by an automated computer system (Model RXYZCM-8; Accuscan Instruments, Columbus, OH, USA), where there were no significant differences in recorded parameters between TCE- and vehicle-treated animals (data not shown).
Neurotoxicity of TCE may be mediated by mitochondrial dysfunction, oxidative stress and inflammation
To study the mechanism of TCE-induced dopaminergic neuronal death, we performed mitochondrial enzyme activity measurements, oxidative stress marker detection, and immunohistochemistry for 3-nitrotyrosine, cleaved caspase 3 and OX-42 in the substantia nigra after 1000 mg/kg TCE treatment. At 2 weeks, we found a significant reduction in mitochondrial complex I enzyme activity, measured by NADH substrate oxidation, in the substantia nigra of TCE-treated rats (Fig. 4a); and also found the similar result at 6 weeks of TCE treatment, but no significant differences were measured in flavin adenine dinucleotide-linked, complex II-driven respiration in TCE compared with vehicle-treated animals (Gash et al. 2008). The mitochondrial bioenergetics data showed TCE administration appeared to be specifically inhibiting complex I activity. Dysfunction of mitochondrial complex I activity will induce ATP depletion, and ultimately lead to cell death, which may be apoptotic or necrotic in nature. We know that caspase 3 is the ultimate executioner caspase protein for the nuclear changes associated with apoptosis. In this study, cleaved caspase 3 immunostaining showed strong nuclear and diffuse cytosolic-positive signals in TCE-treated rats (Fig. 4b). There was a significantly higher level of caspase 3-positive cells in TCE group (5010 ± 316) when compared with vehicle control (1040 ± 135) (p < 0.001), suggesting that apoptosis is involved in the pathological changes of cell death (also see Fig. S1b). In addition, slot blot technology was used to quantify the formation of protein carbonyls (OxyBlot), the lipid peroxidation product (4HNE) and protein tyrosine nitration (3-NT) in our experiment. Compared with the vehicle group, the level of protein carbonyls was significantly increased at 2 weeks, 4HNE was increased at 6 weeks, and 3-NT was increased and sustained at both time-points after TCE treatment (Fig. 4c, left panel). Moreover, 3-NT immunostaining and counter-staining with hematoxylin demonstrated that tyrosine nitration was mostly localized in the neuron-like cells in the substantia nigra of TCE-treated rats (Fig. 4c, right panel). Together, oxidative and nitrative stresses are also involved in the pathological process of TCE treatment. In addition, our studies found activated microglia with large cell bodies and stout processes (Hunter et al. 2007) in the substantia nigra after TCE treatment (Fig. 4d). Activated microglia can contribute to neurodegeneration by releasing neurotoxic factors such as phagocyte oxidase-induced superoxide and several proinflammatory cytokines (e.g. tumor necrosis factor-alpha and interleukin-1 beta). Therefore, inflammation may also be involved in the TCE-induced neurotoxicity.
Fig. 4.
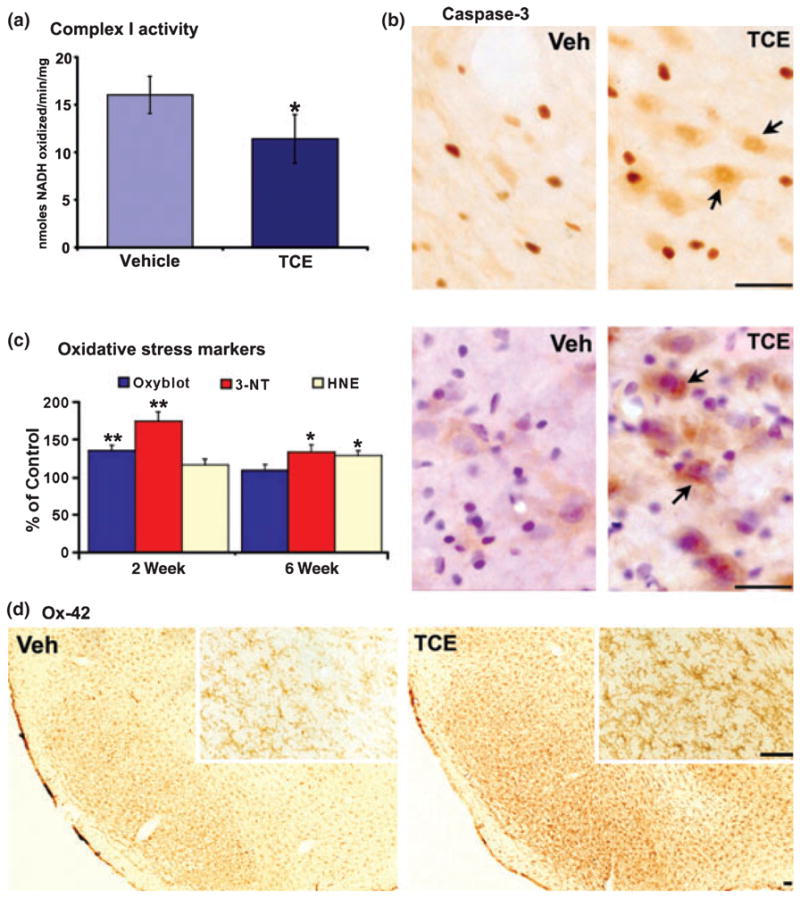
Mitochondrial dysfunction, oxidative stress and inflammation were found in the substantia nigra following 1000 mg/kg TCE treatment. (a) Mitochondrial complex I enzyme activity as measured by NADH substrate oxidation was significantly reduced in the TCE-treated rats as compared with vehicle treatment. (b) Cleaved caspase 3 immunostaining showed more positive signals (arrows) in TCE-treated rats (scale bar = 100 μm). (c) The level of protein carbonyls was significantly increased at 2 weeks, 4-HNE was increased at 6 weeks and 3-NT was increased at both time-points after TCE treatment in comparison with vehicle treatment (left panel). 3-NT immunostaining and counter-staining with hematoxylin indicated that tyrosine nitration was mostly localized in the neuron-like cells (arrows) in TCE-treated rats (right panel, scale bar = 100 μm). (d) Representative OX-42 staining revealed activated microglia with large cell bodies and stout processes at 2 weeks after TCE (the inset depicts a high-magnification image, scale bar = 200 μm). *p < 0.05, **p < 0.01 vs. vehicle, n = 9 per group.
Intracellular α-synuclein accumulation was found in PD-related region following TCE treatment
Sporadic PD is a multisystem disorder that involves only a few susceptible types of nerve cells in specific regions of the human nervous system. The pathological hallmark of PD is the formation of Lewy neurites in cellular processes and Lewy bodies in neuronal perikarya. A major component of Lewy neurites and Lewy bodies is an aggregated form of the normally presynaptic protein α-synuclein. Recently, Braak et al. (2003) analyzed post-mortem data of 168 cases with α-synuclein immunohistochemistry, and suggest pathological changes of PD follow a coherent sequence, which begins in the dorsal motor nucleus of the glossopharyngeal and vagus nerves and/or adjoining intermediate reticular zone (stages 1 and 2), from there reaching additional lower brainstem nuclei (stages 3 and 4) while assuming an essentially upward course and eventually extending into the cerebral cortex (stages 5 and 6). To test these PD pathological changes in our TCE-treated rat model, we performed α-synuclein immunostaining and counter-staining with hematoxylin in sections from brainstem, substantia nigra and cerebral cortex following 1000 mg/kg TCE treatment for 6 weeks. Our data showed that there was marked intracellular α-synuclein accumulation in the dorsal motor nucleus of the vagus nerve, with some in nigral cells of substantia nigra pars compacta, but little accumulation in neurons of cerebral cortex (Fig. 5). In the substantia nigra, fluorescence-based double-immunostaining confirmed the co-localization of TH and α-synuclein immunostaining using confocal microscopy (see Fig. S3). According to Braak’s pathological staging of PD, TCE only affected the early stage and middle stage in our rodent model. This could be interpreted as a TCE-induced moderate injury in which there are no significant decreases of dopamine content in the striatum and no marked deficit of spontaneous locomotor activity in TCE-treated animals.
Fig. 5.

Intracellular α-synuclein accumulation was observed in brainstem and substantia nigra pars compacta at 6 weeks after 1000 mg/kg TCE treatment. Immunohistochemistry for α-synuclein and counter-staining with hematoxylin showed marked intracellular α-synuclein protein accumulation in the dorsal motor nucleus of the vagus nerve (a, outlined area, scale bar = 200 μm), some in nigral cells (b, arrows, scale bar = 10 μm) and little in neurons of cerebral cortex (c, scale bar = 200 μm) of TCE-treated animals in comparison of vehicle treatment.
Discussion
This study confirmed selective dopaminergic neuron death by systemic exposure of animals to TCE. We found that the level of DOPAC had a decrease of about 30% in the striatum, but no significant depletion of dopamine. The data that TCE inhibited mitochondrial complex I enzyme activity, increased oxidative stress markers, activated microglia and induced intracellular α-synuclein accumulation in some specific neurons have helped elucidate the underlying mechanisms for TCE-induced dopaminergic neurodegeneration. TCE treatment can produce some features of Parkinsonism in the rodent model and it may recapitulate features of early to middle stages of PD.
Our findings show that selective dopaminergic neuron degeneration, one of the most important hallmarks of PD, is also a characteristic of TCE neurotoxicity. However, the depletion of dopamine was not noted, even in the presence of significant nigral cell loss. There is a possibility that TCE may preferentially target dopaminergic cell bodies rather than terminals; similar to the paraquat PD model where dopamine levels remained relatively unchanged, even in the presence of a 33% loss of dopaminergic neurons (McCormack et al. 2002). The level of DOPAC decreased in the striatum, which is consistent with previously published studies that found a single intranigral injection of one by-product of TCE induced a progressive decline in DOPAC levels in the striatum of rats (Grote et al. 1995; Wesemann et al. 1994). Released DA is converted to DOPAC by intraneuronal monoamine oxidase after reuptake by the nerve terminal and released DA is also converted to HVA probably at an extraneuronal site, through the sequential action of catechol-O-methyltransferase and monoamine oxidase (Cooper et al. 2003). So it is possible that the decrease in DOPAC may be due to TCE inhibiting either or both dopamine reuptake and monoamine oxidase activity.
TCE is a volatile, lipophilic chemical. There are three routes of exposures to TCE for humans and laboratory animals: inhalation, oral and dermal. The 1985 United States EPA health assessment document on TCE reported that the metabolic pathways for TCE were qualitatively similar in mice, rats and humans. Absorbed TCE is distributed to different target organs (e.g. lungs, liver, kidney and nervous system) via the circulatory system. Most TCE taken into the body is metabolized and metabolites from TCE are thought to be responsible for the toxicity and carcinogenicity observed in different organ systems. There are two irreversible pathways, the oxidative and glutathione-dependent pathways (Lash et al. 2000). The oxidative pathway is the major pathway for TCE metabolism. TCE is metabolized primarily by the cytochrome P-450s isoform CYP2E1 to a trichloroethylene oxide intermediate, which spontaneously rearranges into chloral. Chloral, a major metabolite produced in the oxidative pathway, is a chemically highly reactive aldehyde that is speculated to spontaneously condense with the biogenic amine tryptamine by a Pictet-Spengler cyclization, to afford the alkaloid-type neurotoxin (Bringmann and Hille 1990). Some laboratories have reported one product, 1-trichloromethyl-1,2,3,4-tetrahydro-β-carboline (TaClo), was readily formed under quasi-physiological conditions (buffered water, pH 7.4, 37°C) from the biogenic amine tryptamine (Ta) and chloral (Clo) (Bringmann et al. 1995a,b). The interest was stimulated by the recognition of the structural similarity of the tetrahydro-β-carboline molecular framework in TaClo with that of MPTP, a well-known neurotoxin that elicits Parkinsonism in primates, including man. In vitro studies have reported that TaClo is a strong cytotoxin toward dopaminergic cells (Bringmann et al. 2000; Akundi et al. 2004). The mechanism of TaClo-induced cell death included caspase 3-mediated apoptosis, an increase in the production of reactive oxygen species, and inhibition of mitochondrial respiration (Bringmann et al. 2000; Akundi et al. 2004). It should be noted that TaClo, its potency to inhibit mitochondrial complex I in rat brain homogenate, is greater than the same concentration of MPP+ (Riederer et al. 2002). In addition, in vivo studies found that TaClo possess a lipophilic nature and has the ability to cross biological membranes including the blood brain barrier (Bringmann et al. 2006). Following [14C] TaClo administration, high levels of radioactivity were found in the kidneys and liver, while low levels were detected in the brain, heart and muscle of Wistar rats (Bringmann et al. 2006). Although a small amount of TaClo was found in the brain, radioactivity was eliminated from brain about 2.5 times more slowly than from plasma (Bringmann et al. 2006). The accumulation or storage of continuously formed TaClo, particularly in the brain, as a consequence of chronic exposure to TCE may play a major role in TCE neurotoxicity and dopaminergic neurodegeneration.
Environmental toxins play an important role in the development of sporadic form of PD, where it is suggested that various toxins can initiate a cascade of events in the cell body that induces dopaminergic neuron degeneration and ultimately causes Parkinsonism. These events mainly include mitochondrial dysfunction, inflammation, oxidative/nitrative stress and ubiquitin–proteasome system dysfunction (Lotharius and Brundin 2002; Andersen 2004). Mitochondrial complex I is the first and the most complex of the three energy-transducing enzyme complexes in the mitochondrial electron transport chain. Dysfunction of the mitochondria is one of several major pathophysiological changes observed in PD (Mann et al. 1994; Dawson and Dawson 2003). Our study found TCE significantly reduced mitochondrial complex I activity in the substantia nigra. Therefore, mitochondrial dysfunction is likely involved in the pathogenesis of neuronal death induced by TCE. In addition, oxidation and nitration of proteins, DNA and lipids are markers of neurodegeneration in post-mortem tissues; however, it is impossible to determine using post-mortem analysis whether oxidative stress has a primary role in neurodegeneration or is a secondary end-stage epiphenomenon (Ischiropoulos and Beckman 2003). We observed a significant increase of oxidative/nitrative stress markers in the substantia nigra of animals following TCE treatment. It is suggested that the oxidative/nitrative damage may contribute to the neuronal death as a result of TCE exposure. Recently, there is increasing recognition of the possible role of inflammation as a major factor in the pathogenesis of PD. In both patients and experimental models of PD, they present with all of the classical features of inflammation including phagocyte activation, increased synthesis and release of proinflammatory cytokines and complement activation in their pathology (McGeer et al. 2001). Del Rio Hortega initially described microglia as a separate cell type from the other glial cells such as astrocytes and oligodendrocytes. The microglia is the resident immunocompetent and phagocytic cells in the central nervous system, and is thought to mediate the innate defense system, which serves a critical role in the nervous system. When various stimuli occur and continue, the inner cytoskeleton of the microglia changes, the cell body becomes enlarged and displays a macrophage-like appearance. These activated microglia are thought to act as contributors of proinflammatory and neurotoxic factors (Whitton 2007). Our data showed increased microglia activation in the substantia nigra of TCE-treated animals using immunostaining with a specific marker, OX-42. As a toxin, TCE may also stimulate microglia activation, leading to subsequent release of neurotoxic factors such as PHOX-induced superoxide and cytokines that finally exacerbate cell death. In general, neurodegeneration induced by TCE may be mediated by mitochondrial dysfunction, oxidative/nitrative stress and inflammation. We speculate these events are not separate, but interact to form a vicious cycle, which ultimately leads to dopaminergic cell death. The mitochondria dysfunction and activated microglia generate free radicals to induce oxidative stress. Likewise, the generation of oxidation/nitration byproducts can further damage mitochondrial function and stimulate microglia activation. Eventually, these events may result in protein mishandling and the degeneration of dopaminergic neurons that with time will lead to the features of Parkinsonism.
As a common environmental contaminant, TCE produces toxicity to different organ systems. This study demonstrates the potential link between TCE exposure and the development of PD; and in part explores the underlying mechanism of neurodegeneration that may be mediated through mitochondrial dysfunction, oxidative/nitrative stress and inflammation. Furthermore, the significance of our finding is not only for workplace safety but also with respect to the general environmental hygiene.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The authors greatly thank Laura E. Peters for technical assistance with the HPLC assays. This study was supported by grant AG13494 (to DMG), AG17963 (to WAC), NS044157 (to GYB) and Brain Research Center from 21st Century Frontier Program funded by the Ministry of Science and Technology (2009K001253), Republic of Korea (to HCK).
Abbreviations used
- 3-NT
nitrotyrosine
- ChAT
choline acetyltransferase
- DA
dopamine
- DARPP-32
dopamine and cAMP-regulated phosphoprotein 32
- DOPAC
3, 4-dihydroxyphenylacetic acid
- HVA
homovanillic acid
- PD
Parkinson’s disease
- TaClo
1-trichloromethyl-1,2,3,4-tetrahydro-β-carboline
- TCE
trichloroethylene
- TH
tyrosine hydroxylase
Footnotes
Additional Supporting Information may be found in the online version of this article:
Figure S1. Nissl staining was in the substantia nigra.
Figure S2. Representative TH immunostaining was in the locus coeruleus (LC) of 1000 mg/kg TCE and vehicle-treated animal (a, scale bar = 500 μm).
Figure S3. Double immunofluorescent staining for α-synuclein (α-syn) and TH was in the substantia nigra of TCE-treated animal.
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
References
- Akundi RS, Macho A, Munoz E, Lieb K, Bringmann G, Clement HW, Hull M, Fiebich BL. 1-Trichloromethyl-1,2,3,4-tetrahydro-beta-carboline-induced apoptosis in the human neuroblastoma cell line SK-N-SH. J Neurochem. 2004;91:263–273. doi: 10.1111/j.1471-4159.2004.02710.x. [DOI] [PubMed] [Google Scholar]
- Andersen JK. Oxidative stress in neurodegeneration: cause or consequence? Nat Med. 2004;10(Suppl):S18–S25. doi: 10.1038/nrn1434. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- Bringmann G, Hille A. Endogenous alkaloids in man, VII: 1-trichloromethyl-1,2,3,4-tetrahydro-beta-carboline – a potential chloral-derived indol alkaloid in man. Arch Pharm. 1990;323:567–569. doi: 10.1002/ardp.19903230903. [DOI] [PubMed] [Google Scholar]
- Bringmann G, God R, Feineis D, Janetzky B, Reichmann H. TaClo as a neurotoxic lead: improved synthesis, stereo-chemical analysis, and inhibition of the mitochondrial respiratory chain. J Neural Transm. 1995a;46:245–254. [PubMed] [Google Scholar]
- Bringmann G, God R, Feineis D, Wesemann W, Riederer P, Rausch WD, Reichmann H, Sontag KH. The TaClo concept: 1-trichloromethyl-1,2,3,4-tetrahydro-beta-carboline (TaClo), a new toxin for dopaminergic neurons. J Neural Transm. 1995b;46:235–244. [PubMed] [Google Scholar]
- Bringmann G, Feineis D, Bruckner R, et al. Bromal-derived tetrahydro-beta-carbolines as neurotoxic agents: chemistry, impairment of the dopamine metabolism, and inhibitory effects on mitochondrial respiration. Bioorg Med Chem. 2000;8:1467–1478. doi: 10.1016/s0968-0896(00)00073-0. [DOI] [PubMed] [Google Scholar]
- Bringmann G, Feineis D, Bruckner R, God R, Grote C, Wesemann W. Synthesis of radiolabelled 1-trichloromethyl-1,2,3,4-tetrahydro-beta-carboline (TaClo), a neurotoxic chloral-derived mammalian alkaloid, and its biodistribution in rats. Eur J Pharm Sci. 2006;28:412–422. doi: 10.1016/j.ejps.2006.03.009. [DOI] [PubMed] [Google Scholar]
- Brown MR, Geddes JW, Sullivan PG. Brain region-specific, age-related, alterations in mitochondrial responses to elevated calcium. J Bioenerg Biomembr. 2004;36:401–406. doi: 10.1023/B:JOBB.0000041775.10388.23. [DOI] [PubMed] [Google Scholar]
- Bruning T, Pesch B, Wiesenhutter B, Rabstein S, Lammert M, Baumuller A, Bolt HM. Renal cell cancer risk and occupational exposure to trichloroethylene: results of a consecutive case–control study in Arnsberg, Germany. Am J Ind Med. 2003;43:274–285. doi: 10.1002/ajim.10185. [DOI] [PubMed] [Google Scholar]
- Cass WA, Harned ME, Peters LE, Nath A, Maragos WF. HIV-1 protein Tat potentiation of methamphetamine-induced decreases in evoked overflow of dopamine in the striatum of the rat. Brain Res. 2003;984:133–142. doi: 10.1016/s0006-8993(03)03122-6. [DOI] [PubMed] [Google Scholar]
- Committee. Committee on Human Health Risks of Trichloroethylene, Assessing the Human Health Risks of Trichloroethylene; Key Scientific Issues. The National Academies Press; Washington, DC: 2006. [Google Scholar]
- Cooper JR, Boom FE, Roth RH. The Biochemical Basis of Neurophamacology. Oxford University Press; London, New York: 2003. [Google Scholar]
- Dawson TM, Dawson VL. Molecular pathways of neurodegeneration in Parkinson’s disease. Science. 2003;302:819–822. doi: 10.1126/science.1087753. [DOI] [PubMed] [Google Scholar]
- Gash DM, Rutland K, Hudson NL, et al. Trichloroethylene: Parkinsonism and complex 1 mitochondrial neurotoxicity. Ann Neurol. 2008;63:184–192. doi: 10.1002/ana.21288. [DOI] [PubMed] [Google Scholar]
- Grote C, Clement HW, Wesemann W, Bringmann G, Feineis D, Riederer P, Sontag KH. Biochemical lesions of the nigrostriatal system by TaClo (1-trichloromethyl-1,2,3,4-tetrahydro-beta-carboline) and derivatives. J Neural Transm. 1995;46:275–281. [PubMed] [Google Scholar]
- Guehl D, Bezard E, Dovero S, Boraud T, Bioulac B, Gross C. Trichloroethylene and parkinsonism: a human and experimental observation. Eur J Neurol. 1999;6:609–611. doi: 10.1046/j.1468-1331.1999.650609.x. [DOI] [PubMed] [Google Scholar]
- Hunter RL, Dragicevic N, Seifert K, Choi DY, Liu M, Kim HC, Cass WA, Sullivan PG, Bing G. Inflammation induces mitochondrial dysfunction and dopaminergic neuro-degeneration in the nigrostriatal system. J Neurochem. 2007;100:1375–1386. doi: 10.1111/j.1471-4159.2006.04327.x. [DOI] [PubMed] [Google Scholar]
- Ischiropoulos H, Beckman JS. Oxidative stress and nitration in neurodegeneration: cause, effect, or association? J Clin Invest. 2003;111:163–169. doi: 10.1172/JCI17638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochen W, Kohlmuller D, De Biasi P, Ramsay R. The endogeneous formation of highly chlorinated tetrahydro-beta-carbolines as a possible causative mechanism in idiopathic Parkinson’s disease. Adv Exp Med Biol. 2003;527:253–263. doi: 10.1007/978-1-4615-0135-0_29. [DOI] [PubMed] [Google Scholar]
- Lash LH, Fisher JW, Lipscomb JC, Parker JC. Metabolism of trichloroethylene. Environ Health Perspect. 2000;108(Suppl 2):177–200. doi: 10.1289/ehp.00108s2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Hunter R, Nguyen XV, Kim HC, Bing G. Microsomal epoxide hydrolase deletion enhances tyrosine hydroxylase phosphorylation in mice after MPTP treatment. J Neurosci Res. 2008;86:2792–2801. doi: 10.1002/jnr.21725. [DOI] [PubMed] [Google Scholar]
- Lotharius J, Brundin P. Pathogenesis of Parkinson’s disease: dopamine, vesicles and alpha-synuclein. Nat Rev. 2002;3:932–942. doi: 10.1038/nrn983. [DOI] [PubMed] [Google Scholar]
- Mann VM, Cooper JM, Daniel SE, Srai K, Jenner P, Marsden CD, Schapira AH. Complex I, iron, and ferritin in Parkinson’s disease substantia nigra. Ann Neurol. 1994;36:876–881. doi: 10.1002/ana.410360612. [DOI] [PubMed] [Google Scholar]
- McCormack AL, Thiruchelvam M, Manning-Bog AB, Thiffault C, Langston JW, Cory-Slechta DA, Di Monte DA. Environmental risk factors and Parkinson’s disease: selective degeneration of nigral dopaminergic neurons caused by the herbicide paraquat. Neurobiol Dis. 2002;10:119–127. doi: 10.1006/nbdi.2002.0507. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Yasojima K, McGeer EG. Inflammation in Parkinson’s disease. Adv Neurol. 2001;86:83–89. [PubMed] [Google Scholar]
- Nagatsu T, Sawada M. Cellular and molecular mechanisms of Parkinson’s disease: neurotoxins, causative genes, and inflammatory cytokines. Cell Mol Neurobiol. 2006;26:781–802. doi: 10.1007/s10571-006-9061-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantucharoensri S, Boontee P, Likhitsan P, Padungtod C, Prasartsansoui S. Generalized eruption accompanied by hepatitis in two Thai metal cleaners exposed to trichloroethylene. Ind Health. 2004;42:385–388. doi: 10.2486/indhealth.42.385. [DOI] [PubMed] [Google Scholar]
- Patel SP, Sullivan PG, Pandya JD, Rabchevsky AG. Differential effects of the mitochondrial uncoupling agent, 2,4-dinitrophenol, or the nitroxide antioxidant, Tempol, on synaptic or nonsynaptic mitochondria after spinal cord injury. J Neurosci Res. 2009;87:130–140. doi: 10.1002/jnr.21814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesch B, Haerting J, Ranft U, Klimpel A, Oelschlagel B, Schill W. Occupational risk factors for renal cell carcinoma: agent-specific results from a case-control study in Germany. MURC Study Group Multicenter urothelial and renal cancer study. Int J Epidemiol. 2000;29:1014–1024. doi: 10.1093/ije/29.6.1014. [DOI] [PubMed] [Google Scholar]
- Phoon WH, Chan MO, Rajan VS, Tan KJ, Thirumoorthy T, Goh CL. Stevens-Johnson syndrome associated with occupational exposure to trichloroethylene. Contact dermatitis. 1984;10:270–276. doi: 10.1111/j.1600-0536.1984.tb00145.x. [DOI] [PubMed] [Google Scholar]
- Rasmussen K, Arlien-Soborg P, Sabroe S. Clinical neurological findings among metal degreasers exposed to chlorinated solvents. Acta Neurol Scand. 1993;87:200–204. doi: 10.1111/j.1600-0404.1993.tb04101.x. [DOI] [PubMed] [Google Scholar]
- Riederer P, Foley P, Bringmann G, Feineis D, Bruckner R, Gerlach M. Biochemical and pharmacological characterization of 1-trichloromethyl-1,2,3,4-tetrahydro-beta-carboline: a biologically relevant neurotoxin? Eur J Pharmacol. 2002;442:1–16. doi: 10.1016/s0014-2999(02)01308-0. [DOI] [PubMed] [Google Scholar]
- Rozas G, Guerra MJ, Labandeira-Garcia JL. An automated rotarod method for quantitative drug-free evaluation of overall motor deficits in rat models of parkinsonism. Brain Res Brain Res Protoc. 1997;2:75–84. doi: 10.1016/s1385-299x(97)00034-2. [DOI] [PubMed] [Google Scholar]
- Ruijten MW, Verberk MM, Salle HJ. Nerve function in workers with long term exposure to trichloroethene. British journal of industrial medicine. 1991;48:87–92. doi: 10.1136/oem.48.2.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott CS, Cogliano VJ. Trichloroethylene health risks – state of the science. Environ Health Perspect. 2000;108(Suppl 2):159–160. [PMC free article] [PubMed] [Google Scholar]
- Sullivan PG, Dube C, Dorenbos K, Steward O, Baram TZ. Mitochondrial uncoupling protein-2 protects the immature brain from excitotoxic neuronal death. Ann Neurol. 2003;53:711–717. doi: 10.1002/ana.10543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele DL, Eigenbrodt EH, Ware AJ. Cirrhosis after repeated trichloroethylene and 1,1,1-trichloroethane exposure. Gastroenterology. 1982;83:926–929. [PubMed] [Google Scholar]
- Vamvakas S, Bruning T, Thomasson B, et al. Renal cell cancer correlated with occupational exposure to trichloroethene. J Cancer Res Clin Oncol. 1998;124:374–382. doi: 10.1007/s004320050186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesemann W, Blaschke S, Solbach M, Grote C, Clement HW, Riederer P. Intranigral injected iron progressively reduces striatal dopamine metabolism. J Neural Transm Park Dis Dement Sect. 1994;8:209–214. doi: 10.1007/BF02260941. [DOI] [PubMed] [Google Scholar]
- Whitton PS. Inflammation as a causative factor in the aetiology of Parkinson’s disease. Br J Pharmacol. 2007;150:963–976. doi: 10.1038/sj.bjp.0707167. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.