

Abstract
Previously it was demonstrated that amphipathic isoxazolidines are able to functionally replace the transcriptional activation domains of endogenous transcriptional activators. In addition, in vitro binding studies suggested that a key binding partner of these molecules is the Creb Binding Protein (CBP), more specifically the KIX domain with this protein. Here we show that CBP and the KIX domain play an essential role in the ability of isoxazolidine transcriptional activation domains to activate transcription in cells. Consistent with this model, isoxazolidines are able to function as competitive inhibitors of the activators MLL and Jun, both of which utilize a binding interaction with KIX to up-regulate transcription. Further, modification of the N2 side chain produced two analogs with enhanced potency against Jun-mediated transcription, although increased cytotoxicity was also observed. Collectively these small KIX-binding molecules will be useful tools for dissecting the role of the KIX domain in a variety of pathological processes.
Introduction
Transcription factors, often characterized as undruggable, are nonetheless emerging as potentially attractive therapeutic targets due to their fundamental role in human disease.1–3 One class of transcription factors, transcriptional activators, plays a key regulatory role by binding to DNA and assembling the transcriptional machinery at a particular gene, thus stimulating gene expression. Miscued transcription caused by malfunctioning activators has been implicated in the onset of many cancers and this has spurred widespread interest in the development of small and large molecules that directly target misregulated genes.2 The development of small molecule transcriptional activation domain (TAD) mimics is an advantageous strategy to modulate transcription because, similar to natural TADs, these molecules can function as activators when tethered to a DNA-binding domain (DBD) and as inhibitors when not localized to DNA (Figure 1a).3 TAD mimics are thus promising candidates for modulating gene transcription, for use as mechanistic probes, and as transcription-targeted therapeutics.3
Figure 1.
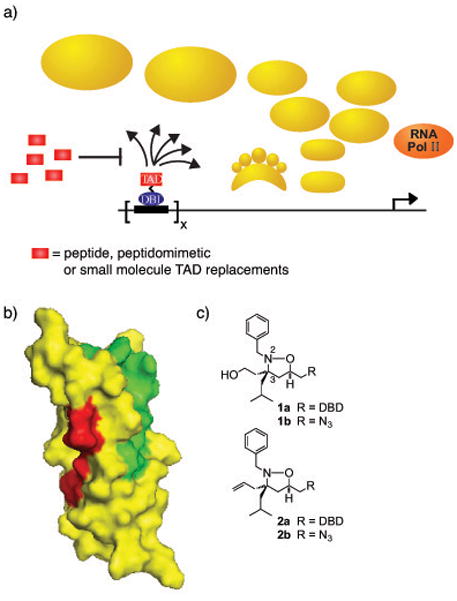
a) Transcriptional activators, minimally comprised of a DNA-binding domain (DBD; blue oval) and a transcriptional activation domain (TAD; red rectangle), upregulate transcription by stimulating chromatin remodeling and facilitating the assembly of the RNA polymerase II holoenzyme at a promoter.14 The TAD is primarily responsible for the direct binding interactions with the transcriptional machinery (gold) in order to accomplish this. Potential binding partners of TADs within the transcriptional machinery include enzymes that modify chromatin, the proteasome, and/or coactivators. Small or large molecule mimics of TADs can serve as competitive inhibitors of activators, thus down-regulating transcription. b) The two activator-binding sites within the KIX domain of CBP/p300. Highlighted in green in the space-filling diagram of the KIX domain are the residues that experience the greatest chemical shift perturbation upon interacting with the TAD of MLL as measured by 1H-15N-HSQC experiments;8b the TADs of Tat, Tax and Jun occupy a similar binding site.8d–f In red are the amino acids that change when interacting with Myb.15 Pymol figures were generated from 1 kdx. c)Isoxazolidine-based mimics of transcriptional activation domains. DBD = OxDex conjugated to an ethylene glycol linker (AEEA).13
Like their natural counterparts, small molecule TADs must interact with coactivators in the transcriptional machinery in order to function. This presents a formidable challenge since the number of putative coactivator targets of activators is large, with few validated as physiologically relevant.4 One target of importance is the CREB binding protein (CBP), a large (265 kDa) multidomain coactivator and histone acetyl transferase.5 CBP integrates transcriptional signals from >100 transcription factors and is an essential protein for cell growth and development. In contrast to most coactivators, the individual domains of CBP have proven amenable to structural characterization and, concomitantly, have been attractive targets for the development of small and large molecule transcriptional regulators.6 A domain that has attracted much attention in this regard is the KIX domain, an 87-residue module whose solution structure has been elucidated.7 Despite its relatively small size, the KIX domain contains at least two activator binding sites, the CREB/Myb site and the MLL/Jun/Tat/Tax site (Figure 1b).8 Screening against the KIX domain has lead to the discovery of both peptides and peptidomimetic TADs that function well in cells;6b,9 derivatives of these peptides have been useful tools for defining characteristics of activator binding sites.10 Not just useful for creating activators, a small molecule screen against the KIX domain has also yielded naphthol AS-E phosphate and related derivatives that competitively inhibit the KIX-CREB binding interaction.6e,6h Complementary to genetic strategies, compounds that inhibit KIX-binding activators will be useful tools in dissecting the role of the KIX domain in physiological processes such as cell differentiation and hematopoiesis.11 Furthermore, both MLL and Jun have been implicated in several cancers, including leukemias and solid tumors;12 thus compounds that block these proteins from forming key interactions with CBP may prove useful in the development of transcription-targeted therapeutics.1–5, 15
We identified several isoxazolidine-based TADs through a top-down discovery strategy that reconstitute the function of a natural activator when localized to a promoter(e.g. 1a, Figure 1c).13 Subsequent in vitro binding studies revealed that one target of these small molecules is the KIX domain of CBP, more specifically the MLL/Jun/Tat/Tax site within that module (Figure 1b).6c What was not clear from these studies was if this interaction is essential for function in a cellular context. Here we provide evidence that the isoxazolidine·KIX interaction is required for cellular activity. Consistent with this model, in the absence of DNA binding, isoxazolidine 1 is able to competitively inhibit transcription mediated by the KIX-dependent activators MLL and c-Jun. Based on natural peptide ligands for KIX, alteration of the aromatic side chain of 1 lead to the identification of two molecules with enhanced potency, although increased cytotoxicity was also observed.
Results and Discussion
Cellular activity of 1 depends upon CBP
Previously we demonstrated that amphipathic isoxazolidine 1 activates transcription and interacts with the KIX domain of CBP, while 2, bearing an alkene in place of the hydroxyl functional group at C3, does not activate nor does it interact with KIX.6c,13 To assess if CBP is required for 1-mediated transcriptional activity, the impact of lowering CBP concentrations via shRNA knockdown was examined in the presence of 1 conjugated to the glucocorticoid receptor (GR) ligand OxDex (1a).16 In these experiments, cells were transfected with a luciferase reporter bearing five Gal4 binding sites and a plasmid encoding a fusion protein of the Gal4 DBD and the minimal ligand binding domain of GR in the presence of 1a. In this two-hybrid assay, molecules tagged with the GR ligand OxDex will interact with the fusion protein and thus be localized to DNA.6b The knockdown of CBP with shRNA resulted in an 86 ± 8% decrease (from 70-fold to 12-fold) in 1a-driven luciferase expression, while a scrambled shRNA had no effect (see SI Figure S1). Additionally, the expression of exogenous CBP partially rescued the activity of 1a (from 12-to 50-fold, see SI Figure S3. In contrast, increased CBP concentration had no impact on the ability of isoxazolidine 2a to activate transcription, suggesting there is a direct link between CBP concentration and 1a-driven activation. Additionally, a change in CBP concentration had no effect on expression of a luciferase gene driven by a CMV promoter that does not contain Gal4 binding sites (see SI Figure S2). Taken together, these data reveal that the activity of 1a is directly impacted by CBP concentration in the cell.
Another common strategy for altering cellular CBP availability is to use the viral protein E1A to sequester it.17 As shown in Figure 2, there is an 80 ± 3% decrease (from 70-fold to 15-fold) in isoxazolidine 1a-mediated activation when E1A is present. As outlined earlier, our previous in vitro studies revealed that 1b interacts with the KIX domain in particular;6c to further probe the importance of the isoxazolidine·KIX interaction for transcriptional activity, the activity experiments were carried out in the presence of over-expressed KIX domain and this led to a 60 ± 3% decrease (to 30-fold) in activation by 1a (Figure 2). This is consistent with previously reported squelching experiments of Tax, a viral KIX-targeting activator, with a 50% decrease in Tax-driven transcription observed when KIX is over-expressed.18 These squelching data indicate that that KIX binding is critical for the transcriptional activity of 1a. However, the inability of over-expressed KIX to completely abrogate the activity of 1a suggests that other binding partners are likely also important.6c In sum, modulation of transcriptional activity by affecting the availability of CBP supports the model that 1a functions as a transcriptional activator through binding the KIX domain of CBP.
Figure 2. Isoxazolidine-driven gene expression requires CBP and the KIX domain.
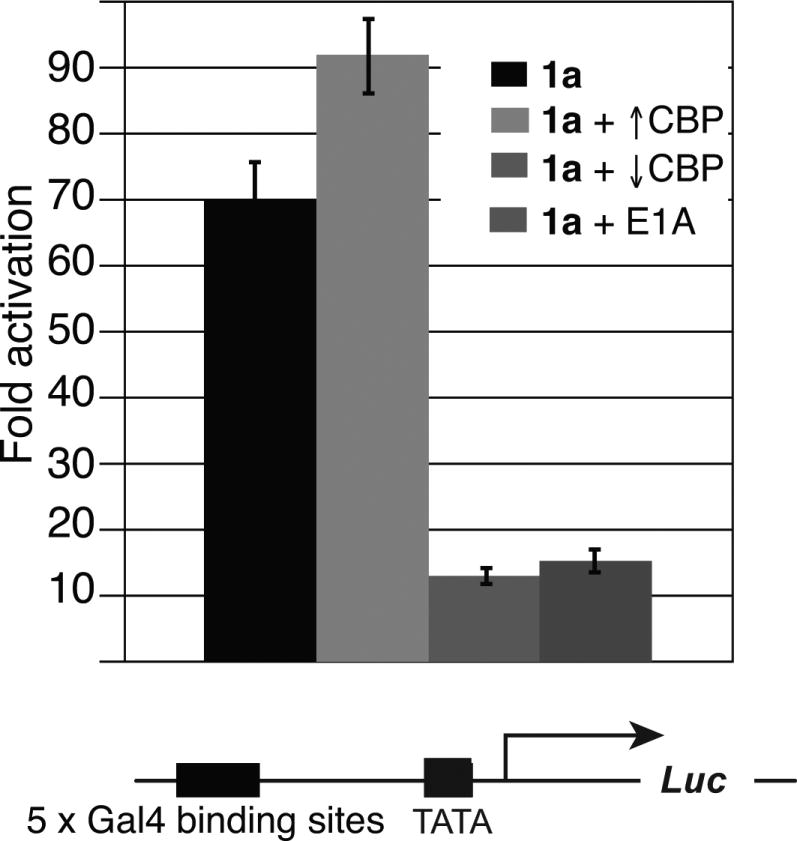
HeLa cells were transfected with a plasmid encoding the DBD of Gal4 fused to the ligand-binding domain of the glucocorticoid receptor, a firefly luciferase reporter plasmid containing five Gal4 binding sites, and a Renilla transfection control. Additional plasmids were transfected as indicated for CBP, CBP shRNA, KIX, and E1A experiments. Four hours post-transfection, 10 μM 1a was added as a solution in DMSO such that the final concentration of DMSO was 1% (v/v). Luciferase output was measured 24h after addition of compound; fold activation is a ratio of firefly and Renilla luminescence divided by the ratio of firefly and Renilla signal observed with DBD alone. Each value represents at least three individual experiments with the indicated error (standard deviation of the mean, SDOM).
Inhibition of MLL and Jun
Although a number of natural TADs interact with the KIX domain,19,21,44,45 the isoxazolidine TADs were the first examples of small molecules that interact with the site utilized by the amphipathic TADs of MLL, Jun, Tat and Tax.6c The overlapping binding footprint of 1b with that of MLL and Jun should enable the small molecule to function as a competitive inhibitor of these activators. Towards this end, a series of activator competition studies were performed using proteins composed of the MLL and Jun TADs and the DBD of Gal4. Although 1b is considerably smaller than the minimal TADs for MLL and Jun, it was able to inhibit the activation of both MLL and Jun in a dose-dependent manner (Figure 3a). The EC50 for inhibition of Jun by 1 is 19 μM and for inhibition of MLL it is 36 μM. In addition, ESX, an activator that is not known to bind the KIX domain was not inhibited in a similar series of experiments. This is consistent with previous results performed in two ESX-driven cell lines where 1 exhibits inhibition only at high concentrations (≥ 50 μM).19
Figure 3. Inhibition of KIX-targeting TADs by 1b.

a) Isoxazolidine 1b decreased Jun and MLL minimal TAD-driven activation in a dose-dependent manner while not affecting ESX, an activator that is not known to bind the KIX domain.20 Cells were transfected with Firefly and Renilla luciferase plasmids in addition to plasmids encoding the DBD of Gal4 fused to the minimal activation domains of either MLL, Jun, or ESX. Four hours post-transfection cells were treated with increasing concentration of 1b (0 → 50 μM) as a solution in DMSO such that the final DMSO concentration was 1% (v/v). Percent inhibition is the ratio of fold activation of Jun at each concentration of compound and the fold activation of Jun in a DMSO-treated control. Each bar represents at least three independent experiments with the indicated error (SDOM). b) MCF-7 cells were treated with 40 μM 1b (as a solution in DMSO such that the final concentration of DMSO was 1% v/v) for 24 h, at which time the cells were lysed and the lysates separated by SDS-PAGE. Western blots were performed using commercial antibodies against GAPDH, Jun, and cyclin D1.
The previous reporter gene experiments demonstrated that 1b inhibits the minimal TAD of Jun. It was unclear, however, if the isoxazolidine would function in a similar manner with the full-length Jun protein in an endogenous promoter context. To assess if inhibition of native Jun could be accomplished with isoxazolidine 1b, we chose to examine expression of the cyclin D1 protein, a key component of the cell cycle machinery.21 Cyclin D1 expression is regulated by the AP-1 transcription factor that is a Jun homo-or a Jun-Fos heterodimer.12f,22 As illustrated in Figure 3b, treatment of Jun-expressing MCF-7 breast cancer cells with 40 μM 1b (a concentration two-fold higher than the EC50) led to a significant decrease in cyclin D1 expression. In the same time course, expression of Jun was minimally impacted while the control, GAPDH, was unchanged. Thus, isoxazolidine 1b exerts an inhibitory effect on expression of a Jun-regulated gene not only in an artificial reporter system, but also in a native context. The demonstration of isoxazolidine 1b-mediated inhibition of a Jun-regulated gene is a key step in developing a new chemical tool for probing the Jun pathway.
Inhibition of MLL and Jun with N2 Analogs
Although the results of Figure 3a illustrate that isoxazolidine 1b serves as a transcriptional inhibitor, concentrations >20 μM are required. For these molecules to serve as probes of KIX function, an increase in potency is highly desirable. Like a peptide, the isoxazolidine is a modular scaffold, amenable to functionalization at the N2, C3, C4, and C5 positions. Thus, to design new molecules with increased potency, we looked to natural KIX-binding peptides for functional groups. The sequences of several natural TADs that bind the MLL/Jun/Tat/Tax site of the KIX domain (Figure 4a) possess diverse amphipathic sequences. Whereas Tax and Jun only possess aliphatic hydrophobic side chains, Tat and MLL TADs contain aromatic side chains from tryptophan and phenylalanine, respectively. These sequence differences may contribute to the range of dissociation constants observed for these activators(MLL = 2.5 μM,19 Tat = 11 μM,20 Jun = 30 μM54), though this has not been determined. In the case of the isoxazolidine scaffold, an isoxazolidine bearing a biphenyl substituentat the N2 position (3) does not bind the KIX domain or activate transcription;6c, 23 thus we used biphenyl as an upper limit on steric modification. Early in our studies, we observed that a smaller substituent, p-hydroxyphenyl (4), did not negatively impact KIX binding by 1H-15N-HSQC binding experiments (SI Figure S4). Thus, to assess the functional impact of aromatic and aliphatic substituents at the N2 position of 1, a small series of isoxazolidines was prepared bearing both functionality similar to that of natural activators (4, 7–9) and small steric (5) and electronic (6) modifications.
Figure 4.

a) Sequences of KIX-binding TADs. b) Analogs of 1b tested for MLL and Jun inhibition; see SI for complete details of synthesis and characterization. c) Inhibition of Jun with isoxazolidine analogs. Inhibition assays were carried out as described in Figure 3a.
While 1b and 4 exhibit no measurable toxicity at concentrations ≥ 50 μM, the remaining analogs inhibited cell growth at concentrations above 10 μM, limiting the range of concentrations that could be examined (see SI Figure S3). As expected, biphenyl 3 did not affect Jun-driven transcription; in addition, compounds 4, 8, and 9 exhibited little activity. However, as shown in Figure 4c and d, isoxazolidines 5, 6 and 7 inhibited Jun transcriptional activity in a dose-dependent manner. Indeed, these three compounds are more effective than 1 (1.3–2 fold greater inhibition at 10 μM). The levels of inhibition achieved by these molecules is higher for Jun than for MLL, perhaps a reflection of the >10 fold difference in the dissociation constants of these two activators (SI Figure S5).
Despite the improved activity of isoxazolidines 5–7 relative to 1, none of the three exhibit sub-micromolar EC50s; this highlights one of the challenges of designing small molecule transcriptional modulators, which possess a much smaller surface area relative to their peptidic counterparts.24 While the isoxazolidine shares an overlapping binding surface with that of Jun and MLL, the peptidic ligands for KIX interact with a significantly larger area of the domain and make more intermolecular contacts that contribute both to binding affinity and specificity.6c,8b,8d To more substantially increase potency, it may be necessary to utilize a small molecule framework that can incorporate additional functional groups for interaction with the KIX domain, such as dimeric isoxazolidines, for example.6c In addition, we note that recent advances with cell-permeable, constrained peptides have shown promising results in inhibiting activators that interact with other domains of CBP.6f
In summary, here we have demonstrated that the isoxazolidine transcriptional activation domain mimic 1 requires the KIX domain of CBP for cellular function and, consistent with this mechanism, 1 can competitively inhibit the ability of the native KIX-dependent activators Jun and MLL to function. Investigation of analogs of 1 lead to the identification of two additional Jun and MLL-inhibitors with improved potency. These inhibitors will be useful tools for further dissecting the role of the KIX domain in a variety of physiological and pathological processes either alone or in combination with molecules that target other sites within CBP and/or its HAT activity. 5a,6e,25 The answers to these questions will further define the role of CBP in small molecule-mediated gene expression and provide a platform for development of new classes of small molecule transcriptional modulators.
Materials and Methods
Plasmids
The murine-CBP expression plasmid pRC/RSV-m CBP-HA26 and the 12S E1A expression plasmid, pBabe 12S E1A27 were purchased from Add gene. The pCI-KIX-NLS plasmid was constructed by PCR amplification of the KIX domain (residues 586-672) from the previously described pRSET-B-His6-KIX(586-672) expression plasmid by using the PCR primers (5′-CTGTCGACGGTGTTCGAAAAGGCTGGCA-3′ and 5′-CAAGGCGGCCGCCTATAAACGTGACCTCCGC-3′).6c The PCR product was digested with Not1 and Sal1 and ligated into the multiple cloning site of the pCI mammalian expression vector (Promega) using standard molecular biology techniques. The SV40 nuclear localization sequence (126-132) was subsequently incorporated into pCI-KIX plasmid C-terminal to the KIX domain via site-directed mutagenesis (Quik-change kit, Stratagene). Plasmid Gal4-MLL(2829-2883) was constructed by PCR amplification of the MLL minimal activation domain (residues 2829-2883)17d from a full length MLL plasmid containing an N-terminal flag tag, (F-MLL, a gift from Dr. Jay Hess) using PCR primers (5′-GACTGGATCCCTGAAATCAGATTCAGACAATAAC-3′ and 5′-CAAGGCGGCCGCAAGACCCAATCCTTCACCAAG-3′. The PCR product was digested with BamH1 and Not1 and ligated into the plasmid pCMV-Gal4 using standard molecular biology techniques. The plasmid pTKGG encoding the DNA binding domain of Gal4 fused to the minimal ligand binding domain of the glucocorticoid receptor was the generous gift of Dr. Tom Kodadek. The plasmid encoding the Jun TAD fused to the Gal4 DBD and the luciferase reporter plasmids, pG5luc and pRLSV40, were purchased from Promega. The CBP shRNA construct was prepared as previously described.16
Transcriptional inhibition assay
HeLa cells (10,000 per well) were transfected with luciferase reporter and Renilla control plasmids in addition to a plasmid encoding for either the MLL or Jun activation domain fused to the DNA-binding domain of Gal4. Compound was added four hours post-transfection and the luminescence was assessed after 24h as described above.
Western blot
HeLa cells (10000 per well) were lysed using passive lysis buffer (Promega) with Complete®, EDTA-free protease inhibitors (Invitrogen) for 10 minutes at room temperature on an orbital shaker. Lysates were centrifuged at 14000 rpm for 20 minutes and the supernatant was mixed with 4× loading dye (Invitrogen) and BME (final concentration of 1%). The samples were then heated at 95°C for 10 minutes, separated by SDS-PAGE, transferred to a PVDF membrane and probed using standard conditions. Jun (sc-1694, 1:1000), cyclin D1 (sc-718, 1:1000), CBP (sc-7300, 1:500), and GAPDH (sc-47724, 1:2000) antibodies were all purchased from Santa Cruz Biotechnology.
Supplementary Material
Acknowledgments
A.K.M. is grateful to the NIH (CA140667), Novartis (Novartis Young Investigator Award), and the NSF (PECASE) for support of this work. W.C.P. is thankful to the NIH for a postdoctoral fellowship (F32 GM090550). C.A.B. was supported by the Pharmacological Sciences Training Program (GM007767). We thank Dr. Ryan Casey for the gift of compound 5 and Mr. Chris Taylor for a sample of isoxazolidine 3. We appreciate Jeff Kampf’s assistance with obtaining the solid state structure of 4.
References
- 1.Verdine GL, Walensky LD. Clin Cancer Res. 2007;13:7264–7270. doi: 10.1158/1078-0432.CCR-07-2184. [DOI] [PubMed] [Google Scholar]
- 2.(a) Pandolfi PP. Oncogene. 2001;20:3116–3127. doi: 10.1038/sj.onc.1204299. [DOI] [PubMed] [Google Scholar]; (b) Darnell JE., Jr Nat Rev Cancer. 2002;2:740749. doi: 10.1038/nrc906. [DOI] [PubMed] [Google Scholar]; (c) Koh JT, Zheng J. ACS Chem Biol. 2007;2:599–601. doi: 10.1021/cb700183s. [DOI] [PubMed] [Google Scholar]; (d) Koehler AN. Curr Opin in Chem Bio. 2010;14:331–340. doi: 10.1016/j.cbpa.2010.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee LW, Mapp AK. J Biol Chem. 2010;285:11033–11038. doi: 10.1074/jbc.R109.075044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mapp AK, Ansari AZ. ACS Chem Biol. 2007;2:62–75. doi: 10.1021/cb600463w. [DOI] [PubMed] [Google Scholar]
- 5.(a) Vo N, Goodman RH. J Biol Chem. 2001;276:13505–13508. doi: 10.1074/jbc.R000025200. [DOI] [PubMed] [Google Scholar]; (b) Goodman RH, Smolik S. Genes Dev. 2000;14:1553–1577. [PubMed] [Google Scholar]
- 6.(a) Kung AL, Zabludoff SD, France DS, Freedman SJ, Tanner EA, Vieira A, Cornell-Kennon S, Lee J, Wang B, Wang J, Memmert K, Naegeli HU, Petersen F, Eck MJ, Bair KW, Wood AW, Livingston DM. Cancer Cell. 2004;6:33–43. doi: 10.1016/j.ccr.2004.06.009. [DOI] [PubMed] [Google Scholar]; (b) Liu B, Alluri PG, Yu P, Kodadek T. J Am Chem Soc. 2005;127:8254–8255. doi: 10.1021/ja0515295. [DOI] [PubMed] [Google Scholar]; (c) Buhrlage SJ, Bates CA, Rowe SP, Minter AR, Brennan BB, Majmudar CY, Wemmer DE, Al-Hashimi H, Mapp AK. ACS Chem Biol. 2009;4:335–344. doi: 10.1021/cb900028j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Cebrat M, Kim CM, Thompson PR, Daugherty M, Cole PA. Bioorg Med Chem. 2003;11:3307–3313. doi: 10.1016/s0968-0896(03)00265-7. [DOI] [PubMed] [Google Scholar]; (e) Best JL, Amezcua CA, Mayr B, Flechner L, Murawsky CM, Emerson B, Zor T, Gardner KH, Montminy M. Proc Natl Acad Sci USA. 2004;101:17622–17627. doi: 10.1073/pnas.0406374101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Henchey LK, Kushal S, Dubey R, Chapman RN, Olenyuk BZ, Arora PS. J Am Chem Soc. 2010;132:941–943. doi: 10.1021/ja9082864. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Li BBX, Xiao XS. Chem Bio Chem. 2009;10:2721–2724. [Google Scholar]
- 7.Radhakrishnan I, Perez-Alvarado GC, Parker D, Dyson HJ, Montminy MR, Wright PE. Cell. 1997;91:741–752. doi: 10.1016/s0092-8674(00)80463-8. [DOI] [PubMed] [Google Scholar]
- 8.(a) Radhakrishnan I, Perez-Alvarado GC, Parker D, Dyson HJ, Montminy MR, Wright PE. J Mol Biol. 1999;287:859–865. doi: 10.1006/jmbi.1999.2658. [DOI] [PubMed] [Google Scholar]; (b) Goto NK, Zor T, Martinez-Yamout M, Dyson HJ, Wright PE. J Biol Chem. 2002;277:43168–43174. doi: 10.1074/jbc.M207660200. [DOI] [PubMed] [Google Scholar]; (c) Zor T, De Guzman RN, Dyson HJ, Wright PE. J Mol Biol. 2004;337:521–534. doi: 10.1016/j.jmb.2004.01.038. [DOI] [PubMed] [Google Scholar]; (d) Campbell KM, Lumb KJ. Biochemistry. 2002;41:13956–13946. doi: 10.1021/bi026222m. [DOI] [PubMed] [Google Scholar]; (e) Vendel AC, Lumb KJ. Biochemistry. 2004;43:904–908. doi: 10.1021/bi035612l. [DOI] [PubMed] [Google Scholar]; (f) Vendel AC, McBryant SJ, Lumb K. J Biochemistry. 2003:12481–7. doi: 10.1021/bi0353023. [DOI] [PubMed] [Google Scholar]
- 9.Frangioni JV, LaRiccia LM, Cantley LC, Montminy MR. Nat Biotechnol. 2000;18:1080–1085. doi: 10.1038/80280. [DOI] [PubMed] [Google Scholar]
- 10.Rowe SP, Mapp AK. Biopolymers. 2008;89:578–581. doi: 10.1002/bip.20946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Iyer NG, Ozdag H, Caldas C. Oncogene. 2004;23:4225–4231. doi: 10.1038/sj.onc.1207118. [DOI] [PubMed] [Google Scholar]; (b) Kasper LH, Fukuyama T, Biesen MA, Boussouar F, Tong CL, de Pauw A, Murray PJ, van Deursen JMA, Brindle PK. Mol Cell Biol. 2006;26:789–809. doi: 10.1128/MCB.26.3.789-809.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Iyer NG, Xian J, Chin SF, Bannister AJ, Daigo Y, Aparicio S, Kouzarides T, Caldas C. Oncogene. 2007;26:21–29. doi: 10.1038/sj.onc.1209771. [DOI] [PubMed] [Google Scholar]; (d) Kimbrel EA, Lemieux ME, Xia XB, Davis TN, Rebel VI, Kung AL. Blood. 2009;114:4804–4812. doi: 10.1182/blood-2009-04-217794. [DOI] [PubMed] [Google Scholar]
- 12.(a) Kinoshita I, Leaner V, Katabami M, Manzano RG, Dent P, Sabichi A, Birrer MJ. Oncogene. 2003;22:2710–2722. doi: 10.1038/sj.onc.1206371. [DOI] [PubMed] [Google Scholar]; (b) Hanson RD, Yu BD, Hess J, Korsmeyer SJ. Blood. 1997;90:184–194. [PubMed] [Google Scholar]; (c) Li JJ, Cao Y, Young MR, Colburn NH. Mol Carcinogen. 2000;29:159–169. doi: 10.1002/1098-2744(200011)29:3<159::aid-mc5>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]; (d) Ayton PM, Cleary ML. Gene Dev. 2003;17:2298–2307. doi: 10.1101/gad.1111603. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Leaner VD, Kinoshita I, Birrer MJ. Oncogene. 2003;22:5619–5629. doi: 10.1038/sj.onc.1206644. [DOI] [PubMed] [Google Scholar]; (f) Wisdom R, Johnson RS, Moore C. Embo J. 1999;18:188–197. doi: 10.1093/emboj/18.1.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Minter AR, Brennan BB, Mapp AK. J Am Chem Soc. 2004;126:10504–10505. doi: 10.1021/ja0473889. [DOI] [PubMed] [Google Scholar]; (b) Buhrlage SJ, Brennan BB, Minter AR, Mapp AK. J Am Chem Soc. 2005;127:12456, 12457. doi: 10.1021/ja0536567. [DOI] [PubMed] [Google Scholar]; (c) Rowe SP, Casey RJ, Brennan BB, Buhrlage SJ, Mapp AK. J Am Chem Soc. 2007;129:10654–10655. doi: 10.1021/ja0736865. [DOI] [PubMed] [Google Scholar]
- 14.(a) Ptashne M, Gann A. Nature. 1997;386:569–577. doi: 10.1038/386569a0. [DOI] [PubMed] [Google Scholar]; (b) Ansari AZ, Mapp AK, Nguyen DH, Dervan PB, Ptashne M. Chem Biol. 2001;8:583–592. doi: 10.1016/s1074-5521(01)00037-0. [DOI] [PubMed] [Google Scholar]
- 15.De Guzman RN, Goto NK, Dyson HJ, Wright PE. J Mol Biol. 2006;355:1005–1013. doi: 10.1016/j.jmb.2005.09.059. [DOI] [PubMed] [Google Scholar]
- 16.Naidu SR, Love IM, Imbalzano AN, Grossman SR, Androphy EJ. Oncogene. 2009;28:2492–2501. doi: 10.1038/onc.2009.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Bannister AJ, Kouzarides T. Embo J. 1995;14:4758–4762. doi: 10.1002/j.1460-2075.1995.tb00157.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Green M, Panesar NK, Loewenstein PM. Oncogene. 2008;27:4446–4455. doi: 10.1038/onc.2008.85. [DOI] [PubMed] [Google Scholar]; (c) Peter Pelka JNGA, Torchia Joseph, Turnell Andrew S, Grand Roger JA, Joe S. Mymryk Nuc Acids Res. 2009;37(12):1095–1106. doi: 10.1093/nar/gkn1057. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ernst P, Wang J, Huang M, Goodman RH, Korsmeyer SJ. Mol Cell Biol. 2001;21:2249–2258. doi: 10.1128/MCB.21.7.2249-2258.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giebler HA, Loring JE, van Orden K, Colgin MA, Garrus JE, Escudero KW, Brauweiler A, Nyborg JK. Mol Cell Biol. 1997;17:5156–5164. doi: 10.1128/mcb.17.9.5156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee LW, Taylor CEC, Desaulniers JP, Zhang MC, Hojfeldt JW, Pan Q, Mapp AK. Bioorg Med Chem Lett. 2009;19:6233–6236. doi: 10.1016/j.bmcl.2009.08.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chang CH, Scott GK, Kuo WL, Xiong XH, Suzdaltseva Y, Park JW, Sayre P, Erny K, Collins C, Gray JW, Benz CC. Oncogene. 1997;14:1617–1622. doi: 10.1038/sj.onc.1200978. [DOI] [PubMed] [Google Scholar]
- 21.(a) Kim JK, Diehl JA. J of Cell Phys. 2009;220:292–296. doi: 10.1002/jcp.21791. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fu MF, Wang CG, Li ZP, Sakamaki T, Pestell RG. Endocrinology. 2004;145:5439–5447. doi: 10.1210/en.2004-0959. [DOI] [PubMed] [Google Scholar]; (c) Arnold A, Papanikolaou A. J Clin Oncol. 2005;23:4215–4224. doi: 10.1200/JCO.2005.05.064. [DOI] [PubMed] [Google Scholar]; (d) Tashiro E, Tsuchiya A, Imoto M. Cancer Sci. 2007;98:629–635. doi: 10.1111/j.1349-7006.2007.00449.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Stacey DW. Curr Opin in Cell Biol. 2003;15:158–163. doi: 10.1016/s0955-0674(03)00008-5. [DOI] [PubMed] [Google Scholar]
- 22.(a) Albanese C, D’Amico M, Reutens AT, Fu M, Watanabe G, Lee RJ, Kitsis RN, Henglein B, Avantaggiati M, Somasundaram K, Thimmapaya B, Pestell RG. J Biol Chem. 1999;274:34186–34195. doi: 10.1074/jbc.274.48.34186. [DOI] [PubMed] [Google Scholar]; (b) Bakiri L, Lallemand D, Bossy-Wetzel E, Yaniv M. Embo J. 2000;19:2056–2068. doi: 10.1093/emboj/19.9.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Amanatullah DF, Zafonte BT, Albanese C, Fu MF, Messiers C, Hassell J, Pestell RG. Methods in Enzymology. 2001;333:116–127. doi: 10.1016/s0076-6879(01)33050-1. [DOI] [PubMed] [Google Scholar]
- 23.Casey RJ, Desaulniers JP, Hojfeldt JW, Mapp AK. Bioorgan Med Chem. 2009;17:1034–1043. doi: 10.1016/j.bmc.2008.02.045. [DOI] [PubMed] [Google Scholar]
- 24.(a) Cochran AG. Chem Biol. 2000;7:R85–R94. doi: 10.1016/s1074-5521(00)00106-x. [DOI] [PubMed] [Google Scholar]; (b) Berg T. Angewandte Chemie-International Edition. 2003;42:2462–2481. doi: 10.1002/anie.200200558. [DOI] [PubMed] [Google Scholar]; (c) Arkin MR, Wells JA. Nat Rev Drug Disc. 2004;3:301–317. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]; (d) Fletcher S, Hamilton AD. Curr Opin Chem Biol. 2005;9:632–638. doi: 10.1016/j.cbpa.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 25.(a) Bannister AJ, Kouzarides T. Nature. 1996;384:641–643. doi: 10.1038/384641a0. [DOI] [PubMed] [Google Scholar]; (b) Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y. Cell. 1996;87:953–959. doi: 10.1016/s0092-8674(00)82001-2. [DOI] [PubMed] [Google Scholar]; (c) Martinez-Balbas Marian A, AJB, Martin Klaus, Haus-Seuffert Phillip, Meisterernst Michael, Kouzarides Tony. EMBO J. 1998;17(6):2886–2893. doi: 10.1093/emboj/17.10.2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chrivia JC, Kwok RPS, Lamb N, Hagiwara M, Montminy MR, Goodman RH. Nature. 1993;365:855–859. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- 27.Samuelson AV, Lowe SW. Proc Nat Acad Sci USA. 1997;94:12094–12099. doi: 10.1073/pnas.94.22.12094. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.