

Abstract
The design and synthesis of hydroxyethylamine isosteres as inhibitors of cathepsin D based on SAR data has been accomplished. A library of 96 of these hydroxyethylamine isosteres are described and many have proven to be very potent inhibitors of human cathepsin D activity as measured using a fluorometric assay technique, via peptide substrate Ac-Glu-Glu(Edans)-Lys-Pro-Ile-Cys-Phe-Phe-Arg-Leu-Gly-Lys(Methyl Red)-Glu-NH2. Compounds showing strongest inhibition of cathepsin D activity were those that contain a hydroxyethyl-N’-2- or N’-(4-chlorophenyl)piperazine moiety (IC50 values range from 0.55 to 8.5 nM), with N’-(2-pyrimidyl)piperizine (IC50 values range from 0.5 to 21.6 nM), with N-N’-L-piperazinocolinamide (IC50 values range from 0.001 – 0.25 nM), or N-N’-L-piperazinocolin-N-methylamide (IC50 values range from 0.015 – 7.3 nM) .
Keywords: Aspartyl protease, Cathepsin D, Inhibitors, Hydroxyethylamine, Piperazine, Pipecolinate, Piperazinocolinate
INTRODUCTION
Cathepsin D, an aspartyl protease, is clearly involved in the process of tumor invasion and metastasis [1-5] and has been linked to bladder cancer [5], hepatocellular carcinoma [6], breast cancer [1,7-10], and prostate cancer [4,11]. Cathepsin D is a key mediator of apoptosis caused by the lysosomal release of mature cathepsin D into the cytosol, leading to the mitochondrial release of cytochrome c into the cytosol, and activation of procaspases [12-15]. In fact, cathepsin D has recently emerged as a prognostic indicator in several cancers, including breast cancer [1,16], lung cancer [17,18], colon cancer [19] and cervical cancer [20]. The use of heptocellular carcinoma suppressor 1 (HCCS1) as a tumor suppressor [21], or another chemotherapeutic drug, etoposide (VP-16), was found to result in the release of cathepsin D into the cytosol, which then brings about a cascade of cytochrome-c release, caspase activation, and cell death [12].
Aspartyl proteases, unlike most proteolytic enzymes, catalyze the cleavage of the peptide bond without the direct use of nucleophilic attack by a functional group of the enzyme [22]. The catalysis and linear free-energy relationships of aspartic proteases, including cathepsin D, were investigated by Bjelic and Aqvist [23]. Although pepstatin A was found to be a potent inhibitor of the HIV-1 aspartyl protease, the peptidic nature of the inhibitor resulted in poor bioavailability [24]. In order to improve bioavailability and improve in vivo half-life, recent research has focused on smaller inhibitors that contain non-peptide functionalities in place of the peptide bond cleavage site of the substrate [25, 26].
The use of hydroxyethyl isosteres with cyclic tertiary amines have led to compounds with enhanced oral absorption [25, 26]. Similarly, hydroxyethylamine isosteres have been used as potent inhibitors of the aspartyl protease plasmepsin [27, 28]. Hydroxyethyl amine isosteres have also been utilized in the design of cathepsin D inhibitors for a structure based combinatorial library [29, 30]. Utilizing information gathered through crystallographic and magnetic resonance experiments, Kick and Roe [29] generated a combinatorial library of cathepsin D inhibitors through molecular modeling. Ellman and Kuntz [31], who played a role in Kick and Roe’s paper [29], used a similar approach to generate a library of 1039 inhibitors of both cathepsin D and the malarial aspartyl protease plasmepsin II [31]. These inhibitors also utilize the hydroxyethyl amine isostere in their basic structure. The basic structure of Kick and Roe’s cathepsin D inhibitors (Figure 1) shows the S-hydroxyethyl amine epimer to be the more active epimer, rather than the R-epimer preferred by the HIV-1 aspartyl protease. Due to the close similarity of cathepsin D and plasmepsin II, several compounds were found to be potent inhibitors of both aspartyl proteases. Recently, the importance of the hydroxyethyl group and the ‘S’ configuration to the HE transition state was demonstrated with the discovery of a new aspartyl protease BACE-1 and cathepsin D inhibitors containing hydroxyethylene scaffold and an aminoethylene central core [32]. Kuntz further demonstrated through molecular dynamics and free-energy analysis of cathepsin D inhibitors interactions that S-hydroxyethylene with tertiary amines or amides show good calculated binding in the active site loop [33]. However, neither Kick and Roe’s [29, 30] or the Ellman and Kuntz [31] combinatorial libraries contained any compounds with cyclized tertiary amines, similar to those found in the orally bioavailable protease inhibitors, such as palinovir (Figure 2), an HIV-1 aspartyl protease inhibitor [34].
Figure 1.
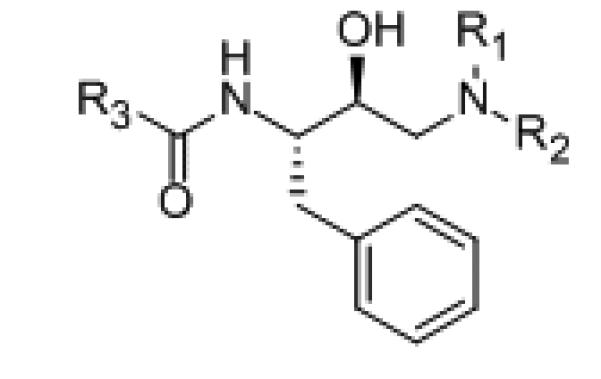
Basic structure of Kick and Roe’s [29] cathepsin D inhibitors with preferred S,S-stereochemistry
Figure 2.
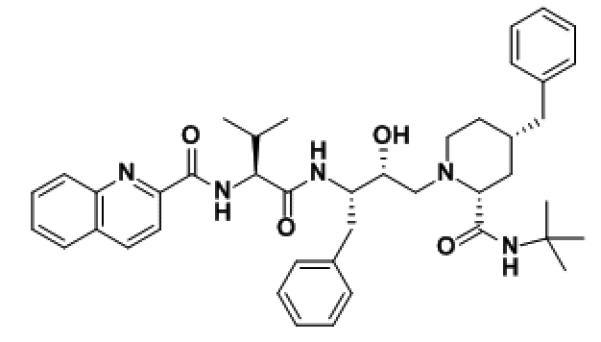
Palinavir, a HIV-1 aspartyl protease inhibitor with cyclic tertiary amine [34]
Our earlier work [35, 36] involved the development of compounds similar in structure to those created in the combinatorial libraries of Kick’s and Ellman’s [29-31] but which contain cyclized tertiary amines, like those in palinavir. Model studies [37] utilizing AMBER 8 [38] demonstrated that N-piperazine (S)-hydroxyethyl amine with a 2-carboxylic amide in the axial position (Figure 3) would hold the compound into a preferred conformation for cathepsin D inhibition. In this paper we describe the preparation and assay of a library of 96 compounds containing a cyclic tertiary amine as part of the hydroxyethyl isostere.
Figure 3.
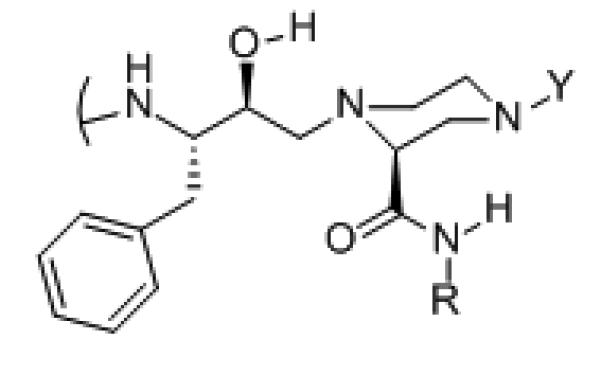
Model studies utilizing AMBER 8 [38] show that a 2-carboxylic amide in the piperazine axial position is perferred
MATERIALS AND METHODS
Synthesis
Anhydrous solvents were “anhydrous grade” from Aldrich Chemical Company. Dry solvents were distilled from sodium immediately prior to use. All other solvents were HPLC grade. All reagents were purchased from Sigma-Aldrich Chemical Company. Thin layer chromatography (TLC) was run on Whatman PE SIL G/UV 250 μm silica gel plates. Column chromatography was run on either Aldrich TLC-grade silica gel 2-25μ particle size BET surface area ~500 m2/g, average pore diameter 60 anstroms, or Sigma Sephadex LH-20, lipophilic, bead size 20-100μm. The high resolution mass spectra were determined by high capacity trap (HCT) electrospray ion-trap mass spectrometry and mass spectra recorded using a QTOF-2 micromass spectrometer. The 1H and 13C NMR spectra were collected either on a JEOL Eclipse 300 MHz spectrometer or on a Bruker 400 MHz NMR superconducting spectrometer. Elemental analyses demonstrating compound purities of more than 95% were performed by Galbraith Laboratories, Knoxville, TN. Butoxy-carbonyl (BOC)-protected chiral amino epoxides (Precursor C) were prepared as described in our earlier work [35, 36, 41] using a slight modification of a procedure reported by Evans and coworkers [40].
General procedure for preparation of Precursor D (Scheme 1)
Scheme 1.
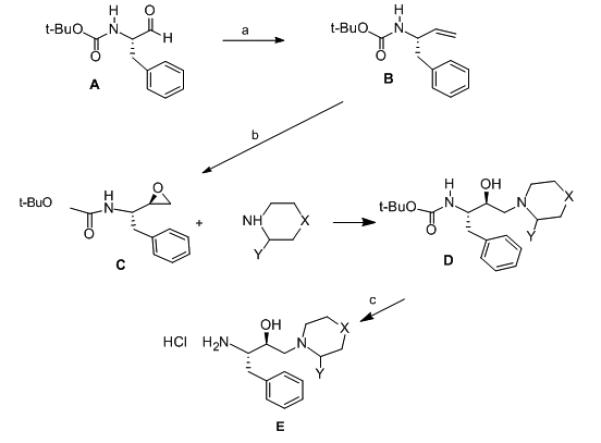
Reagents and conditions: (a) i: triphenylmethylponium bromide (35 mmol), n-butyl lithium (32 mmol) in THF at 0° C under N2 atoms. ii: 0.01M HCl and ether extraction. (b) mepba (32 mmol) in CH2Cl2 at 250 C. (c) non-aqueous HCl (2 M in chloroform) at 0 °C for 1 hr.
A solution of Precursor C (1.0 g, 3.1 mmol) in 100 mL dry THF was treated with 10 mmol appropriate cyclic amine. The solution was refluxed for 48 hrs. The mixture was then cooled to room temperature, concentrated under reduced pressure to about one half its volume, and partitioned between ethyl acetate (200 mL) and 5% aqueous sodium potassium tartarate (200 mL) containing 1.0 g NaCl. The organic layer was washed with distilled water (100 mL) and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure to give a white solid (0.867 g). The crude product was purified by silica gel column chromatography (2.5 cm × 60 cm length) using 50% ethyl acetate/hexanes as the mobile phase to give Precursor D in yields ranging from 60 – 75%. Products were analyzed by TLC (50% ethyl acetate/hexanes); 1H NMR (CDCl3/TMS, 400MHz); 13C NMR (CDCl3, 400 MHz); and ESI-MS.
General procedure for removal of BOC protecting group to form compounds Precursor E
1.5 mmol of the appropriate BOC-protected compound (Precursor D in Scheme 1) was dissolved in 50 mL cold (0° C) 2M HCl in chloroform. The mixture was stirred at 0 °C for 1 hour. Cold diethyl ether (250 mL) was added to induce precipitation of the product. The liquid was decanted, and the precipitant was washed twice with cold ether (100 mL). The crude solid was dissolved in 20 mL methanol and then recrystallized by the addition of 250 mL cold ether. The white solid was again washed twice with cold ether (100 mL) and dried under reduced pressure to produce precursor E.
General Procedure for Coupling Cbz-dipeptide to Precursor E
A precooled solution (−15 °C) of an appropriate carbobenzoxy-dipeptide (Sigma), (0.35 mmol) in 10 mL anhydrous DMF was treated with 56 μL (0.40 mmol) triethyl amine. The mixture was allowed to react at −15 °C for 30 minutes and was then treated with 34 μL (0.35 mmol) ethyl chloroformate. The mixture was stirred under N2 atmosphere for 1 hour at −15 °C. A precooled (0 °C) solution containing 0.32 mmole of appropriate Precursor E in 25 mL anhydrous DMF and 125 μL (1.0 mmole) triethyl amine was then added to the mixed anhydride of the Cbz-dipeptide. The combined mixture was stirred under N2 at 0 °C for 4 hours, allowed to warm to room temperature, and stirred overnight at room temperature. The mixture was partitioned between the layers of ethyl acetate (250 mL) and 0.01 M aqueous NaOH. The organic layer was removed and saved. The aqueous layer was extracted again with another 250 mL ethyl acetate. The organic layers were pooled, washed with distilled water (100 mL), dried over anhydrous magnesium sulfate, and evaporated under reduced pressure to produce carbobenzoxy protected inhibitors. The product structures were verified by TLC (10% ethanol/ethyl acetate); 1H NMR (methanol-d4 400 MHz); and 13C NMR (methanol-d4 400 MHz)
General Procedure for preparation of compounds 1-96 (Scheme 2) from compounds from carboxy protected compounds
Scheme 2.
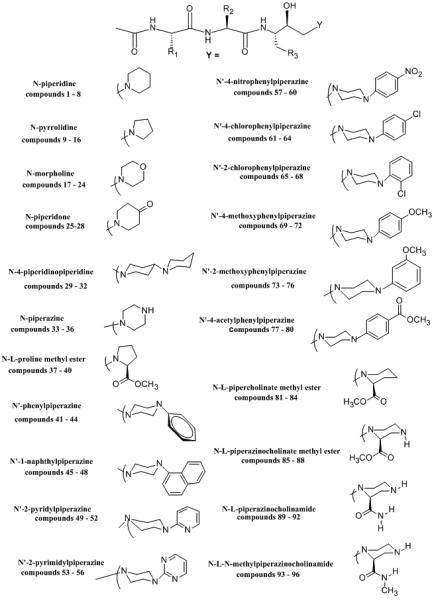
Structures of synthetic inhibitor compounds
A solution of the carbobenzy protected compound (0.20 mmol) in 250 mL methanol and 1 mL 0.01 M aqueous HCl was treated with 0.050 g pre-moistened 10% Pd-C to form a slurry in a 3 neck flask. H2 gas was bubbled (1 atm) through the rapidly stirring mixture at room temperature for 3 hours. The mixture was then filtered to remove the catalyst, and the solvent was evaporated under reduced pressure. The crude amine hydrochloride was dissolved in 10 mL DMSO and treated with 125 μL (0.10 mole) triethyl amine. The mixture was stirred at room temperature for 30 minutes. Acetic anhydride (95 μL, 1.0 mmol) was added and the mixture was stirred overnight at room temperature. Cold diethyl ether (200 mL) was added to precipitate the product. The liquid was decanted and the white solid was washed three times with cold ether (100 mL). The crude product was purified by Sephadex LH-20 column chromatography (column size 5 cm dia. × 80 cm) using methanol as the mobile phase to give 29-81% yield of Compounds 1-96. The structure and purity of the products were verified by TLC, NMR, HRMS, and C, H, N combustion analysis of products (Provided in Supplementary Materials).
Model Studies
The spatial model was based on the crystal structure of cathepsin D (Protein Data Bank ID 1LYA). Swiss-PdViewer was utilized for model building [37]. AMBER 8 (MMX) was used for molecular dynamics simulations and energy minimizations. [38]
Enzyme Assay
Inhibition data with human liver cathepsin D was gathered by continuous fluorometric assay technique, via commercially available (Sigma) fluorometric peptide substrate Ac-Glu-Glu(Edans)-Lys-Pro-Ile-Cys-Phe-Phe-Arg-Leu-Gly-Lys(Methyl Red)-Glu-NH2. Typically 5 μL inhibitor (DMSO) was mixed with 489 μL formate buffer (50 mM, pH 3.5) and then treated with 1 μL human liver cathepsin D in 0.05% Triton X-100. The enzyme/inhibitor solution was pre-incubated for 4 minutes at 37 °C and the reaction was initiated by the addition of 5 μL substrate solution (250 μM in DMSO). The final concentration of the enzyme was 0.04 nM. Increase in fluorescence intensity at emission maximum of 430 nm (excitation wavelength = 340 nm) was reported as a function of time. Nonlinear regression analysis was used as described by Erickson, et al. [39]. The substrate Km was estimated to be 4.2 μM. Four simultaneous repetitions at six or more inhibitor concentrations were used in each trial and compared to a blank containing no enzyme and a standard that contained enzyme and substrate but no inhibitor. Standard error values were calculated and determined to be 3 to 5%.
RESULTS AND DISCUSSION
Synthesis
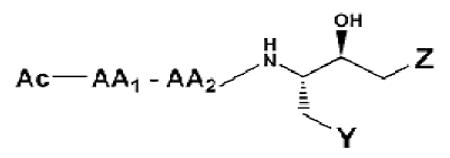
The synthetic inhibitors containing hydroxyethyl amine isosteres (1 - 96) were prepared from a BOC-chiral amino aminoalkyl epoxide (Precursor C) shown in Scheme 1. The novel chiral aminoalkyl epoxides (Precursor C) were first reported by Evans and coworkers [40] and have been subsequently used in the preparation of several HIV-1 aspartyl protease inhibitors with hydroxyethyl amine isosteres. In the earlier phase of this project [35, 36, 41] we successfully prepared BOC-protected chiral amino epoxides starting from commercially available corresponding BOC-amino acids. In this paper, we utilized a similar approach for the preparation of the aminoalkyl epoxides with only slight modifications. BOC-amino esters were reduced directly to the aldehyde (Precursor A) with diisobutyl aluminum hydride (DIBAL). The BOC-amino aldehyde product was reacted with the ylide (triphenylphosphonium methylide) without prior purification in order to avoid potential epimerization of the amino aldehyde prior to formation of the epoxide (Scheme 1). This reaction takes place with retention of stereochemical configuration at the α-carbon of the protected amino aldehyde for us as it did for Evans and coworkers [40]. The ylide attack on the diastereotopic faces of the aldehyde is nonspecific, and the epoxide that was obtained is a chromatographically separable mixture of isomers easily distinguished by NMR adsorption on the C2 proton (δ 3.7 and 4.1). Because the S-hydroxyethyl amine isostere is the more active isomer for cathepsin D inhibition [29,30] we utilized the 2R,3S protected amino epoxide in our synthesis (Scheme 1). Optical rotatory dispersion spectra, as well as specific rotation measurements were documented for each BOC-protected hydroxyethyl amine isostere (Precursor D), as well as for each of the final products (1 – 96). Substituted piperdine, pyrrolidine, piperazine, and pipecolinamides, etc. were used as nucleophiles in the preparation of the cyclized tertiary amines intermediates.
Activity
The synthetic inhibitors were screened for their inhibition of cathepsin D (Table 1) by fluorometric methods [36, 39] using a fluorometric assay of human liver cathepsin D with picomolar accuracy. The commercially available peptide substrate Ac-Glu-Glu(Edans)-Lys-Pro-Ile-Cys-Phe-Phe-Arg-Leu-Gly-Lys(Methyl Red)-Glu-NH2 was used in the fluorometric assays of cathepsin D at an excitation wavelength of 340 nm with a 430 nm cutoff filter for emission.
Table 1.
Inhibition of Cathepsin D Activity in Nanomolar Inhibitor Concentrations.
 | ||||||
|---|---|---|---|---|---|---|
| No. | AA1 | AA2 | Y | Z | IC50, nM |
Ki, nM |
| 1* | Ala | Phe | phenyl | N-piperidine | 45.4 | 44.0 |
| 2* | Val | Phe | ” | ” | 4.6 | 6.30 |
| 3* | Leu | Phe | ” | ” | 3.5 | 7.72 |
| 4 | Leu | Leu | ” | ” | 10.9 | 15.52 |
| 5 | Ala | Phe | cyclohexyl | N-piperidine | 80.5 | 95.32 |
| 6 | Val | Phe | ” | ” | 19.5 | 32.63 |
| 7 | Leu | Phe | ” | ” | 15.3 | 21.45 |
| 8 | Leu | Leu | ” | ” | 33.6 | 41.02 |
| 9* | Ala | Phe | phenyl | N-pyrrolidine | 110.0 | 115.0 |
| 10* | Val | Phe | ” | ” | 65.0 | 67.2 |
| 11* | Leu | Phe | ” | ” | 27.7 | 35.14 |
| 12 | Leu | Leu | ” | ” | 307.4 | 388.51 |
| 13 | Ala | Phe | cyclohexyl | N-pyrrolidine | 355.4 | 498.65 |
| 14 | Val | Phe | ” | ” | 298.5 | 378.22 |
| 15 | Leu | Phe | ” | ” | 275.4 | 325.42 |
| 16 | Leu | Leu | ” | ” | 308.4 | 388.32 |
| 17* | Ala | Phe | phenyl | N-morpholine | 40.4 | 42.63 |
| 18* | Val | Phe | ” | ” | 9.31 | 11.62 |
| 19* | Leu | Phe | ” | ” | 6.99 | 9.52 |
| 20* | Leu | Leu | ” | ” | 27.61 | 26.52 |
| 21 | Ala | Phe | cyclohexyl | N-morpholine | 98.9 | 112.32 |
| 22 | Val | Phe | ” | ” | 78.3 | 96.02 |
| 23 | Leu | Phe | ” | ” | 60.5 | 75.81 |
| 24 | Leu | Leu | ” | ” | 82.7 | 98.53 |
| 25 | Ala | Phe | phenyl | N-4-piperidinone | 65.3 | 69.94 |
| 26 | Val | Phe | ” | ” | 55.0 | 61.04 |
| 27 | Leu | Phe | ” | ” | 54.2 | 35.62 |
| 28 | Leu | Leu | ” | ” | 57.5 | 64.53 |
| 29 | Ala | Phe | phenyl | N-4-piperidinopiperidine | 245.4 | 357.4 |
| 30 | Val | Phe | ” | ” | 157.3 | 199.5 |
| 31 | Leu | Phe | ” | ” | 148.2 | 175.1 |
| 32 | Leu | Leu | ” | ” | 165.4 | 209.5 |
| 33* | Ala | Phe | phenyl | N-piperazine | 44.4 | 53.0 |
| 34* | Val | Phe | ” | ” | 10.0 | 22.4 |
| 35* | Leu | Phe | ” | ” | 5.8 | 7.03 |
| 36* | Leu | Leu | ” | ” | 6.9 | 10.3 |
| 37* | Ala | Phe | phenyl | N-L-proline methyl ester | 105.0 | 98.0 |
| 38* | Val | Phe | ” | ” | 58.0 | 59.9 |
| 39* | Leu | Phe | ” | ” | 32.0 | 34.6 |
| 40* | Leu | Leu | ” | ” | 41.3 | 49.8 |
| 41* | Ala | Phe | phenyl | N’-phenyl piperazine | 22.8 | 18.02 |
| 42* | Val | Phe | ” | ” | 14.5 | 15.32 |
| 43* | Leu | Phe | ” | ” | 10.8 | 14.5 |
| 44* | Leu | Leu | ” | ” | 39.5 | 48.6 |
| 45 | Ala | Phe | phenyl | N’-(1-naphthyl) piperazine | 35.7 | 42.54 |
| 46 | Val | Phe | ” | ” | 21.5 | 26.03 |
| 47 | Leu | Phe | ” | ” | 2.1 | 3.52 |
| 48 | Leu | Leu | ” | ” | 23.5 | 27.12 |
| 49 | Ala | Phe | phenyl | N’-pyridyl piperazine | 25.3 | 28.34 |
| 50 | Val | Phe | ” | ” | 9.4 | 8.32 |
| 51 | Leu | Phe | ” | ” | 10.6 | 12.54 |
| 52 | Leu | Leu | ” | ” | 15.6 | 18.94 |
| 53 | Ala | Phe | phenyl | N’-(2-pyrimidyl) piperazine | 21.6 | 15.83 |
| 54 | Val | Phe | ” | ” | 8.5 | 7.53 |
| 55 | Leu | Phe | ” | ” | 0.75 | 0.16 |
| 56 | Leu | Leu | ” | ” | 10.2 | 7.91 |
| 57* | Ala | Phe | phenyl | N’-(4-nitrophenyl) piperazine | 39.9 | 38.5 |
| 58* | Val | Phe | ” | ” | 15.4 | 11.5 |
| 59* | Leu | Phe | ” | ” | 22.6 | 19.4 |
| 60* | Leu | Leu | ” | ” | 65.9 | 56.4 |
| 61 | Ala | Phe | phenyl | N’-(4-chlorophenyl) piperazine | 8.5 | 9.83 |
| 62 | Val | Phe | ” | ” | 6.5 | 4.55 |
| 63 | Leu | Phe | ” | ” | 7.5 | 7.41 |
| 64 | Leu | Leu | ” | ” | 5.5 | 7.94 |
| 65 | Ala | Phe | phenyl | N’-(2-chlorophenyl) piperazine | 2.6 | 2.33 |
| 66 | Val | Phe | ” | ” | 1.8 | 0.19 |
| 67 | Leu | Phe | ” | ” | 2.0 | 0.11 |
| 68 | Leu | Leu | ” | ” | 0.55 | 1.60 |
| 69 | Ala | Phe | phenyl | N’-(4-methoxyphenyl) piperazine | 76.0 | 75.4 |
| 70 | Val | Phe | ” | ” | 55.0 | 45.6 |
| 71 | Leu | Phe | ” | ” | 58.5 | 48.7 |
| 72 | Leu | Leu | ” | ” | 65.3 | 57.8 |
| 73 | Ala | Phe | phenyl | N’-(2-methoxyphenyl) piperazine | 8.5 | 9.84 |
| 74 | Val | Phe | ” | ” | 6.5 | 4.56 |
| 75 | Leu | Phe | ” | ” | 7.5 | 7.42 |
| 76 | Leu | Leu | ” | ” | 10.9 | 15.63 |
| 77 | Ala | Phe | phenyl | N’-(4-acetylphenyl) piperazine | 46.3 | 37.3 |
| 78 | Val | Phe | ” | ” | 5.6 | 6.70 |
| 79 | Leu | Phe | ” | ” | 4.5 | 3.80 |
| 80 | Leu | Leu | ” | ” | 6.5 | 6.15 |
| 81* | Ala | Phe | phenyl | N-L-pipecolinate methyl ester | 109.1 | 94.3 |
| 82* | Val | Phe | ” | ” | 67.0 | 63.5 |
| 83* | Leu | Phe | ” | ” | 41.5 | 23.1 |
| 84* | Leu | Leu | ” | ” | 73.3 | 89.7 |
| 85* | Ala | Phe | phenyl | N-N’-L-piperazine-2-carboxylic acid methyl ester |
11.0 | 4.56 |
| 86* | Val | Phe | ” | ” | 6.4 | 6.9 |
| 87* | Leu | Phe | ” | ” | 1.5 | 1.02 |
| 88* | Leu | Leu | ” | ” | 6.6 | 5.32 |
| 89 | Ala | Phe | phenyl | N-N’-L-piperazine-2-carboxylic amide | 0.25 | 0.18 |
| 90 | Val | Phe | ” | ” | 0.11 | 0.07 |
| 91 | Leu | Phe | ” | ” | .001 | ------ |
| 92 | Leu | Leu | ” | ” | 0.15 | 0.04 |
| 93 | Ala | Phe | phenyl | N-N’-L-piperazine-2-carboxylic- N-methyl amide |
0.015 | 0.01 |
| 94 | Val | Phe | ” | ” | 7.30 | 6.25 |
| 95 | Leu | Phe | ” | ” | 0.65 | 0.22 |
| 96 | Leu | Leu | ” | ” | 0.95 | 0.32 |
Modifications in the ring of the hydroxy ethyl tertiary amine appeared to have a significant effect on the potency of the inhibitors. Those compounds with N’-substituted aromatic piperizine groups were better inhibitors of cathepsin D activity (Table 1). These compounds included compounds 41 - 80 in Table 1. The compounds showing strongest inhibition of cathepsin D activity were those that contain a N’-2- or (4-chlorophenyl)piperazine (compounds 65-72 in Table 1), or with N’-(2-pyridyl)piperizine (compounds 49-52 in Table 1) or N’-(2-pyrimidyl)piperizine (compounds 52-56 in Table 1). Examinations of the enzyme-inhibitor interactions by computer-assisted molecular modeling were based on 3-D structures from X-ray crystallography [42] and molecular dynamics data [37, 43]. Estimates for the enthalpic and entropic components of the binding free energy of the enzyme-inhibitor complex were used to predict binding affinities of the compounds, using AMBER 8 (MMX) molecular dynamics simulations [38, 43]. Molecular modeling of inhibitors showed that a 2(S)-carboxylic amide in an axial piperizine position (Figure 3) can, through hydrogen bonding, help stabilize the backbone structure of the inhibitor into the desired conformation for tight binding in the cathepsin D active site. Therefore, we prepared compounds containing a 2(S)-N,N’-piperazinocolinamide (Compounds 89 – 96 in Table 1). These compounds showed potent inhibition of cathepsin D activity.
CONCLUSIONS
A library of 96 newly synthesized compounds has been established as effective cathepsin D inhibitors by in vitro fluorometric assay. The use of molecular dynamic symulations to predict inhibition activity has led to the development of potent cathepsin D inhibitors with IC50 values in the nanomolar range. Compounds showing strongest inhibition of cathepsin D activity were those that contain a hydroxyethyl-N’-2- or N’-(4-chlorophenyl)piperazine moiety (IC50 values range from 0.55 to 8.5 nM), with N’-(2-pyrimidyl)piperizine (IC50 values range from 0.5 to 21.6 nM), with N-N’-L-piperazinocolinamide (IC50 values range from 0.001 – 0.25 nM), or N-N’-L-piperazine-2-carboxylic-N-methyl amide (IC50 values range from 0.015 – 7.3 nM) .
Supplementary Material
ACKNOWLEDGMENT
This work was supported by a NIH: National Cancer Institute grant: 3R15 CA086933-04 and 3R15CA086933-04A2S1.
REFERENCES
- [1].Kulić A, Sirotković-Skerlev M, Jelisavac-Cosić S, Herceg D, Kovac Z, Vrbanec D. Anti-p53 antibodies in serum: Relationship to tumor biology and prognosis of breast cancer patients. Med. Oncol. 2010;27:887–93. doi: 10.1007/s12032-009-9301-1. [DOI] [PubMed] [Google Scholar]
- [2].Masson O, Prebois C, Derocq D, Meulle A, Dray C, Daviaud D, Quillot D, Valet P, Muller C, Liaudet-Coopman E. Cathepsin D, a key protease in breast cancer, is up-regulated in obese mouse and human adipose tissue, and controls adipogenesis. Public Library of Science. 2011;6(2):e16452. doi: 10.1371/journal.pone.0016452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Mazouni C, Romain S, Bonnier P, Quafik L, Martin PM. Prognostic significance of tumor-related proteases as a function of estrogen receptor status. Cancerl Biol. Ther. 2011;11(2):277–83. doi: 10.4161/cbt.11.2.13964. [DOI] [PubMed] [Google Scholar]
- [4].Hu S, Delorme N, Liu Z, Velasco-Gonzalez C, Garai J, Pullikuth A, Koochekpour S. Prosaposin down-modulation decreases metastatic prostrate cancer adhesion, migration, and invasion. Mol. Cancer. 2010;9:30–1. doi: 10.1186/1476-4598-9-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Makridakis M, Gagos S, Petrolekas A, Roubelakis MG, Bitsika V, Stravodimos K, Pavlakis K, Anagnou NP, Coleman J, Vlahou A. Chromosomal and proteome analysis of new T24-base cell line model for aggressive bladder cancer. Proteomics. 2009;9:287–98. doi: 10.1002/pmic.200800121. [DOI] [PubMed] [Google Scholar]
- [6].Tsai CC, Huang KW, Chen HF, Zhan BW, Lai YH, Lee FH, Lin CY, Ho YC, Chao YW, Su YC, Jane SW, Chen YC, Hsu CI, Suzuki Y, Sugano S, Lin JY. Gene expression analysis of human hepatocellular carcinoma by using full length cDNA library. J. Biomed. Sci. 2006;13(2):241–249. doi: 10.1007/s11373-005-9062-6. [DOI] [PubMed] [Google Scholar]
- [7].Beaujouin M, Prébois C, Derocq D, Laurent-Matha V, Masson O, Pattingre S, Coopman P, Bettache N, Grossfield J, Hollingsworth RE, Zhang H, Hyman BT, ven der Geer P, Smith GK, Liaudet-Coopman E. Pro-cathepsin D interacts with the extracellular domain of the beta chain of LRP1 and promotes LRP1-dependent fibroblast outgrowth. J. Cell. Sci, 2010;123(Pt 19):3336–46. doi: 10.1242/jcs.070938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ma Y, Zhao M, Zhong J, Shi L, Liu J, Wang J, Yuan X, Huang C. Proteomic profiling of proteins associated with lymph node metastasis in colorectal cancer. J. Cell. Biochem. 2010;110(6):1512–9. doi: 10.1002/jcb.22726. [DOI] [PubMed] [Google Scholar]
- [9].Guo P, Dong XY, Zhao KW, Sun X, Li Q, Dong JT. Estrogen-induced interaction between KLF5 and estrogen receptor (ER) suppresses the function of ER in ER-positive breast cancer cells. Int. J. Cancer. 2010;126(1):81–9. doi: 10.1002/ijc.24696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Alvarez RH, Price JE. Biomarkers for breast cancer: The search continues. Cancer. Biol. Ther. 2010;9(1):31–2. doi: 10.4161/cbt.9.1.10917. PMID: 20150755. [DOI] [PubMed] [Google Scholar]
- [11].Chen L, Liu W, Zhu J, Zhao X, Wright E, Cao L, Ding I, Rodgers GP. Olfactomedin 4 surpresses prostate cancer cell growth and metastatis via negative interaction with cathepsin D and SDF-1. Carcinogenesis. 2011;32(7):986–94. doi: 10.1093/carcin/bgr065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Emert-Sedlak L, Shangary S, Rabinovitz A, Miranda M, Delach SM, Johnson DE. Involvement of cathepsin D in chemotherapy-induced cytochrome c release, caspase activation, and cell death. Mol. Cancer Ther. 2005;4(5):733–742. doi: 10.1158/1535-7163.MCT-04-0301. [DOI] [PubMed] [Google Scholar]
- [13].Beaujouin M, Liaudet-Coopman E. Cathepsin D overexpressed by cancer cells can enhance apoptosis-dependent chemo-sensitivity independently of its catalytic activity. Adv. Exp. Med. Bio. 2008;617:453–461. doi: 10.1007/978-0-387-69080-3_44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Miura Y, Sakurai Y, Hayakawa M, Shimada Y, Zempel H, Sato Y, Hisanaga S, Endo T. Translocation of lysosomal cathepsin D caused by oxidative stress or proteasome inhibition in primary cultured neurons and astrocytes. Biol. Pharm. Bull. 2010;33(1):22–8. doi: 10.1248/bpb.33.22. [DOI] [PubMed] [Google Scholar]
- [15].Minarowska A, Gacko M. Regulatory role of cathepsin D in apoptosis. Folia Histochemica et Cytobiologica. 2007;45(3):159–163. [PubMed] [Google Scholar]
- [16].Mazouni C, Bonnier P, Romain P, Martin PM. A nomogram predicting thw probability of primary breast cancer survival at 2- and 5-years using pathological and biolo0gical tumor parameters. J. Surg. Oncol. 2011;103(8):746–50. doi: 10.1002/jso.21712. [DOI] [PubMed] [Google Scholar]
- [17].Vetvicka V, Vetvickova J. Procathepsin D and cytokines influence the proliferation of lung cancer cells. Anticancer Res. 2011;31(1):47–51. PMID: 21273579. [PubMed] [Google Scholar]
- [18].Lou X, Xiao T, Zhao K, Wang H, Zheng H, Lin D, Lu Y, Gao Y, Cheng S, Liu S, Xu N. Cathepsin D is secreted from M-BE cells: its potential role as a biomarker of lung cancer. J. Proteome Res. 2007;6(3):1083–1092. doi: 10.1021/pr060422t. [DOI] [PubMed] [Google Scholar]
- [19].Xie LQ, Zhao C, Cai SJ, Xu Y, Huang LY, Bian JS, Shen CP, Lu HJ, Yang PY. Novel proteomic strategy reveal combined α1 antitrypsin and cathepsin D as biomarkers for colorectal cancer early screening. J. Proteome Res. 2010;9(9):4701–9. doi: 10.1021/pr100406z. [DOI] [PubMed] [Google Scholar]
- [20].Lomnytska MI, Becker S, Hellman K, Hellström AC, Souchelnytskyi S, Hellman U, Andersson S, Auer G. Diagnostic protein marker patterns in squamous cervical cancer. Proteomics Clin. App. 2010;4(1):17–31. doi: 10.1002/prca.200900086. [DOI] [PubMed] [Google Scholar]
- [21].Gan Y, Zhao X, Hu J, Wang ZG, Zhao XT. HCCS1 overexpression induces apoptosis via cathepsin D and intracellular calcium, and HCCS1 disruption in mice causes placental abnormality. Cell. Death Differ. 2008;15(9):1481–90. doi: 10.1038/cdd.2008.73. [DOI] [PubMed] [Google Scholar]
- [22].Cooper JB. Aspartic proteinases in disease: A structural perspective. Curr. Drug Targets. 2002;3(2):155–73. doi: 10.2174/1389450024605382. [DOI] [PubMed] [Google Scholar]
- [23].Bjelic S, Aqvist J. Catalysis and free energy relationships in aspartic proteases. Biochemistry. 2006;45:7709–23. doi: 10.1021/bi060131y. [DOI] [PubMed] [Google Scholar]
- [24].Krauchenco S, Sanches M, Polikapov I. Effectiveness of commercial inhibitors against subtype F HIV-1 protease. J. Enzyme Inhib. Med. Chem. 2009;24(3):638–45. doi: 10.1080/14756360802321740. [DOI] [PubMed] [Google Scholar]
- [25].Smith AB, Hirschman R, Pasternak A, Yao W, Sprengeler PA. An Orally Bioavailable Pyrrolinone Inhibitor of HIV-1 Protease: Computational Analysis, and X-ray Crystal Structure of the Enzyme Complex. J. Med. Chem. 1997;40:2440–2444. doi: 10.1021/jm970195u. [DOI] [PubMed] [Google Scholar]
- [26].Melnick M, Reich SH, Lewis KK, Mitchell LJ, Nguyen D, Trippe AJ, Dawson H, Davies JF, Appelt K, Wu BW, Musick L, Gehlhaar DK, Webber S, Shetty B, Kosa M, Kahil D, Andrada D. Bis-Tertiary Amide Inhibitors of the HIV-1 Protease Generated via Protein Structure-Based Iterative Design. J. Med. Chem. 1996;39:2795–2811. doi: 10.1021/jm960092w. [DOI] [PubMed] [Google Scholar]
- [27].Gupta D, Yedidi RS, Varghese S, Kovari LC, Woster PM. Mechanism-based inhibitors of the aspartyl protease plasmepsin II as potential antimalarial agents. J. Med. Chem. 2010;53(10):4234–47. doi: 10.1021/jm100233b. PMID: 20438064. [DOI] [PubMed] [Google Scholar]
- [28].Miura T, Hidaka K, Uemura T, Kashimoto K, Hori Y, Kawasaki Y, Ruben AJ, Freire E, Kimura T, Kiso Y. Improvement on both plasmepsin inhibitory activity and antimalarial activiy by 2-aminoethyl substitution. Biorg. Med. Chem. Lett. 2010;20(16):4836–9. doi: 10.1016/j.bmcl.2010.06.099. [DOI] [PubMed] [Google Scholar]
- [29].Kick EK, Roe DC, Skillman AG, Liu G, Ewing TJ, Sun Y, Kuntz ID, Ellman JA. Structure-Based Design and Combinatorial Chemistry Yield Low Nanomolar Inhibitors of Cathepsin D. Chem. and Biol. 1997;4:297–307. doi: 10.1016/s1074-5521(97)90073-9. [DOI] [PubMed] [Google Scholar]
- [30].Kick EK, Ellman JA. An Expedient Method for the Solid Phase Synthesis of Aspartic Acid Protease Inhibitors Directed Toward the Generation of Libraries. J. Med. Chem. 1995;38:1427–1430. doi: 10.1021/jm00009a002. [DOI] [PubMed] [Google Scholar]
- [31].Haque TS, Skillman AG, Lee CE, Habashita H, Gluzman IY, Ewing TJ, Goldberg DE, Kuntz ID, Ellman JA. Potent, Low-Molecular weight Non-Peptide Inhibitors of Malarial Aspartyl Protease Plasmepsin II. J. Med. Chem. 1999;42:1428–1440. doi: 10.1021/jm980641t. [DOI] [PubMed] [Google Scholar]
- [32].Björklund C, Adolfsson H, Jansson K, Lindberg J, Vrang L, Hallberg A, Rosenquist S, Samuelsson B. Discovery of potent BACE-1 inhibitors containing a new hydroxyethylene (HE) scaffold: exploration of P1’ alkoxy residues and an aminoethylene (AE) central core. Biorg. Med. Chem. 2010;18(4):1711–23. doi: 10.1016/j.bmc.2009.12.051. PMID: 20122837. [DOI] [PubMed] [Google Scholar]
- [33].Huo S, Wang, Cieplak P, Kollman PA, Kuntz ID. Molecular dynamics and free energy analysis of cathepsin D-inhibitor interactions: insight into structure-based ligand design. J. Med. Chem. 2002;45(7):1412–9. doi: 10.1021/jm010338j. [DOI] [PubMed] [Google Scholar]
- [34].Reich SH, Melnick M, Pino MJ, Fuhry MN, Trippe AJ, Appelt K, Davies JF, Wu BW, Musick L. Structure-Based Design and Synthesis of Substituted 2-Butanols as Nonpeptidic Inhibitors of HIV Protease: Secondary Amide Series. J. Med. Chem. 1996;39:2781–2794. doi: 10.1021/jm960093o. [DOI] [PubMed] [Google Scholar]
- [35].McConnell RM, Godwin WE, Stefan A, Newton C, Myers N, Hatfield SE. Synthesis and cathepsin D inhibition of peptide-hydroxyethylamine isosteres with cyclic tertiary amines. Letters in Peptide Sci. 2003;10:69–78. [Google Scholar]
- [36].McConnell RM, Green AW, Trana CJ, Lindley JF, Godwin WE, Hatfield SE. New cathepsin D inhibitors with hydroxyethylamine isosteres: preparation and characterization. Med. Chem. 2006;2(1):27–38. doi: 10.2174/157340606775197705. [DOI] [PubMed] [Google Scholar]
- [37].Steinfield R, Reinhart K, Schreiber K, Hillebrand M, Kraetzner R, Brück W, Saftig P, Gärtner J. Cathepsin D deficiency is associated with human neurodegenerative disorder. Am. J. Hum. Genet. 2006;78(6):988–98. doi: 10.1086/504159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Case DA, Cheatham TE, III, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. J. Comput. Chem. 2005;26(16):1668–88. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Majer P, Collins JR, Gulnik SV, Erickson JW. Structure-based substrate specificity mapping of human cathepsin D using statine-based inhibitors. Protein Sci. 1997;6:1458–66. doi: 10.1002/pro.5560060710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Evans BE, Rittle KE, Homnick CF, Springer JP, Hirshfield J, Veber DF. A Stereocontrolled Synthesis of Hydroxyethylene Dipeptide Isosteres Using Novel, Chiral Aminoalkyl Epoxides and γ-(aminoalkyl)-γ-Lactones. J. Org. Chem. 1985;50:4615–4625. [Google Scholar]
- [41].McConnell RM, Frizzell D, Camp A, Evans A, Jones W, Cagle C. New Pepstatin Analogues: Synthesis and Pepsin Inhibition. J. Med. Chem. 1991;34:2298–2300. doi: 10.1021/jm00111a054. [DOI] [PubMed] [Google Scholar]
- [42].Baldwin ET, Bhat TN, Gulnik S, Hosur MV, Sowder RC, II, Cachau RE, Collins J, Silva AM, Erickson JW. Crystal structures of native and inhibited forms of human cathepsin D: implications for lysosomal targeting and drug design. Proc. Natl. Acad. Sci. 1993;90(14):6796–800. doi: 10.1073/pnas.90.14.6796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Huo S, Wang J, Cieplak P, Kollman PA, Kuntz ID. Molecular Dynamics and Free Energy Analysis of Cathepsin D-Inhibitor Interactions: Insight into Structure-Based Ligand Design. J. Med. Chem. 2002;45:1412–9. doi: 10.1021/jm010338j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.