

Abstract
Plasmodium falciparum, the causal agent of malaria, continues to evolve resistance to frontline therapeutics such as chloroquine and sulfadoxine-pyrimethamine. Here we study the amino acid replacements in dihydrofolate reductase (DHFR) that confer resistance to pyrimethamine while still binding the natural DHFR substrate, 7,8-dihydrofolate, and cofactor, NADPH. The chain of amino acid replacements that has led to resistance can be inferred in a computer, leading to a broader understanding of the coevolution between the drug and target. This in silico approach suggests that only a small set of specific active site replacements in the proper order could have led to the resistant strains in the wild today. A similar approach can be used on any target of interest to anticipate likely pathways of future resistance for more effective drug development.
Keywords: DHFR, Drug resistance, Phylogeny, Evolution, Plasmodium, Malaria, Coevolution
Introduction
Malaria continues to be a severe health concern, with over half of the world’s population at risk for infection [1]. In Africa, malaria is the second leading cause of death from infectious disease, following only HIV/AIDS. The parasites responsible for malaria come from the genus Plasmodium, and are transferred to humans through the Anopheles mosquito. The parasite Plasmodium falciparum has a long history of human infection dating to between 112,000 and 1,036,000 years ago [2] or at the most 2.5 million years ago [3]. The genus Plasmodium itself dates back at least to the mammalian radiation at 12.8 million years ago. Ayala et al. [4] examined the history of the Plasmodium genus through molecular evolution. Their results demonstrated that P. falciparum, P. malariae, P. ovale, and P. vivax all share a common history that predates hominid evolution, and that the origin of P. falciparum correlates well with the origin of the hominids [4].
Arisue et al. [5] provided additional insight into the evolution of 18 species in the Plasmodium genus through the analysis of the serine repeat antigen (SERA) multigene family. This gene family began in a common ancestor of the genus Plasmodium and, through a series of gene duplications, diverged into two types of SERA genes (either a cysteine or serine-type). This divergence may have coincided with the world-wide expansion of Plasmodium. The resulting phylogeny of the genus [4, 5] has also been adopted as the canonical tree offered by the National Center for Biological Information (NCBI) (Fig. 1). We have adopted this phylogeny to better understand the evolution of dihydrofolate reductase (DHFR) within Plasmodium.
Fig. 1.

Taxonomic tree of 17 Plasmodium species adopted from www.ncbi.nlm.nih.gov/Taxonomy/CommonTree/wwwcmt.cgi
In Plasmodium, DHFR is found as a domain in a complex with thymidylate synthase and catalyzes the reduction of 7,8-dihydrofolate to 5,6,7,8-tetrahydrofolate [6, 7]. Given this is a critical component of purine and amino acid synthesis, it is not surprising that the overall secondary and tertiary structure of DHFR is highly conserved [8]. Inhibition of DHFR effectively blocks thymidine synthesis, leading to cell death [9]. DHFR is also considered as a therapeutic target for cancer [10], and because of its relevance to central components of cellular mechanics, a large quantity of detailed structure and activity data exists.
DHFR remains an attractive therapeutic target given inhibition kills prokaryotes such as S. aureus and M. tuberculosis, unicellular eukaryotes such as P. falciparum as well as cancer cells [9–12]. The underlying mechanisms, dynamics, and kinetics of DHFR-based drug resistance, remains a very active area of current research [12–16]. Extensive structure and activity studies have been performed on DHFR and chromosomal DHFR has a highly conserved structural motif including a well-defined active site and catalytic mechanism [8].
The recent worldwide use of anti-malarial drugs has led to a coevolutionary response of malarial strains to rapidly evolve resistance. Chloroquine, which targets heme metabolism, was first introduced into clinical practice in 1947. It took less than 10 years for resistance to independently develop in Southeast Asia and South America [17]. By the late 1970s resistant strains of malaria were prevalent throughout the world. As a replacement therapy, sulfadoxine-pyrimethamine, targeting dihydropteroate synthase (DHPS) and DHFR respectively, was introduced into clinical practice in the 1960s in areas where chloroquine resistance predominated. Unfortunately, wide-spread resistance developed within 10 years due to key amino acid replacements in both DHPS (A437G, K540E, A581G and A613S) and DHFR (N51I, C59R, S108N and I164L) [18]. In DHFR, these mutations allow P. falciparum the opportunity to bind the natural substrate, 7,8-dihydrofolate and the cofactor NADPH, while simultaneously reducing the binding affinity of therapeutics designed to block the active site [17–19]. Single mutant (S108N), double mutant (C59R, S108N), triple mutant (N51I, C59R, S108N) and quadruple mutant (N51I, C59R, S108N, I164L) drug resistant strains of P. falciparum are now commonplace in the wild primarily in Southeast Asia and the Western Pacific, although resistant genotypes are now beginning to appear in Africa particularly in patients with repeated infections [19].
Here we examine the structural evolution of DHFR to better understand the evolutionary processes that conferred resistance. Towards this goal we first generated a structure-based alignment of wt-DHFR protein sequences from 17 Plasmodium species in order to evaluate the degree of site-specific variability/conservation of both sequence as well as structure. DHFR-specific phylogenetic trees were generated and compared to the canonical Plasmodium phylogeny. Using homology models of potential and actual mutant Pf-DHFR sequences, we then used in silico docking experiments to model the selection pressure based on specific protein–ligand interactions and the resulting evolution of drug resistance in P. falciparum DHFR in response to pyrimethamine.
Materials and methods
Plasmodium DHFR sequences
Following a comprehensive search of the NCBI Protein sequence database, 554 Plasmodium DHFR sequences were downloaded in FASTA format, and imported into BioEdit [20] where they were sorted by species and by their classification as either a wild-type or a mutant sequence. As many of the sequences include thymidylate synthase, all sequences were truncated to include only the DHFR domains. For species having more than one wt sequence in the database, a representative wt sequence was selected at random. The “canonical” phylogenetic tree of the resulting 17 wt sequences was generated from publically available tools at the NCBI (www.ncbi.nlm.nih.gov/Taxonomy/CommonTree/wwwcmt.cgi). The resulting phylogeny was saved (*.phy), and imported into MEGA for 5.0 [21] for formatting (Fig. 1).
Homology modeling
X-ray crystal structures currently exist for wt, double mutant and quadruple mutant P. falciparum DHFR (1J3I.pdb, 1J3 J.pdb and 1J3 K.pdb respectively) as well as for wild-type P. vivax DHFR (2BL9.pdb). Pairwise sequence alignments were performed for each of the remaining 15 wt Plasmodium DHFR sequences using MOE (www.chemcomp.com) in order to determine which of the two wild-type X-ray crystal structure sequences to use as a template for homology modeling: wt-PfDHFR (1J3I.pdb) or wt-PvDHFR (2BL9.pdb). The X-ray structure having the largest value of Pairwise Percentage Identity (indicating the greatest sequence homology) to each sequence was selected. All homology models were generated and refined using the Amber99 potential and default MOE settings. RMSD values for each homology model from the corresponding template X-ray crystal structure ranged between 0.37 and 1.24 Å.
Likewise, 16 “mutant” homology models of fictitious P. falciparum DHFR were built in silico using all 16 combinations of the amino acid substitutions N51I, C59R, S108N, I164L. RMSD values for each homology model from 1J3I.pdb (wt Pf-DHFR) ranged between 0.93 and 1.34 Å.
Structure-based sequence alignments
We recently published a set of structurally aligned wild-type DHFR sequences from X-ray crystal structures of 22 prokaryotic and eukaryotic species with diverse evolutionary history [22]. Using Deepview [23] each of these 22 structures as well as each of the two mutant X-ray crystal structures for P. falciparum (1J3 J.pdb and 1J3 K.pdb) were “cleaned” by removing all waters, ligands, and ions. In addition, each *.pdb file was truncated to include only a monomer of DHFR in each *.pdb file. These were then imported into MOE along with the 15 generated homology models of Plasmodium DHFR wt sequences where they were structurally aligned using MOE’s “Superpose” function (Fig. 2). The overall RMSD for the 39 structures was 1.46 Å.
Fig. 2.
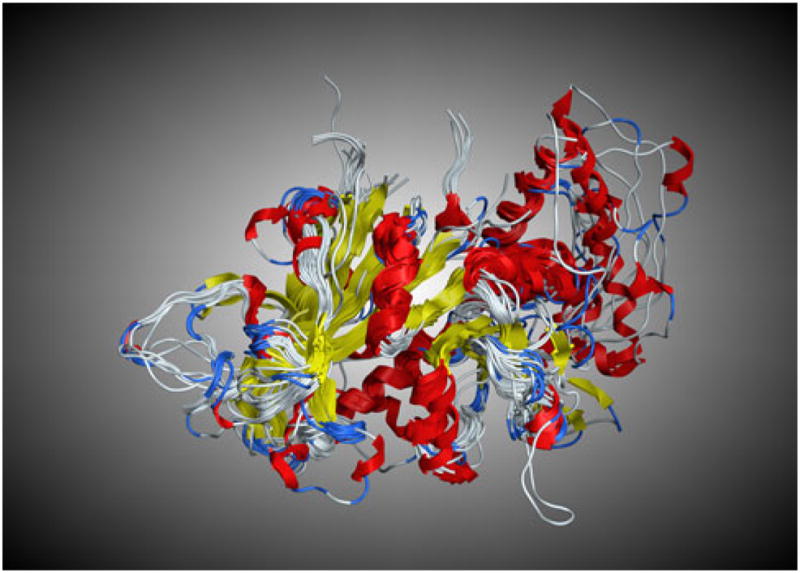
The structural superimposition of DHFR proteins from 24 X-ray crystal structures and 15 homology models (RMSD = 1.46 Å)
The 17 aligned Plasmodium wt DHFR sequences were then exported as a *.fasta file and imported into BioEdit for further alignment. For each sequence, a mask was generated labeling each residue for the type of secondary structure present (alpha helix (H) or beta-sheet (S)). The structure-based sequence alignment was truncated in Bio-Edit to include residues 6 Å from bound NADPH and the active site residues (Fig. 3).
Fig. 3.

Structural alignment of 17 DHFR sequences truncated to residues 6 Å from bound NADPH and the active site with corresponding secondary structure residue masks. Residues >6 Å from bound NADPH and the active site are not shown and are designated by dashed lines. The first 17 rows represent the actual residues in each sequence. The second set of 17 rows are labeled for the type of secondary structure present: H for alpha helix, S for beta-sheet. X-ray crystal structures exist for P. falciparum (1J3I.pdb) and P. vivax (2BL9.pdb), all others sequences represent homology models
Plasmodium DHFR phylogenetics
A phylogenetic tree was generated from the structural alignment with MEGA 5.0 [21] using the UPGMA method with the following parameters: Dayhoff amino acid replacement model; uniform rates of site-specific mutation; complete deletion of gaps and missing data (Fig. 4). The phylogeny was evaluated using the bootstrap method with 1,000 replications. Analyses using the Neighbor–Neighbor joining method as well as non-uniform rates of site-specific mutation were very similar to that presented in Fig. 5.
Fig. 4.

UPGMA tree generated with MEGA5. Parameters: complete deletion, Dayhoff model, uniform rates of substitution. Tested with Bootstrap n = 1,000. Double mutant P. falciparum (C59R, S108N) and quadruple mutant P. falciparum (N51I, C59R, S108N, I164L) are also shown relative to the wt P. falciparum
Fig. 5.

Superposition of X-ray crystal conformations of pyrimethamine and NADPH bound to wt DHFR from 3QGT.pdb (colored in gold) versus docked conformations (in CPK), RMSD values <1.00 Å
TOPD/FMTS [24] with default settings was used to perform pairwise comparisons of this phylogenetic tree to the canonical DHFR phylogenetic tree. Three different algorithms were used to evaluate differences between the trees. These include the Split Difference based on partition metrics [25], the Nodal Distance based on path length metrics [26] and the Disagree Distance which identifies the fraction of taxa that disagree between two trees [27]. 100 comparisons using randomized trees generated with a Markovian algorithm were performed with each method for each pair of trees in order to determine if the similarity was greater than random [24].
Determination of sequence and structure conservation and variability
The structure-based alignment was analyzed in an attempt to quantify the frequency of site-specific amino acid substitutions and the degree of sequence conservation. BioEdit was used to calculate the frequency of occurrence for each of the 20 naturally occurring amino acids at all positions in the aligned sequences. For cases where one or more gaps were present at a particular position in the alignment, the gaps were assigned a null value. For each position in the aligned sequences, the frequency of finding a residue in either an α-helix or β-sheet conformation was also calculated.
The average amino acid frequency of occurrence for each position in the alignment was then determined. The higher this average, the greater the degree of position specific conservation. The lower this average, the greater the variability for amino acid replacement. Likewise, the average frequency for secondary structure and residue class was calculated at each position. These residue classes reflect commonly used classifications of amino acids (e.g., acid, base, glycine, proline, non-aromatic hydrophobic, aromatic, polar) based on both side chain functionality and the underlying codon. Given the critical role that proline and glycine play in secondary structure elements such as turns and helices, they were given separate classifications.
The average of the average frequency per position was then calculated for amino acid frequency, secondary structure amino acid class. These values provided a metric for the overall degree of conservation or variability at each site in the alignment—not only in terms of amino acid residue identity, but also secondary structure and residue class. For purposes of this paper these values will be called Amino Acid Conservation and Secondary Structure Conservation, and Residue Class Conservation respectively.
In silico docking experiments
Docking experiments were performed using GOLD (www.ccdc.cam.ac.uk) with the default GOLD fitness functions and default evolutionary parameters [28]. In GOLD, the default scoring functions are the Pairwise Linear Potential (PLP) Fitness Score (an empirical fitness function optimized for pose prediction termed “PLP Fitness”) and “PLP score” (similar to empirical estimates of the free energy of binding) [29]. The more positive the PLP Fitness score or the more negative the PLP score the better the pose.
For each homology model, docking experiments were first performed with NADPH and subsequently with 7,8-dihydrofolate and pyrimethamine each in the presence of the top scoring NADPH pose. The amide hydrogen on Ser108 was selected as the binding site center for all NADPH binding experiments and the carbonyl oxygen of Ile164 was selected as the binding site center for all pyrimethamine and 7,8-dihydrofolate binding experiments [30, 31]. For the NADPH docking experiments, the side chain of Arg129 was allowed to be flexible in order to avoid a steric clash with NADPH. Ten docking runs were performed per structure unless 3 of the 10 poses were within 1.5 Å RMSD of each other. All poses were output into *.sdf files.
In order to verify that poses resulting from in silico docking represented correctly-bound conformations, each pose was inspected visually and compared to the experimentally determined binding modes and conformations from the X-ray crystal structure of wt-PfDHFR with bound pyrimethamine and NADPH (3QGT.pdb). This process is illustrated in Fig. 5. Poses inconsistent with experimentally determined binding modes and conformations were discarded. Only the top scoring pose (based on PLP Fitness and PLP Score) for each compound was retained.
Results and discussion
DHFR phylogenetics and insights into Plasmodium evolution
In a recent analysis of Plasmodium phylogeny based on mitochondria encoded proteins: cytochrome c oxidase subunits I and III and cytochrome b (all three of which are not under positive selection) [32], Hayakawa et al. [33] argued for coevolution of the malaria parasites with their hosts: rodent; bird; primate group 1; and primate group 2.
The phylogenetic tree generated from our structure-based Plasmodium DHFR sequence alignments (Fig. 4) clustered into 4 main clades corresponding well with the host organism: bird (P. galliniceum); rodent (Vinckeia); primate group 1 (Plasmodium); and primate group 2 (Laverania). This clustering is also consistent with phylogenetic trees generated using ribosomal RNA sequences [34], circumsporozite protein genes [35], as well as genes for cytochrome b [36].
However significant differences remain relative to the canonical tree (Fig. 1) as well as phylogenies based on mitochondrial, ribosomal as well as circumsporozite protein genes [32, 34–36]. Within the DHFR phylogeny, these differences included grouping the Laverania clade with P. gallinaceum in addition to placement of the Vinckeia clade basal to all other taxa.
A pairwise comparison was performed using three different metrics to evaluate these phylogenetic differences. The calculated Split Difference had a value of 0.692 and the Disagree Difference had a value of 0.750 compared to the calculated “random” trees, producing values of 0.977 and 0.952 respectively. For these metrics, two truly random trees would provide a value of 1. The Nodal Difference had a value of 2.249 compared to the calculated “random” value of 2.799. For the Nodal Difference, the larger the value, the greater the tree dissimilarity.
These differences imply that DHFR is under a different selection pressure than the mitochondrial, ribosomal as well as circumsporozite protein genes used to generate the other trees. There are many possibilities for this observed difference that may include selection pressures targeting regulation of the catalytic function as well as selection pressures targeting the protein–protein interactions of DHFR with thymidylate synthase domains. This will be further explored in future studies.
Determination of sequence and structure conservation and variability
For the structure-based alignment of Plasmodium DHFR, the Amino Acid Conservation (in terms of residue identity) was 51.95 % ± 5.63 %. This ranges from a value of 5.88 % representing the case where each of the Plasmodium sequences have a different amino acid at each position to a value of 100.00 % representing complete conservation at each and every position. The calculated value of 51.95 % is indicative of a high degree of site-specific sequence conservation.
The value for Secondary Structure Conservation was higher, 80.14 ± 3.95 %. This is to be expected given the 1.46 Å RMSD calculated from the structural alignment (Fig. 2). Likewise the value for Residue Class Conservation was also higher, 64.27 ± 5.56 %. Although a substantial degree of site-specific sequence variability exists across the 17 Plasmodium species, there is low tolerance for amino acid replacements that result in changes to secondary structure or residue class for DHFR.
A similar analysis was performed for the structure alignments of the active site residues. Not surprisingly, the value of Amino Acid Conservation for the active site, 74.42 ± 11.46 %, was almost 50 % higher than the Amino Acid Conservation for the entire sequence alignment. The value for Secondary Structure Conservation was also higher, 95.19 ± 5.32 % indicating minimal tolerance for changes in secondary structure type. Likewise the value for Residue Class Conservation was also higher than the total sequence alignment, 86.93 ± 8.96 %. From analysis of the positional conservation for the active site residues (Fig. 3) it appears that 30 out of 53 (roughly 57 %) of the active site positions are completely conserved. Only 7 out of 53 (roughly 13 %) of the active site positions had variations with more than 3 different amino acids.
The structure-based alignment was then analyzed to determine the degree of conservation for the positions corresponding to the four amino acid replacements found in drug resistant strains of P. falciparum. Of these, only active site position 59, corresponding to the point mutation C59R, was not highly conserved. In 13 of the other wt species, serine, threonine or valine is present in position 59. However, positions 51, 108, and 164 corresponding to amino acid replacements N51I, S108N, I164L respectively, are highly conserved and are found only in resistant P. falciparum DHFR sequences.
A similar analysis was performed using our previously published structure-based alignment of X-ray crystal structures of 22 prokaryotic and eukaryotic species [23]. Unlike the alignment of Plasmodium DHFR sequences, this structure based alignment had a much greater degree of variation in active site residue identities: 27.08 ± 7.53 % Amino Acid Conservation [22]. Only 3 active site positions out of 53 (5.7 %) were completely conserved. These correspond to glycine in positions 105, 165, and 166. Only one species, Bacillus anthracis, was found to have asparagine in position 108. As mentioned previously, the amino acid substitution S108N is thought to have introduced drug resistance to P. falciparum [32]. Candida glabrata, Mycobacterium tuberculosis, Lactobacillus casei and Thermotoga maritima all have arginine in position 59. Haloferax volcanii and Babesia bovis have isoleucine and arginine in positions 51 and 59 respectively. Additionally, Babesia bovis has leucine in position 164. Although these species share one or more residues in common with mutant Pf-DHFR sequences, their resistance to pyrimethamine remains unknown.
Given that there are 53 positions in the active site (defined in our studies) and 20 possible naturally occurring amino acids, this amounts to a space of 2053 or ~9 × 1068 possible amino acid replacement combinations. However, there is a high degree of conservation in the active site of DHFR throughout the evolutionary history of the genus Plasmodium over the last 12.8 million years. The relatively recent evolution of resistance in Pf-DHFR due to changes in 4 active site positions indicates the presence of a strict selection pressure from drugs like pyrimethamine. In the next section we mimicked this selection pressure through in silico docking experiments.
Docking studies: fitness and selection
Docking studies were performed with each of the 17 wt Plasmodium DHFR X-ray crystal and homology structures (Table 1) as well as with each of the 16 P. falciparum wt and mutant homology models (Table 2). As described in the methods section, a subjective, fuzzy objective function was created to evaluate the ability of each sequence to maintain binding affinity for both the co-factor NADPH and the substrate 7,8-dihydrofolate while reducing affinity for the drug pyrimethamine. This fitness function was defined using the relative binding affinities for NADPH and 7,8-dihydrofolate evaluated through a ratio of the corresponding docking scores for each DHFR structure versus those of wt P. falciparum (1J3I.pdb). Values close to 1 indicated “similar” affinity for each ligand as the wt sequence and low values indicated “lesser” affinity. To evaluate the relative resistance of each sequence to pyrimethamine, the ratio of the docking scores for pyrimethamine versus that of 7,8-dihydrofolate were calculated for each DHFR structure. A “low” value for this ratio indicated reduced binding affinity relative to the substrate 7,8-dihydrofolate, thus implying “enhanced” resistance.
Table 1.
PLP Fitness and PLP score docking scores of NADPH, dihydrofolate, and pyrimethamine for all 17 homology models of Plasmodium wt DHFR sequences
| NADPH
|
Dihydrofolate (DHF)
|
Pyrimethamine
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PLP Fitness | Ratio versus Pf | PLP Score | Ratio versus Pf | PLP Fitness | Ratio versus Pf | PLP Score | Ratio versus Pf | PLP Fitness | Ratio versus DHF | PLP Score | Ratio versus DHF | |
| P. falciparum | 83.66 | N/A | −63.49 | N/A | 72.71 | N/A | −66.18 | N/A | 58.77 | 0.8083 | 51.55 | 0.7789 |
| P. billbrayi | 93.32 | 1.1155 | −76.92 | 1.2115 | 73.33 | 1.0085 | −63.08 | 0.9532 | 64.20 | 0.8755 | 57.03 | 0.9041 |
| P. berghei | 96.08 | 1.1485 | −70.92 | 1.1170 | 59.26 | 0.8150 | −62.50 | 0.9444 | 58.39 | 0.9853 | 52.14 | 0.8342 |
| P. billcollinsi | 90.48 | 1.0815 | −68.98 | 1.0865 | 72.57 | 0.9981 | −65.97 | 0.9968 | 63.04 | 0.8687 | 55.43 | 0.8402 |
| P. brasilianum | 68.20 | 0.8152 | −54.18 | 0.8534 | 68.49 | 0.9420 | −62.77 | 0.9485 | 64.28 | 0.9385 | 58.26 | 0.9282 |
| P. chabaudi | 68.25 | 0.8158 | −56.19 | 0.8850 | 54.49 | 0.7494 | −50.87 | 0.7687 | N/A | N/A | N/A | N/A |
| P. cynomolgi | 59.94 | 0.7165 | −45.47 | 0.7162 | 61.12 | 0.8406 | −59.27 | 0.8956 | 62.47 | 1.0221 | 54.03 | 0.9116 |
| P. gallinaceum | 78.36 | 0.9366 | −57.85 | 0.9112 | 77.57 | 1.0668 | −74.62 | 1.1275 | 61.57 | 0.7937 | 54.15 | 0.7257 |
| P. inui | 98.91 | 1.1823 | −71.33 | 1.1235 | 69.81 | 0.9601 | −63.25 | 0.9557 | 66.15 | 0.9476 | 56.59 | 0.8947 |
| P. knowlesi | 72.26 | 0.8637 | −61.45 | 0.9679 | 76.14 | 1.0472 | −64.49 | 0.9745 | 63.59 | 0.8352 | 57.33 | 0.8890 |
| P. knowlesi H | 88.20 | 1.0543 | −70.86 | 1.1161 | 62.18 | 0.8552 | −55.47 | 0.8382 | 50.99 | 0.8200 | 44.39 | 0.8003 |
| P. malariae | 76.91 | 0.9193 | −63.56 | 1.0011 | 75.69 | 1.0410 | −68.43 | 1.0340 | 62.08 | 0.8202 | 56.68 | 0.8283 |
| P. ovale | 79.66 | 0.9522 | −59.45 | 0.9364 | 63.68 | 0.8758 | −57.33 | 0.8663 | 67.50 | 1.0600 | 64.46 | 1.1244 |
| P. reichenowi | 85.93 | 1.0271 | −68.71 | 1.0822 | 70.01 | 0.9629 | −66.84 | 1.0100 | 62.03 | 0.8860 | 56.34 | 0.8429 |
| P. vinckei | 78.24 | 0.9352 | −59.44 | 0.9362 | 64.47 | 0.8867 | −60.12 | 0.9084 | 67.94 | 1.0538 | 60.61 | 1.0082 |
| P. yoelli yoelli | 28.44 | 0.3399 | −19.27 | 0.3035 | 64.14 | 0.8821 | −57.27 | 0.8654 | N/A | N/A | N/A | N/A |
| P. vivax | 104.40 | 1.2479 | −82.27 | 1.2958 | 66.06 | 0.9085 | −59.25 | 0.8953 | 67.33 | 1.0192 | 56.85 | 0.9595 |
Included are ratios of NADPH and dihydrofolate docking scores for each Plasmodium species versus P. falciparum. Also included are the ratios of docking scores of pyrimethamine versus dihydrofolate. Bolded values represent possible resistance to pyrimethamine. N/A indicates inability to bind. Bolded values indicate potential resistance to pyrimethamine
Table 2.
PLP Fitness and PLP score docking scores of NADPH, dihydrofolate, and pyrimethamine for all 16 homology models of wt, single, double, triple and quadruple mutant P. falciparum DHFR sequences
| NADPH
|
Dihydrofolate (DHF)
|
Pyrimethamine
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PLP Fitness | Ratio versus Pf | PLP Score | Ratio versus Pf | PLP Fitness | Ratio versus Pf | PLP Score | Ratio versus Pf | PLP Fitness | Ratio versus DHF | PLP Score | Ratio versus DHF | |
| P. falciparum—wt | 83.66 | N/A | −63.49 | N/A | 72.71 | N/A | −66.18 | N/A | 58.77 | 0.8083 | 51.55 | 0.7789 |
| C59R | 48.45 | 0.5791 | −27.29 | 0.4298 | 63.22 | 0.8695 | −59.11 | 0.8932 | 51.79 | 0.8192 | −46.22 | 0.7819 |
| I164L | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| N51I | 51.78 | 0.6189 | −39.45 | 0.6214 | 65.05 | 0.8946 | −64.24 | 0.9707 | 56.12 | 0.8627 | − 48.41 | 0.7536 |
| S108N | 80.04 | 0.9567 | −61.93 | 0.9754 | 65.12 | 0.8956 | −59.91 | 0.9053 | N/A | N/A | N/A | N/A |
| C59R, I164L | 49.42 | 0.5907 | −35.92 | 0.5658 | 52.66 | 0.7242 | −50.38 | 0.7613 | 51.49 | 0.9778 | −54.29 | 1.0776 |
| C59R, S108N | 73.76 | 0.8817 | −59.20 | 0.9324 | 73.94 | 1.0169 | −70.48 | 1.0650 | N/A | N/A | N/A | N/A |
| C59R, N51I | 52.10 | 0.6228 | −35.65 | 0.5615 | 67.35 | 0.9263 | −63.65 | 0.9618 | 64.43 | 0.9566 | −55.57 | 0.8931 |
| N51, I164L | 70.02 | 0.8370 | −55.13 | 0.8683 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| S108N, N51I | 67.92 | 0.8119 | −47.85 | 0.7537 | 60.76 | 0.8356 | −57.58 | 0.8701 | N/A | N/A | N/A | N/A |
| S108N, I164L | 74.12 | 0.8860 | −58.72 | 0.9249 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| C59R, S108N, N51I | 50.25 | 0.6006 | −39.84 | 0.6275 | 78.93 | 1.0855 | −75.78 | 1.1451 | N/A | N/A | N/A | N/A |
| C59R, S108N, I164L | 77.43 | 0.9255 | −56.14 | 0.8842 | 58.34 | 0.8024 | −54.60 | 0.8250 | N/A | N/A | N/A | N/A |
| C59R, I164L, N51I | 50.80 | 0.6072 | −48.18 | 0.7589 | 81.31 | 1.1183 | −81.24 | 1.2276 | 62.39 | 0.7673 | −58.21 | 0.7165 |
| S108N, N51I, I164L | 55.69 | 0.6657 | −36.43 | 0.5738 | 59.56 | 0.8191 | −58.73 | 0.8874 | N/A | N/A | N/A | N/A |
| N51I, C59R, S108N, I164L | 85.41 | 1.0209 | −63.67 | 1.0028 | 63.07 | 0.8674 | −62.56 | 0.9453 | N/A | N/A | N/A | N/A |
Included are ratios of NADPH and dihydrofolate docking scores for each mutant versus wt. Also included are the ratios of docking scores of pyrimethamine versus dihydrofolate. Bolded values represent possible resistance to pyrimethamine. N/A indicates inability to dock. Bolded values indicate potential resistance to pyrimethamine
In order to verify that poses resulting from in silico docking represented correctly-bound conformations, each pose was inspected visually and compared to the experimentally determined binding modes and conformations from the X-ray crystal structure of wt-PfDHFR with bound pyrimethamine and NADPH (3QGT.pdb). This process is illustrated in Fig. 5.
Poses of NADPH, pyrimethamine and 7,8-dihydrofolate that were inconsistent with the experimentally determined binding modes and conformations from the X-ray crystal structure of wt-PfDHFR with bound pyrimethamine and NADPH (3QGT.pdb) and/or poses that were displaced from the appropriate binding site were considered not to bind and were designated by “N/A” in Tables 1 and 2. Using these metrics, we defined a drug “resistant” DHFR model as one with NADPH and 7,8-dihydrofolate ratios approximating 1 and a pyrimethamine pose that was scored as “N/A.” If the docked pose of pyrimethamine was consistent with that of the X-ray crystal structure, the DHFR model was classified as “non-resistant”—even if the docking score ratio was “low.” Models where the NADPH pose and/or dihydrofolate pose were scored as “N/A” were eliminated as organisms with these amino acid replacements would be culled in nature.
The results presented in Table 1 show that only two DHFR sequences were predicted to be resistant, P. yoelli yoelli and P. chabaudi. At first we considered these to represent possible ‘false positives’. However, upon further inspection we discovered that the P. yoelli yoelli sequences in the NCBI Protein database, including the one used in these studies: gi# 82540717, are all identical to that of one coming from a pyrimethamine resistant strain [37, 38]. Likewise the P. chabaudi DHFR sequence used in this analysis (gi# AAA29587.1) also came from a pyrimethamine resistant strain [39]. To our surprise, it turned out that these sequences were correctly identified as being pyrimethamine resistant.
The results of the docking studies of the 16 models of wt and mutant Pf-DHFR sequences presented in Table 2 are summarized in Fig. 6. In these studies we identified the naturally occurring single mutant (S108N), double mutant (C59R, S108N), triple mutant (C59R, S108N, N51I) and quadruple mutant (C59R, S108N, N51I, I164L) all as “resistant.” However, the single-mutants N51I and C59R were classified as being not resistant as pyrimethamine bound to the active site. The I164L mutant was also eliminated as the cofactor NADPH and substrate dihydrofolate did not bind well.
Fig. 6.

Pathways of resistance to pyrimethamine by DHFR. Starting with the wild type amino acid sequence for DHFR (green), several one-step, two-step, and even three-step amino acid replacements provide no resistance to pyrimethamine (pathways with crossed circles). The only viable pathways to the known double, triple, and quadruple mutant DHFR sequences (shown in red) are through either C59R or S108N replacements, leading to a cascade of alternate replacement sets that provide further resistance
In the literature, it has been proposed that the first mutation conveying resistance to pyrimethamine may have been S108N resulting from a steric clash between the side chain of Asn 108 and pyrimethamine [30, 31, 40–43]. In our docking studies we observed that while this mutation does indeed result in a steric clash with pyrimethamine [31, 32], it does not affect binding of the substrate dihydrofolate (Fig. 7).
Fig. 7.

Superposition of bound X-ray crystal of dihydrofolate bound to S. aureus wt-DHFR (3RD.pdb) versus conformations from docking experiments to P. falciparum DHFR: (A) wt and (B) double mutant (C59R, S108N)
Interestingly, we observed that the docked pose of dihydrofolate forms hydrogen bonds with the side chain of Arg 59 in the mutant C59R, S108N structure that are not possible with Cys 59 (Fig. 7). This provides a structure-based explanation for the observed synergistic enhancement of resistance in mutant DHFR containing the S108N as well as C59R mutations.
We also predicted 3 additional replacement sets (S108N, N51I); (C59R, S108N, I164L); and (S108N, N51I, I164L) to be resistant to pyrimethamine. To our knowledge, these replacements have not yet been observed in the wild. It is possible that some/all of these may represent intermediate steps in the evolution of resistant strains of P. falciparum that have been lost over time due to competition with the genotypes that exist in the wild today.
Pathways to pyrimethamine resistance
In recent studies Lozovsky et al. and Brown et al. [44, 45] performed similar analyses in which assays were performed measuring inhibition of bacterial growth with plasmids containing all 16 mutants. From the resulting IC50 values, evolutionary models were generated and predicted pathways to pyrimethamine resistance were predicted and evaluated. Interestingly, in these studies they observed that the S108N mutation lead to resistance (as expected) and that the N51I as well as the I164L mutations did not. Our results are in complete agreement (Fig. 7). However, in their studies, the C59R mutation did lead to resistance, whereas in our analysis it did not. It is possible that resistance from the C59R mutation may arise from a greater binding affinity for the substrate over pyrimethamine due to increased hydrogen bonding. Based on this discrepancy, we plan to refine our fitness function to model these interactions more accurately. Overall, our predicted pathway to pyrimethamine resistance closely mirrors that of Lozovsky et al. including a (S108, N51I) branch.
Conclusions
Over the last 50 years, P. falciparum has demonstrated the ability to rapidly evolve resistance to front-line therapeutics such as chloroquine and sulfadoxine-pyrimethamine. This poses a grand challenge: how can we combat a parasite that rapidly evolves in response to therapeutics that take years to develop at significant cost? We believe the answer is to include models as a component of drug development that can faithfully recapitulate coevolutionary processes. A portfolio of potential therapeutics can result, all in anticipation of likely possible future amino acid replacements.
In this paper, we have demonstrated that in silico modeling of evolution can simulate the evolutionary trajectories of the clinically relevant therapeutic target dihydrofolate reductase (DHFR) and identify amino acid replacements that maintain 7,8-dihydrofolate and NADPH binding while simultaneously reducing drug affinity, thus providing increased “resistance” for the host. Furthermore, our model has helped elucidate the likely pathways of amino acid replacement leading to double, triple, and quadruple mutant DHFR in the wild. The simulation also provides a means to better understand those pathways that are highly unlikely to lead to resistance to pyrimethamine while still maintaining an affinity to its natural substrate. These results are consistent with the growing consensus that there are a limited number of pathways to drug resistance in P. falciparum [44–46].
In future studies, we propose to couple this methodology with in silico and in vitro screening [47] as well as computational intelligence based de novo ligand design [48] in order to identify compounds active against future drug resistant strains of malaria parasites. This same approach can be used on any target of interest to better understand previous pathways to resistance, leading to the opportunity to predict likely pathways of future resistance as an additional benefit to future drug development.
Acknowledgments
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number SC3GM100791. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors would like to thank Patrick Rose and Doug McKenzie as well as the reviewers for valuable suggestions and insightful comments that helped make this a much more complete manuscript.
Contributor Information
David Hecht, Email: dhecht@swccd.edu, Southwestern College, 900 Otay Lakes Rd., Chula Vista, CA 91910, USA.
Gary B. Fogel, Email: gfogel@natural-selection.com, Natural Selection, Inc., 9330 Scranton Rd., San Diego, CA 92121, USA.
References
- 1. [Accessed Jan 10, 2012];Center of Disease Control. http://www.cdc.gov/malaria/about/facts.html.
- 2.Baron JM, Higgins JM, Dzik WH. A revised timeline for the origin of Plasmodium falciparum as a human pathogen. J Mol Evol. 2010;73:297–304. doi: 10.1007/s00239-011-9476-x. [DOI] [PubMed] [Google Scholar]
- 3.Ricklefs RE, Outlaw DC. A molecular clock for malaria parasites. Science. 2010;329:226–229. doi: 10.1126/science.1188954. [DOI] [PubMed] [Google Scholar]
- 4.Ayala FJ, Escalante AA, Rich SM. Evolution of Plasmodium and the recent origin of the world populations of Plasmodium falciparum. Parassitologia. 1999;41:55–68. [PubMed] [Google Scholar]
- 5.Arisue N, Kawai S, Hirai M, Palacpac NM, Jia M, Kaneko A, Tanabe K, Horii T. Cluster to evolution of the SERA mutigene family in 18 Plasmodium species. PLoS ONE. 2011;6:e17775. doi: 10.1371/journal.pone.0017775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blakely RL, editor. Folates and Pterins. Wiley; New York: 1985. Dihydrofolate Reductases; pp. 191–253. [Google Scholar]
- 7.Blakely RL. In: Advances in enzymology and related areas of molecular biology. Meister A, editor. Wiley; New York: 1995. pp. 23–102. [Google Scholar]
- 8.Wallace L, Matthews CR. Highly divergent dihydrofolate reductases conserve complex folding mechanisms. J Mol Biol. 2002;315:193–211. doi: 10.1006/jmbi.2001.5230. [DOI] [PubMed] [Google Scholar]
- 9.Ferone R. Folate metabolism in malaria. Bull World Health Organ. 1977;55:291. [PMC free article] [PubMed] [Google Scholar]
- 10.Huennekens FM. The methotrexate story: a paradigm for development of cancer chemotherapeutic agents. Adv Enzyme Regul. 1994;34:397. doi: 10.1016/0065-2571(94)90025-6. [DOI] [PubMed] [Google Scholar]
- 11.Rollo IM. Dihydrofolate reductase inhibitors as antimicrobial agents and their potentiation by sulfonamides. CRC Crit Rev Clin Lab Sci. 1970;1:565. doi: 10.3109/10408367009043919. [DOI] [PubMed] [Google Scholar]
- 12.Bertino JR. Cancer research: from folate antagonism to molecular targets. Best Pract Res Clin Haematol. 2009;22:577–582. doi: 10.1016/j.beha.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 13.Mita T, Tanabe MT, Kita K. Spread and evolution of Plasmodium falciparum drug resistance. Parasit Int. 2009;58:201–209. doi: 10.1016/j.parint.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 14.Volpato JP, Pelletier JN. Mutational ‘hot-spots’ in mammalian, bacterial and protozoal resistance: sequence and structural comparison. Drug Resist Updat. 2009;12:28–41. doi: 10.1016/j.drup.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 15.Zhanel GG, Wang X, Nichol K, Nikulin A, Wierzbowski AK, Mulvey M, Hoban DJ. Molecular characterization of Canadian pediatric multi-drug resistant Streptococcus pneumonia from 1998–2004. Int J Antimicrob Agents. 2006;28:465–471. doi: 10.1016/j.ijantimicag.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 16.Zhao S, McDermott PF, White DG, Qaiyumi S, Friedman SL, Abbott JW, Glenn A, Ayers SL, Post KW, Fales WH, Wilson RB, Reggiardo C, Walker RD. Characterization of multidrug resistant Salmonella recovered from diseased animals. Vet Microbiol. 2007;123:122–132. doi: 10.1016/j.vetmic.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 17.Payne D. Spread of chloroquine resistance in Plasmodium falciparum. Parasitol Today B. 1987;3:241–246. doi: 10.1016/0169-4758(87)90147-5. [DOI] [PubMed] [Google Scholar]
- 18.Wernsdorfer WH. The development and spread of drug resistant malaria. Parasitol Today. 1991;7:297–303. doi: 10.1016/0169-4758(91)90262-m. [DOI] [PubMed] [Google Scholar]
- 19.Saito-Nakano Y, Tanabe K, Kamei K, Iwagami M, Komaki-Yasuda K, Kawazu S, Kano S, Ohmae H, Endo T. Genetic evidence for Plasmodium falciparum resistance to chloroquine and pyrimethamine in Indochina and the Western Pacific between 1984 and 1998. Am J Trop Med Hyg. 2008;79(4):613–619. [PubMed] [Google Scholar]
- 20.Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser. 1999;41:95–98. [Google Scholar]
- 21.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hecht D, Tran J, Fogel GB. Structure-based anaylsis of dihydrofolate reductcase evolution. Mol Phylo Evol. 2011;61:212–230. doi: 10.1016/j.ympev.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 23.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 24.Puigbò P, Garcia-Vallvé S, McInerney JO. TOPD/FMTS: a new software to compare phylogenetic trees. Bioinformatics. 2007;23:1556–1558. doi: 10.1093/bioinformatics/btm135. [DOI] [PubMed] [Google Scholar]
- 25.Robinson DF, Foulds LR. Comparison of phylogenetic trees. Math Biosci. 1981;53:131–147. [Google Scholar]
- 26.Steel MA, Penny D. Distribution of tree comparison metrics—some new results. Syst Biol. 1993;42:126–141. [Google Scholar]
- 27.Penny D, Hendy MD. The use of tree metrics. Syst Zool. 1985;34:75–82. [Google Scholar]
- 28.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 29.Korb O, Stützle T, Exner TE. Empirical scoring functions for advanced protein-ligand docking with PLANTS. J Chem Inform Model. 2009;49:84–96. doi: 10.1021/ci800298z. [DOI] [PubMed] [Google Scholar]
- 30.Fogel GB, Cheung M, Pittman E, Hecht D. In Silico screening against wild-type and mutant Plasmodium falciparum Dihydrofolate Reductase. J Mol Graph Model. 2008;26:1145–1152. doi: 10.1016/j.jmgm.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 31.Fogel GB, Cheung M, Pittman E, Hecht D. Modeling the inhibition of quadruple mutant plasmodium falciparum dihydrofolate reductase by pyrimethamine derivatives. J Comput Aided Mol Des. 2008;22:29–38. doi: 10.1007/s10822-007-9152-9. [DOI] [PubMed] [Google Scholar]
- 32.Joy DA, Feng X, Mu J, Furuya T, Chotivanich K, Kretti AU, Ho M, Wang A, White NJ, Suh E, Berrli P, Su X-Z. Early origin and recent expansion of Plasmodium falciparum. Science. 2003;300:318–321. doi: 10.1126/science.1081449. [DOI] [PubMed] [Google Scholar]
- 33.Hayakawa T, Culleton R, Otani H, Horii T, Tanabe K. Big bang in the evolution of extant malaria parasites. Mol Biol Evol. 2008;25:2233–2239. doi: 10.1093/molbev/msn171. [DOI] [PubMed] [Google Scholar]
- 34.Escalante AA, Ayala FJ. Phylogeny of the malarial genus Plasmodium, derived from rRNA gene sequences. Proc Natl Acad Sci USA. 1994;91:11373–11377. doi: 10.1073/pnas.91.24.11373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Escalante AA, Barrio E, Ayala FJ. Evolutionary origin of human and primate malarias: evidence from the circumsporozoite protein gene. Mol Biol Evol. 1995;12:616–626. doi: 10.1093/oxfordjournals.molbev.a040241. [DOI] [PubMed] [Google Scholar]
- 36.Escalante AA, Freeland DE, Collins WE, Lal AA. The evolution of primate malaria parasites based on the gene encoding cytochrome b from the linear mitochondrial genome. Proc Natl Acad Sci USA. 1998;95:8124–8129. doi: 10.1073/pnas.95.14.8124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walliker D, Sanderson A, Yoeli M, Hargreaves BJ. A genetic investigation of virulence in a rodent malaria parasite. Parasitology. 1976;72:183–194. doi: 10.1017/s0031182000048484. [DOI] [PubMed] [Google Scholar]
- 38.Pattaradilokrat S, Cheesman SJ, Carter R. Congenicity and genetic polymorphism in cloned lines derived from a single isolate of a rodent malaria parasite. Mol Biochem Parasitol. 2008;157:244–247. doi: 10.1016/j.molbiopara.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 39.Cowman AF, Lew AM. Antifolate drug selection results in duplication and rearrangement of chromosome 7 in Plasmodium chabaudi. Mol Cell Biol. 1989;9:5182–5188. doi: 10.1128/mcb.9.11.5182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yuvaniyama J, Chitnumsub P, Kamchonwongpaison S, Vanichtanankul J, Sirawaraporn W, Taylor P, Walkinshaw M, Yuthavong Y. Insights into antifolate resistance from malarial DHFR–TS structures. Nat Struct Biol. 2003;10:357–365. doi: 10.1038/nsb921. [DOI] [PubMed] [Google Scholar]
- 41.Sirawaraporn W, Sathikul T, Sirawaraporn R, Yuthavong Y, Santi DV. Antifolate-resistant mutants of Plasmodium falciparum dihydrofolate reductase. Proc Natl Acad Sci USA. 1997;94:1124–1129. doi: 10.1073/pnas.94.4.1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kongsaeree P, Khongsuk P, Leartsakulpanich U, Chitnumsub P, Tarnchombpoo B, Walkinshaw MD, Yuthavong Y. Crystal structure of dihydrofolate reductase from Plasmodium vivax: pyrimethamine displacement linked with mutation-induced resistance. Proc Natl Acad Sci USA. 2005;102:13046–13051. doi: 10.1073/pnas.0501747102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peterson DS, Walliker D, Wellems TE. Evidence that a point mutation in dihydrofolate reductase-thymidylate synthase confers resistance to pyrimethamine in falciparum malaria. Proc Natl Acad Sci USA. 1988;85:9114–9118. doi: 10.1073/pnas.85.23.9114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lozovsky ER, Chookajorn T, Brown KM, Imwong M, Shaw PJ, Kamchonwongpaisan S, Neafsey DE, Weinreich DM, Hartl DL. Stepwise acquisition of pyrimethamine resistance in the malaria parasite. Proc Natl Acad Sci USA. 2009;106:12025–12030. doi: 10.1073/pnas.0905922106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brown KM, Costanzo MS, Xu W, Roy S, Lozovsky ER, Hartl DL. Compensatory mutations restore fitness during the evolution of dihydrofolate reductase. Mol Biol Evol. 2010;27:2682–2690. doi: 10.1093/molbev/msq160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Costanzo MS, Hartl DL. The evolutionary landscape of antifolate resistance in Plasmodium falciparum. J Genet. 2011;90:187–190. doi: 10.1007/s12041-011-0072-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hecht D, Fogel GB. 2012 IEEE computational intelligence in bioinformatics and computational biology. San Diego: 2012. Towards predictive structure-based models of evolved drug resistance; pp. 120–126. [Google Scholar]
- 48.Hecht D, Fogel G. A novel in silico approach to drug discovery via computational intelligence. J Chem Inf Model. 2009;49:1105–1121. doi: 10.1021/ci9000647. [DOI] [PubMed] [Google Scholar]