

Abstract
We measured myosin crossbridge detachment rate and the rates of MgADP release and MgATP binding in mouse and rat myocardial strips bearing one of the two cardiac myosin heavy chain (MyHC) isoforms. Mice and rats were fed an iodine-deficient, propylthiouracil diet resulting in ~100% expression of β-MyHC in the ventricles. Ventricles of control animals expressed ~100% α-MyHC. Chemically-skinned myocardial strips prepared from papillary muscle were subjected to sinusoidal length perturbation analysis at maximum calcium activation pCa 4.8 and 17°C. Frequency characteristics of myocardial viscoelasticity were used to calculate crossbridge detachment rate over 0.01 to 5 mM [MgATP]. The rate of MgADP release, equivalent to the asymptotic value of crossbridge detachment rate at high MgATP, was highest in mouse α-MyHC (111.4±6.2 s−1) followed by rat α-MyHC (65.0±7.3 s−1), mouse β-MyHC (24.3±1.8 s−1) and rat β-MyHC (15.5±0.8 s−1). The rate of MgATP binding was highest in mouse α-MyHC (325±32 mM−1.s−1) then mouse β-MyHC (152±23 mM−1.s−1), rat α-MyHC (108±10 mM−1.s−1) and rat β-MyHC (55±6 mM−1.s−1). Because the events of MgADP release and MgATP binding occur in a post power-stroke state of the myosin crossbridge, we infer that MgATP release and MgATP binding must be regulated by isoform- and species-specific structural differences located outside the nucleotide binding pocket, which is identical in sequence for these four myosins. We postulate that differences in the stiffness profile of the entire myosin molecule, including the thick filament and the myosin-actin interface, are primarily responsible for determining the strain on the nucleotide binding pocket and the subsequent differences in the rates of nucleotide release and binding observed among the four myosins examined here.
Keywords: PTU, sinusoidal analysis, time-on
1. INTRODUCTION
Relative expression of the two cardiac myosin heavy chain (MyHC) isoforms, α- and β-MyHC, in the adult mammalian ventricles is closely related to species, heart rate and heart size [1]. Ventricles of mice, rats and other rodents predominately express α-MyHC, while ventricles of rabbits and larger animals predominately express β-MyHC [1, 2]. Chemically-skinned myocardium containing mainly the α-MyHC isoform demonstrates higher ATPase rate, faster velocity of sarcomere shortening, higher power production and greater rate of force redevelopment after a quick stretch than myocardium containing mainly β-MyHC [3–14]. At the molecular level, isolated α-MyHC is characterized by a higher ATPase rate, faster velocity of actin motility and shorter crossbridge lifetime compared to β-MyHC of the same species [2, 8, 9, 15–21], yet the two cardiac isoforms demonstrate a similar unitary force and lever arm displacement [18, 20]. Differences in ATPase rate and crossbridge lifetimes, which are detected or inferred from assays using isolated myosin, appear to underlie the distinct differences in energy consumption and mechanical performance at the level of the organized sarcomere [17, 22–24].
Significant progress has been made in elucidating structural differences that further distinguish the two cardiac isoforms [8, 25]. However, thus far there is no definitive understanding of the structural bases of cardiac myosin function [8]. To emphasize the difficulty of this problem, consider that there is a very high degree of MyHC isoform homology (>99%) between species (including isoforms of skeletal muscle), yet any given isoform from a small species consistently demonstrates faster crossbridge kinetics compared to the same isoform of a larger species [2, 26–28]. While differences in myosin kinetics observed with isolated myosin may be reasonably considered to be due to myosin amino acid sequence alone, it is not known to what degree the intact sarcomere contributes to isoform-specific and species-specific differences in myosin kinetics. We hypothesized that the constituent proteins and organizational structure of the intact cardiac sarcomere influence ensemble myosin crossbridge kinetics in a manner that would more accurately represent the physiologically relevant kinetic differences due to isoform and species not observable with isolated myosin.
Myosin crossbridge formation underlies in large part the frequency dependent viscoelastic characteristics of vertebrate muscle preparations that preserve myofilament lattice structure [29–34]. The interpretation of viscoelastic characteristics in terms of myosin crossbridge kinetics, however, has not been universal. Kawai and others have utilized a model of strain-dependent myosin crossbridge recruitment that relies upon known biochemistry of the crossbridge cycle [31, 32, 34]. This model does not consider that the deformation of strongly bound myosin crossbridges during length perturbation could significantly contribute to the recorded viscoelasticity. Ter Keurs and colleagues have developed a more extensive model to include calcium activation of the thin filament and passive viscoelasticity [30, 33]. Campbell and colleagues have modeled elastic force due to crossbridge deformation as a contributor to muscle viscoelasticity and have demonstrated that viscoelastic characteristics at the highest frequencies are predominately due to this distortion component [29, 35]. We have produced an explicit model of the mechanical consequences of myosin crossbridges undergoing strain during length perturbation and have demonstrated that the mean myosin crossbridge lifetime can be deduced from analysis of the highest frequencies of the viscoelastic properties of striated muscle [36].
Based on our ability to estimate the mean myosin crossbridge lifetime (ton) at the level of the skinned myocardial strip [36], we investigated differences in MgATP-dependent myosin kinetics and myocardial mechanical performance due to the two cardiac isoforms in mice and rats. We present here the MgATP-dependence of the myosin crossbridge ton in myocardial strips containing predominately α-MyHC or β-MyHC. We found that at saturating MgATP ton is shortest, and therefore MgADP release rate is fastest, for mouse α-MyHC followed by rat α-MyHC, mouse β-MyHC and rat β-MyHC. Our findings for MgADP release rate qualitatively corroborate findings based on laser trap experiments and in vitro motility assays, which suggest a shorter ton for α-MyHC compared with β-MyHC [18]. We report, however, a four-fold difference in crossbridge detachment rate and ton between the isoforms compared to the two-fold difference most often reported for isolated myosin [2, 18, 19]. Our findings suggest that myosin kinetics depend significantly on the presence of an intact myofilament lattice structure and the mechanical stresses borne by the S1 head therein.
2. METHODS
2.1 Animal Models
All procedures were reviewed and approved by the Institutional Animal Care and Use Committee of The University of Vermont College of Medicine and complied with the Guide for the Use and Care of Laboratory Animals published by the National Institutes of Health. Male mice of 129/SvEv strain (SvEv) were purchased from Taconic Farms (Germantown, NY). Male Wistar-Kyoto rats (WKY) were obtained from Harlan Sprague-Dawley (Indianapolis, IN). Half of these animals were fed an iodine-deficient 0.15% propylthiouracil (PTU) diet (Harlan Teklan, Indianapolis, IN) for at least 10 weeks, resulting in hypothyroidism and a complete shift to β-MyHC expressed in the LV. Control SvEv mice, which normally express α-MyHC in the LV, were untreated. Control WKY rats were injected with 6 mg/kg L-thyroxine (T-2501, Sigma Aldrich) daily for 7 days, which induced hyperthyroidism and up-regulation of α-MyHC. Transgenic mice overexpressing β-MyHC in the FVB strain background (FVBβTG) were provided by Dr. Jeffrey Robbins at the University of Cincinnati Children’s Hospital [8].
2.2 Myosin Isoform and Protein Phosphorylation
Myosin isoform content in the LV was determined by the method of Reiser and Kline [37] with the use of Fluormax-2 Imaging analysis (Bio-Rad, Hercules CA). Relative isoform content was quantified by densitometry using ImageJ v1.38 (National Institutes of Health, USA). Protein phosphorylation stain (Pro-Q diamond, Invitrogen) and Western blots for myosin regulatory light chain (MLC2) content (cat. ab92721, Abcam, San Francisco, CA), MLC2 phosphorylation at Serine-19 (cat. ab2480, Abcam), cardiac troponin-I phosphorylation (cTnI) at Serines-23,24 (cat. #4404, Cell Signaling) were performed using tissue homogenates prepared in (mmol/L) 300 KCl, 25 Imidazole, 5 MgCl2, 2 EGTA, 10 DTT and pH 7.4. A myofilament fraction was then extracted by centrifugation or using chemically skinned myocardial strips described below.
2.3 Solutions for Skinned Strips
Chemicals and reagents were obtained from Sigma Aldrich (St. Louis, MO) and concentrations are given in mM unless otherwise noted. Relaxing solution: pCa 8.0, 5 EGTA, 5 MgATP, 1 Mg2+, 35 phosphocreatine (PCr), 300 U/mL creatine kinase (CK), ionic strength 200, pH 7.0. Activation solution: same as relaxing with pCa 4.0. Rigor solution: same as relaxing with pCa 4.8 and no MgATP, PCr and CK. Skinning solution: same as relaxing with 30 2,3-butanedione monoxime (BDM), 1% Triton-X100 wt/vol and subsequently 50% glycerol wt/vol. Storage solution: same as relaxing with 30 BDM, 10 μg/mL leupeptin and 50% glycerol wt/vol. Alkaline phosphatase bathing solution consisted of relaxing solution with 6 U/mL recombinant alkaline phosphatase (P-4252, Sigma Aldrich).
2.4 Skinned Myocardial Strips
Skinned myocardial strips were studied as previously described [36, 38]. Immediately before mechanical analysis, strips underwent 20 min incubation in 6 U/mL alkaline phosphatase at room temperature. Strip were then mounted between a piezoelectric motor and a strain gauge, lowered into a 30 μL droplet of relaxing solution (pCa 8.0) maintained at 17°C, and incrementally stretched to 2.2 μm sarcomere length measured by digital Fourier Transform. Strips were calcium activated from pCa 8.0 to pCa 4.8, and various MgATP concentrations were applied at pCa 4.8 by adding rigor solution.
Recorded forces were normalized to cross-sectional area to provide isometric tension (T). Individual recordings of T minus relaxed tension (Tmin) were normalized to maximum tension (Tmax) minus Tmin and fit to the Hill equation:
| (Equation 1) |
where [Ca2+]50 = calcium concentration at half activation, and nHill = Hill coefficient using a nonlinear least squares algorithm. We also report pCa50 = −log [Ca2+]50.
2.5 Sinusoidal Length Perturbation Analysis
Sinusoidal length perturbations of amplitude 0.125% strip length were applied at 0.125–100 Hz as previously described [36, 38]. The elastic and viscous moduli, E(ω) and V(ω), respectively, were measured from the in-phase and out-of-phase portions of the tension response to length perturbation. A complex modulus, Y(ω), was defined as E(ω) + iV(ω), where i = √−1. The fitting of the frequency dependence of the complex moduli to a mathematical model (Equation 2) provided estimates of six model parameters (A, k, B, 2πb, C, 2πc), and myosin ton as was calculated as (2πc)−1 [36]. Refer to Supplemental Material for further explanation and interpretation of the other parameters.
| (Equation 2) |
2.6 Myosin Enzyme Kinetics
We assume the total myosin crossbridge lifetime, ton, is represented adequately as the sum of two time periods: time to release ADP, t−ADP, and time to bind ATP, t+ATP after Warshaw and colleagues [18, 24]. Noting the myosin crossbridge detachment rate, 2πc, is the reciprocal of the mean ton and the two intermediate time periods can be represented as the reciprocals of the respective rate constants, we can represent the dependence of the myosin crossbridge detachment rate in terms of MgATP concentration ([MgATP]) as follows:
| (Equation 3) |
where k−ADP = rate of ADP release and also the asymptotically maximum myosin crossbridge detachment rate (s−1), k+ATP = rate of MgATP binding per [MgATP] (M−1.s−1), and the ratio (k−ADP/k+ATP) = concentration of MgATP producing half the maximum detachment rate ([MgATP]50).
2.7 Analysis
At least two skinned strips were examined per condition per animal. Multiple measurements from the same heart were averaged to provide a single measure for that heart. All data are presented as mean ± SEM, and n refers to number of hearts. Statistical significance is reported at P<0.05 and P<0.01. Trends at P<0.1 are also reported if in support of similarly statistically significant parameters. Student’s t-test was used to compare values between groups defined by myosin isoform (e.g., α-MyHC vs. β-MyHC). Two-way analysis of variance was used to test for differential effects of alkaline phosphatase between groups on parameters of tension-pCa relationships.
3. RESULTS
3.1 Animal Characteristics
Hypothyroidism resulted in the complete replacement of α-MyHC with β-MyHC in the LV of both mice and rats (Figure 1). Euthyroid mice and hyperthyroid rats exhibited ~100% expression of α-MyHC. The FVBβTG mice expressed ~70% β-MyHC by densitometry (69.6±1.4%) consistent with previous findings [8].
Figure 1.
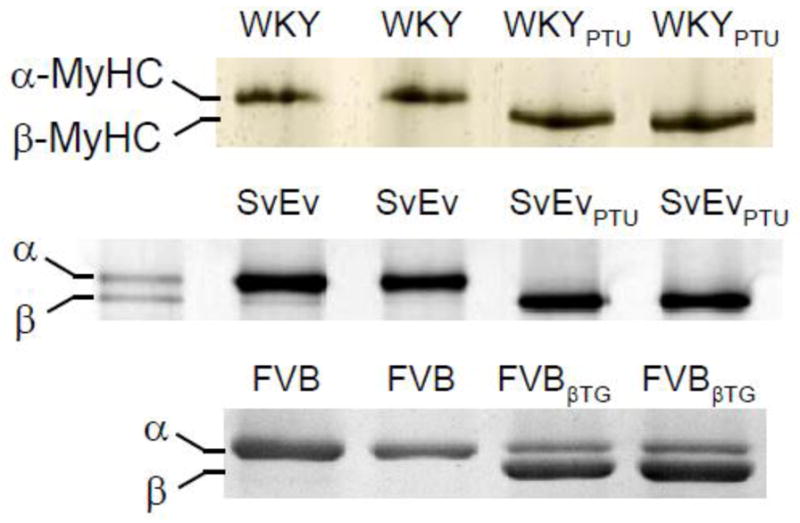
Characteristics of cardiac myofibrillar proteins. Each lane represents one heart, and examples from two hearts are shown for each population. Gel electrophoresis stained with Coomassie-Blue demonstrated ~100% α-MyHC expression in hyperthyroid Wistar Kyoto (WKY) rats and normal 129/SvEv wildtype mice (SvEv), while ~100% β-MyHC was expressed in the PTU-fed WKY rats and SvEv mice. Transgenic mice expressing ~70% β-MyHC (FVBβTG) were compared against non-transgenic FVB controls expressing 100% α-MyHC to test for any potential complications of PTU diet.
3.2 Tension-pCa relationship
Figure 2 illustrates the tension-pCa relationships for SvEv and SvEvPTU mouse myocardial strips with and without AP treatment. Without AP treatment, strips from SvEvPTU mice were less sensitive to calcium activation as indicated by a rightward shift in the tension-pCa relationship for SvEvPTU strips relative to SvEv controls (Figure 2A). A similar reduction in thin filament calcium sensitivity after PTU treatment has been reported previously [22], although no shift in calcium sensitivity has also been reported [6, 9]. With AP treatment, the tension-pCa relationships were similar between the PTU-fed mice and euthyroid controls (Figure 2B).
Figure 2.

Normalized tension-pCa relationships without and with alkaline phosphatase (AP) treatment. A. Thin filament calcium sensitivity was reduced due to PTU diet in myocardial strips not treated with AP. B. After AP treatment, thin filament calcium sensitivity was normalized to that of nonPTU-fed mice. The remainder of this study focused on AP treated myofilaments. C. AP treatment was found to reduce phosphorylation of cardiac troponin-I (cTnI) at Serines-23,24 in both normal and hypothyroid SvEv mice. Ponceau stain is used to detect total protein content on nitrocellulose prior to Western blot. D. Phosphorylation of cardiac regulatory light chain (MLC2) at Serine-19 appeared to be reduced by BDM exposure in the skinned strips compared to homogenate (hom) [51] and reduced further by AP treatment.
Effects of AP treatment on myofilament protein phosphorylation were too subtle or site-specific to be detected by Pro-Q diamond stain (See Supplemental Material), yet AP treatment reduced the phosphorylation content of cTnI at Serines-23,24 and MLC2 at Serine-19 (Figures 2C and 2D). The phosphorylation status of other proteins, such as troponin-T, myosin binding protein-C and titin, and other sites on cTnI were also likely affected by AP treatment, but not investigated here.
The differential effects of AP treatment on mechanical properties of the mouse groups were demonstrated through analysis of variance (Table 1). AP treatment significantly lowered relaxed tension, Tmin, in both SvEv and SvEvPTU groups as indicated by a significant AP main effect. AP treatment also enhanced the developed tension in SvEvPTU but not in the SvEv, as indicated by the significant PTU×AP interaction for Tdev; in other words, only after AP treatment was Tdev higher in the SvEvPTU compared to SvEv. Thin filament calcium sensitivity measured as 50% calcium activation in both absolute or logarithmic terms was significantly lower in SvEvPTU compared to SvEv controls without AP treatment. With AP treatment, however, differences in thin filament calcium sensitivity were abolished (Table 1).
Table 1.
Characteristics of tension-pCa relationships in mouse myocardium due to PTU diet and alkaline phosphatase (−AP vs +AP) treatment. Results of repeated-measures ANOVA are presented as PTU effect, AP effect or PTU×AP interaction at P<0.05 level. Tdev = Tmax–Tmin.
| SvEv (n=7) −AP | SvEvPTU (6) −AP | SvEv (7) +AP | SvEvPTU (6) +AP | ANOVA | |
|---|---|---|---|---|---|
| Tmin (mN.mm−2) | 4.04±0.36 | 3.03±0.52 | 2.67±0.29 | 2.60±0.30 | AP |
| Tmax (mN.mm−2) | 22.6±1.6 | 23.7±1.3 | 19.9±1.5 | 29.5±1.9† | PTU, PTU×AP |
| Tdev (mN.mm−2) | 18.6±1.5 | 20.7±1.4 | 17.3±1.4 | 26.9±1.8† | PTU, PTU×AP |
| [Ca2+]50 (μM) | 1.59±0.10 | 1.87±0.07 * | 1.67±0.07 | 1.63±0.06 | |
| pCa50 | 5.80±0.03 | 5.73±0.02 * | 5.78±0.02 | 5.79±0.02 | |
| nHill | 3.81±0.17 | 4.33±0.18# | 3.79±0.14 | 4.21±0.17# | PTU |
Differences reported at
P<0.10,
P<0.05,
P<0.01 by t-test against nonPTU-fed controls of same AP treatment.
These results suggest that at least some of the differential functional effects of hypothyroidism related to myofilament protein phosphorylation status, most notably that of the cTnI (Figure 2C), were ameliorated by AP treatment. For the remainder of this study, comparisons of the functional and kinetic characteristics between α-MyHC and β-MyHC were performed with strips having undergone AP treatment to better control for any differences in site-specific phosphorylation profiles that may have arisen from manipulation of thyroid hormone.
3.3 Complex Modulus
Sinusoidal length perturbation analysis was used to examine the effects of mouse cardiac myosin isoform on myofilament viscoelastic properties and the mean myosin crossbridge lifetime. The elastic and viscous moduli were examined at relaxed and maximum calcium activated conditions and over several concentrations of MgATP in the range 0.01–5 mM. The elastic and viscous moduli measured under relaxed conditions were subtracted from those measured under activated conditions to focus the analyses on crossbridge-dependent viscoelasticity and the underlying myosin kinetics.
At saturating 5 mM MgATP and at frequencies roughly between 2–10 Hz, the elastic and viscous moduli of the WKYPTU strips containing β-MyHC were significantly higher in magnitude than those of the WKY containing α-MyHC (Figure 4A). These differences arise from the higher magnitude and lower frequency characteristics associated with the β-MyHC in the WKYPTU compared to α-MyHC in the WKY. Our findings for these moduli are qualitatively similar to those reported previously for rat myocardium [29, 39]. Figure 4B illustrates an even stronger distinction in the magnitudes and frequency characteristics of the elastic and viscous moduli between β-MyHC and α-MyHC in mouse myocardium.
Figure 4.

Sensitivity of myosin crossbridge detachment rate, 2πc, and time-on, ton, to MgATP. A–C. At all MgATP concentrations greater than 0.1 mM, myosin detachment rate was significantly higher for α-MyHC compared to β-MyHC in rats (panel A) and mice (panels B and C). Detachment rates for 70% β-MyHC in the FVBβTG mouse were lower than 100% α-MyHC, but not as low as 100% β-MyHC (Panel C, where dotted lines reflect curves from Panel B). D–F. At all MgATP concentrations greater than 0.1 mM, mean myosin crossbridge ton was shorter in the α-MyHC compared to β-MyHC. For the same isoform myosin crossbridge ton was shorter in mice compared to rats.
As MgATP concentration was lowered, moduli for both α- and β-MyHC exhibited lower frequency characteristics compared to moduli at 5 mM MgATP (Figures 4C and 4D). This shift to lower frequencies reflects the longer myosin crossbridge rigor state due to the relative scarcity of MgATP. The complex moduli at each MgATP concentration were fit with Equation 3, and the resultant model parameters are provided in the Supplemental Material. The results for MgATP dependency of the myosin crossbridge detachment rate, 2πc, and its reciprocal, ton, are presented below.
3.4 Rates of MgADP Release and MgATP Binding
Values for the kinetic model parameters of Equation 3 are presented in Table 2. The asymptotic maximum myosin detachment rate, k−ADP, in the WKYPTU was just 20–25% that in the WKY (illustrated in Figure 4A), indicating that rat α-MyHC detachment rate was at least four times faster than that of rat β-MyHC. The maximum myosin detachment rate in the mouse α-MyHC (SvEv) was also at least four times faster than that of mouse β-MyHC (SvEvPTU) (Figure 4B). These results in the intact myofilament lattice suggest a much higher MgADP release rate in α-MyHC compared to β-MyHC in both species than would have been expected from assays using isolated myosin, which exhibit a two-fold difference [2, 8, 15, 19, 21].
Table 2.
Variables of myosin enzyme kinetics model applied to myosin detachment rate 2πc vs MgATP relationships (Equation 3).
| WKY (n=4) 100% α | WKYPTU (4) 100% β | SvEv (7) 100% α | SvEvPTU (6) 100% β | FVB (8) 100% α | FVBβTG (4) 70% β-30% α | predicted100% β | |
|---|---|---|---|---|---|---|---|
| k−ADP (s−1) | 65.0±7.9 | 15.5±0.8* | 111.4±6.2 | 24.3±1.8† | 115.1±5.1 | 48.9±3.1† | 20.5 |
| k+ATP (mM−1.s−1) | 108±10 | 55±6* | 325±32 | 152±23† | 338±41 | 212±31* | 158 |
| t−ADP (ms) | 18.2±1.9 | 64.9±3.3† | 9.1±0.4 | 42.3±2.9† | 9.3±0.4 | 21.3±1.6† | 26.4 (48.7) |
| [MgATP]50 (μM) | 579±66 | 334±14* | 365±43 | 178±25† | 394±49 | 271±31# | 218 |
Mean time period of MgADP state, t−ADP = 1/ k−ADP, and MgATP concentration at half maximum detachment rate, [MgATP]50 = k−ADP/k+ATP, are also provided.
P<0.01,
P<0.05,
P<0.1 against 100% α-MyHC controls. Predicted values for 100% β-MyHC in the FVB strain were calculated as (FVBβTG – 0.3 × FVB) / 0.7; an alternative value for the predicted t−ADP, given in parentheses, is the reciprocal of the predicted k−ADP.
To remove any possible effects of hypothyroidism on myocardial tissue and sarcomeric structure, we examined MgATP dependency of myosin detachment rate using a transgenic mouse expressing ~70% β-MyHC, which preserves normal tissue and sarcomeric structure [8]. Values for k−ADP and k+ATP for α-MyHC in the FVB strain were remarkably similar to those for α-MyHC in the SvEv strain (Table 2). Values for these parameters in the FVBβTG expressing 70% β-MyHC fell between those values measured for 100% α-MyHC and 100% β-MyHC in the SvEv strain and tended to be closer to those for β-MyHC. While we are aware that the FVBβTG sarcomere incorporates roughly 49% β/β-homodimers, 9% α/α-homodimers and 42% α/β-heterodimers, each with kinetics that likely differ [40], we assumed a linear combination to predict values for k−ADP and k+ATP for 100% β-MyHC based on measurements in the FVBβTG and FVB. With an assumption that 70% of the value is due to β-MyHC and 30% due to α-MyHC in the FVBβTG, the predicted value for k−ADP for 100%β-MyHC was at least four-fold lower than that for 100% α-MyHC (Table 2). The predicted value for k+ATP for 100%β-MyHC was about half that for 100% α-MyHC. These results for FVBβTG suggest that the myosin isoform and its relative content were the most significant determinants of MgATP-dependent myosin crossbridge detachment rate observed here using myocardial viscoelasticity.
The MgATP binding rate per [MgATP], k+ATP, in the α-MyHC was at least double that of β-MyHC in both species, although lower in the rat compared to mouse (Table 2). While α-MyHC demonstrated a higher MgATP binding rate compared to β-MyHC, β-MyHC requires less MgATP to achieve its maximum detachment rate, as reflected in the lower MgATP50 for α-MyHC compared to β-MyHC in both species.
4. DISCUSSION
4.1 Isoform- and Species-Dependent Kinetics
Differences in mechanical performance between α-MyHC and β-MyHC have previously been examined using skinned myocardium of mice and rats [3–13] or isolated molecules from mice, rabbits and humans [2, 15, 18, 20, 21, 24]. In all these studies, α-MyHC elicits faster ATP turnover kinetics and higher measures of mechanical performance compared to β-MyHC of the same species. The most notable functional difference between the isoforms at the single molecule level is the myosin crossbridge lifetime [18, 20, 24], which at saturating MgATP concentrations is most dependent upon the MgADP release rate. We are aware of only one study that has made a direct comparison of MgADP release rates between cardiac isoforms [18]. In that study rabbit cardiac myosin was used to facilitate the detection of crossbridge binding events in the laser trap. The crossbridge lifetime of isolated mouse or rat cardiac myosin would presumably be shorter and would make technically difficult the detection of MgADP release rates in the laser trap. Our method of using viscoelastic characteristics of the myocardium to calculate crossbridge lifetime, ton, has no such limitation and has been used to detect ton as short at ~2 ms in mouse cardiac strips examined at 37°C [36].
We found in the present study that MgADP release rate was progressively slowed according to species and isoform in the following order: mouse α-MyHC > rat α-MyHC > mouse β-MyHC > rat β-MyHC. Our results reveal a four-fold difference in MgADP release rate between α-MyHC and β-MyHC in the intact sarcomere for both mouse and rat. Data from Rundell et al [9] in skinned myocardial strips with mixtures of the cardiac isoforms would predict a four-fold difference in ATPase between rat isoforms. Data from Suzuki et al. [38] would predict a 3.5-fold difference in ton between rabbit isoforms. Studies using isolated myosin on the other hand consistently report a two-fold difference in kinetics or ton between the two isoforms [2, 8, 15, 19, 21]. The disparate results between assays such as ours using an intact myofilament lattice and those using isolated myosin suggest that kinetic differences between the isoforms are blunted outside the structural context of the intact sarcomere.
Our data further demonstrate that MgATP binding rate in an intact sarcomere is different between isoforms and species. Previous estimates of MgATP binding rate using isolated myosin were not different between the isoforms [18, 41], yet we report here that the MgATP binding rate of α-MyHC is twice that of β-MyHC in the same species. Furthermore, the MgATP binding rate in mouse is nearly triple that in rat for the same isoform. These data suggest that MgATP binding rate in addition to MgADP release rate contributes to slower crossbridge kinetics in β-MyHC compared to α-MyHC and in the rat compare to mouse. We would expect these rates to be further slowed in species with lower heart rates.
4.2 Sequence Basis of Myosin Function
One might suspect differences in amino acid sequences at or near the nucleotide binding pocket to underlie these differences in rates of MgADP release and MgATP binding. However, the amino acid sequence of the nucleotide binding pocket and neighboring amino acids are thoroughly conserved among the myosins examined here (161 AYQYMLTDRE NQSILITGES GAGKTVNTKR VIQY 194). Nearby structures, such as Loop 1, point to structural differences between isoforms but not between species and therefore cannot explain the differences in MgADP release and MgATP binding rates between species [26]. We propose that constituent proteins and the organization of intact myofilament lattice significantly influence MgADP release and MgATP binding by imposing loads on the myosin crossbridge and thereby deforming the molecular structures within the mysoin head reponsible for nucleotide release and binding.
We would argue that the differences in MgADP release and MgATP binding rates among different myosins bearing a similar nucleotide binding pocket sequence suggest differential structural deformations of the nucleotide binding pocket. The differential structural deformations of the nucleotide binding pocket arise as a consequence of differential stiffness profiles of the larger myosin molecule, including the rod and myosin-actin interface. More specifically, the deformation profile of the myosin molecule in a post power-stroke state is inversely proportional to its stiffness profile. In this way, amino acids at any location in the myosin molecule can influence the deformation of the myosin nucleotide binding pocket and thereby also influence the release and binding kinetics. As discussed previously [15], more than 40 point mutations in the myosin rod have been identified as underlying familial cardiomyopathies in humans and should therefore be considered relevant to influencing myosin function through their effects on the myosin stiffness/deformation profile. A similar argument could be made regarding accessory sarcomeric proteins such as myosin binding protein-C and titin and the mechanical consequences of their post-translational modifications.
4.3 Control of Sarcomeric Protein Phosphorylation
The use of the skinned myocardial strip represents an important model system for detecting the kinetic and mechanical properties of the myosin crossbridge cycle in part because it preserves the myofilament lattice and surrounding myocardial structure, thus lending physiological structural significance to the findings. The myocardial strip is also in some cases preferable to isolated cardiomyocytes, because it is prepared without the use of proteases that can unpredictably undermine sarcomeric protein integrity and content.
The transgenic mouse model currently represents one of the most powerful tools to study the mechanical consequences of differences in amino acid sequences and genetic mutations in cardiac sarcomeric proteins. In the context of mouse models of disease or of protein mutations we have thought it important to control for protein phosphorylation profiles. In the current study, we found that hypothyroidism produced by PTU treatment not only affected MyHC isoform, but also elicited functional differences due to sarcomeric protein phosphorylation that were too subtle to detect by Pro-Q diamond stain, which detects total protein phosphorylation. Cardiac troponin-I and myosin binding protein-C are reportedly less phosphorylated with hypothyroidism [42]. We would infer that site specific phosphorylation of these or other proteins was differentially affected by hypothyroidism, therefore warranting AP treatment in order to compare kinetics between α-MyHC and β-MyHC. Given that AP treatment reduced Tmin in both populations, we suspect titin as at least one additional target protein for AP.
4.4 Limitations
The interpretation of viscoelastic properties of striated muscle in terms of myosin crossbridge kinetics is not complete. Kawai and colleagues have utilized a model based on strain-dependent crossbridge recruitment that provides some basis for interpreting the biochemistry of the crossbridge cycle, but there is no inclusion of elastic distortion of crossbridges as a contributor to viscoelasticity [31, 32]. Campbell’s model and data suggest that that crossbridge distortion contributes significantly to viscoelasticity at high frequencies, which corresponds to the C-process of Kawai [29, 39]. Our interpretation of the C-process is qualitatively similar to that of Campbell’s and allows us to detect the myosin crossbridge detachment rate and therefore mean crossbridge lifetime [36].
Our interpretation of viscoelastic properties of striated muscle relies solely upon consideration of the mechanical consequences of temporarily formed crossbridges during length perturbation, and we have not assigned specific biochemical states to a process or rate constant. The use of Equation 3 provides our link between the crossbridge detachment rate and MgATP-dependent biochemistry [18, 24]. Our subsequent interpretation of MgATP-dependent kinetics is valid to the degree to which Equation 3 represents the crossbridge detachment rate. It is possible, for example, that forcible myosin crossbridge detachment independent of MgADP release and MgATP binding could contribute to our measure of detachment rate and lead to an overestimate of k−ADP
4.5 Conclusion
The expression of α-MyHC is reduced in mammalian ventricles with development of any one of several physiological and pathological conditions including aging, sedentary behavior, hypothyroidism, diabetes, elevated systemic blood pressure and sarcomeric protein mutation or deletion [1, 23, 43, 44]. The mechanical consequences of this MyHC isoform shift includes a higher economy of force production (force/ATPase) [4, 16, 45] at the expense of lower velocity and power of contraction due to the greater amount of β-MyHC. The question remains, however, whether this isoform shift is compensatory or maladaptive to the underlying disease state [46, 47].
It has been suggested that the down-regulation of α-MyHC in human ventricles with disease is compensatory due in part to the higher economy of β-MyHC [16, 46, 48] or maladaptive due to the lower velocity and power, longer crossbridge lifetime and poorer relaxation properties of β-MyHC [8, 49, 50]. It can also be argued that a shift from an initially very low amount (<10%) to an undetectable amount of α-MyHC, as occurs with cardiovascular-linked diseases affecting human ventricular remodeling, is of minor physiological importance compared to the myriad of other means by which cardiac sarcomeric performance can be modified [38]. The present study does not resolve this question. Our study does, nevertheless, provide a mechanistic framework for linking isoform- and species-specific alterations in MgADP release and MgATP binding to differences in constituent proteins and organization structure between the mouse and rat cardiac sarcomere in addition to amino acid sequence differences between the α- and β-MyHC isoforms. Our results suggest that alterations in nucleotide release and binding rates originate not from structural differences near the nucleotide binding pocket, but from differences in deformation of this site that depend on the stiffness of the S1 head relative to that of the remainder of the myosin molecule, thick filament, myosin-actin interface and accessory proteins of the organized sarcomere.
Supplementary Material
Figure 3.

Elastic and viscous moduli vs frequency under maximum activated conditions at 5 mM and 0.25 mM MgATP. A. At 5 mM MgATP the characteristic dips and shoulders of the elastic and viscous moduli occur at lower frequencies due to β-MyHC in the PTU-fed rats compared to α-MyHC in the control rats. B. Mouse myocardial strips at 5 mM MgATP demonstrated similarly differential and exaggerated frequency characteristics between β-MyHC and α-MyHC. C and D. At 0.25 mM MgATP the frequency characteristics are shifted to lower frequencies compared to 5 mM MgATP. These characteristics are still lower in β-MyHC compared to α-MyHC and lower in rat (panel C) compared to mouse (panel D).
Highlights.
Myosin detachment rate of cardiac isoforms was detected in chemically skinned myocardium.
MgADP release rate was highest in mouse α-MyHC > rat α-MyHC > mouse β-MyHC > rat β-MyHC.
MgADP release rate was four-fold faster in α-MyHC compared to β-MyHC of same species.
MgATP binding rate was highest in mouse α-MyHC > mouse β-MyHC > rat α-MyHC > rat β-MyHC.
MgATP binding rate was two-fold faster in α-MyHC compared to β-MyHC of same species.
Acknowledgments
We are grateful to Dr. Susan Lowey for invaluable discussion throughout the completion of this work. This study was funded by NIH grants P01 HL59408 (YW, SMT, DWM, PV, JR, BMP), T32 HL007647 (BCWT) and R01 HL089944 (MML).
Footnotes
6. DISCLOSURES
There are no disclosures.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lompre AM, Mercadier JJ, Wisnewsky C, Bouveret P, Pantaloni C, D’Albis A, et al. Species- and age-dependent changes in the relative amounts of cardiac myosin isoenzymes in mammals. Developmental biology. 1981;84(2):286–90. doi: 10.1016/0012-1606(81)90396-1. [DOI] [PubMed] [Google Scholar]
- 2.Malmqvist UP, Aronshtam A, Lowey S. Cardiac myosin isoforms from different species have unique enzymatic and mechanical properties. Biochemistry. 2004;43(47):15058–65. doi: 10.1021/bi0495329. [DOI] [PubMed] [Google Scholar]
- 3.Pagani ED, Julian FJ. Rabbit papillary muscle myosin isozymes and the velocity of muscle shortening. Circ Res. 1984;54(5):586–94. doi: 10.1161/01.res.54.5.586. [DOI] [PubMed] [Google Scholar]
- 4.Morano I, Bletz C, Wojciechowski R, Ruegg JC. Modulation of crossbridge kinetics by myosin isoenzymes in skinned human heart fibers. Circ Res. 1991;68(2):614–8. doi: 10.1161/01.res.68.2.614. [DOI] [PubMed] [Google Scholar]
- 5.Fitzsimons DP, Patel JR, Moss RL. Role of myosin heavy chain composition in kinetics of force development and relaxation in rat myocardium. J Physiol. 1998;513 (Pt 1):171–83. doi: 10.1111/j.1469-7793.1998.171by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herron TJ, Korte FS, McDonald KS. Loaded shortening and power output in cardiac myocytes are dependent on myosin heavy chain isoform expression. Am J Physiol Heart Circ Physiol. 2001;281(3):H1217–22. doi: 10.1152/ajpheart.2001.281.3.H1217. [DOI] [PubMed] [Google Scholar]
- 7.Herron TJ, McDonald KS. Small amounts of alpha-myosin heavy chain isoform expression significantly increase power output of rat cardiac myocyte fragments. Circ Res. 2002;90(11):1150–2. doi: 10.1161/01.res.0000022879.57270.11. [DOI] [PubMed] [Google Scholar]
- 8.Krenz M, Sanbe A, Bouyer-Dalloz F, Gulick J, Klevitsky R, Hewett TE, et al. Analysis of myosin heavy chain functionality in the heart. The Journal of biological chemistry. 2003;278(19):17466–74. doi: 10.1074/jbc.M210804200. [DOI] [PubMed] [Google Scholar]
- 9.Rundell VL, Manaves V, Martin AF, de Tombe PP. Impact of beta-myosin heavy chain isoform expression on cross-bridge cycling kinetics. Am J Physiol Heart Circ Physiol. 2005;288(2):H896–903. doi: 10.1152/ajpheart.00407.2004. [DOI] [PubMed] [Google Scholar]
- 10.Korte FS, Herron TJ, Rovetto MJ, McDonald KS. Power output is linearly related to MyHC content in rat skinned myocytes and isolated working hearts. Am J Physiol Heart Circ Physiol. 2005;289(2):H801–12. doi: 10.1152/ajpheart.01227.2004. [DOI] [PubMed] [Google Scholar]
- 11.Korte FS, McDonald KS. Sarcomere length dependence of rat skinned cardiac myocyte mechanical properties: dependence on myosin heavy chain. J Physiol. 2007;581(Pt 2):725–39. doi: 10.1113/jphysiol.2007.128199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krenz M, Sadayappan S, Osinska HE, Henry JA, Beck S, Warshaw DM, et al. Distribution and structure-function relationship of myosin heavy chain isoforms in the adult mouse heart. The Journal of biological chemistry. 2007;282(33):24057–64. doi: 10.1074/jbc.M704574200. [DOI] [PubMed] [Google Scholar]
- 13.Stelzer JE, Brickson SL, Locher MR, Moss RL. Role of myosin heavy chain composition in the stretch activation response of rat myocardium. J Physiol. 2007;579(Pt 1):161–73. doi: 10.1113/jphysiol.2006.119719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Locher MR, Razumova MV, Stelzer JE, Norman HS, Moss RL. Effects of low-level α-myosin heavy chain expression on contractile kinetics in porcine myocardium. Am J Physiol Heart Circ Physiol. 2011;300(3):H869–78. doi: 10.1152/ajpheart.00452.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alpert NR, Brosseau C, Federico A, Krenz M, Robbins J, Warshaw DM. Molecular mechanics of mouse cardiac myosin isoforms. Am J Physiol Heart Circ Physiol. 2002;283(4):H1446–54. doi: 10.1152/ajpheart.00274.2002. [DOI] [PubMed] [Google Scholar]
- 16.Alpert NR, Mulieri LA. Heat, mechanics, and myosin ATPase in normal and hypertrophied heart muscle. Fed Proc. 1982;41(2):192–8. [PubMed] [Google Scholar]
- 17.Litten RZ, 3rd, Martin BJ, Low RB, Alpert NR. Altered myosin isozyme patterns from pressure-overloaded and thyrotoxic hypertrophied rabbit hearts. Circ Res. 1982;50(6):856–64. doi: 10.1161/01.res.50.6.856. [DOI] [PubMed] [Google Scholar]
- 18.Palmiter KA, Tyska MJ, Dupuis DE, Alpert NR, Warshaw DM. Kinetic differences at the single molecule level account for the functional diversity of rabbit cardiac myosin isoforms. J Physiol. 1999;519(Pt 3):669–78. doi: 10.1111/j.1469-7793.1999.0669n.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sata M, Sugiura S, Yamashita H, Momomura S, Serizawa T. Dynamic interaction between cardiac myosin isoforms modifies velocity of actomyosin sliding in vitro. Circ Res. 1993;73(4):696–704. doi: 10.1161/01.res.73.4.696. [DOI] [PubMed] [Google Scholar]
- 20.Sugiura S, Kobayakawa N, Fujita H, Momomura S, Omata M. Direct characterization of single molecular kinetics of cardiac myosin in vitro. Heart and vessels. 1997;(Suppl 12):97–9. [PubMed] [Google Scholar]
- 21.VanBuren P, Harris DE, Alpert NR, Warshaw DM. Cardiac V1 and V3 myosins differ in their hydrolytic and mechanical activities in vitro. Circ Res. 1995;77(2):439–44. doi: 10.1161/01.res.77.2.439. [DOI] [PubMed] [Google Scholar]
- 22.Metzger JM, Wahr PA, Michele DE, Albayya F, Westfall MV. Effects of myosin heavy chain isoform switching on Ca2+-activated tension development in single adult cardiac myocytes. Circ Res. 1999;84(11):1310–7. doi: 10.1161/01.res.84.11.1310. [DOI] [PubMed] [Google Scholar]
- 23.Nadal-Ginard B, Mahdavi V. Molecular basis of cardiac performance. Plasticity of the myocardium generated through protein isoform switches. J Clin Invest. 1989;84(6):1693–700. doi: 10.1172/JCI114351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tyska MJ, Warshaw DM. The myosin power stroke. Cell Motil Cytoskeleton. 2002;51(1):1–15. doi: 10.1002/cm.10014. [DOI] [PubMed] [Google Scholar]
- 25.McNally EM, Kraft R, Bravo-Zehnder M, Taylor DA, Leinwand LA. Full-length rat alpha and beta cardiac myosin heavy chain sequences. Comparisons suggest a molecular basis for functional differences. J Mol Biol. 1989;210(3):665–71. doi: 10.1016/0022-2836(89)90141-1. [DOI] [PubMed] [Google Scholar]
- 26.Pereira JS, Pavlov D, Nili M, Greaser M, Homsher E, Moss RL. Kinetic differences in cardiac myosins with identical loop 1 sequences. The Journal of biological chemistry. 2001;276(6):4409–15. doi: 10.1074/jbc.M006441200. [DOI] [PubMed] [Google Scholar]
- 27.Pellegrino MA, Canepari M, Rossi R, D’Antona G, Reggiani C, Bottinelli R. Orthologous myosin isoforms and scaling of shortening velocity with body size in mouse, rat, rabbit and human muscles. J Physiol. 2003;546(Pt 3):677–89. doi: 10.1113/jphysiol.2002.027375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andruchov O, Andruchova O, Wang Y, Galler S. Kinetic properties of myosin heavy chain isoforms in mouse skeletal muscle: comparison with rat, rabbit, and human and correlation with amino acid sequence. American journal of physiology Cell physiology. 2004;287(6):C1725–32. doi: 10.1152/ajpcell.00255.2004. [DOI] [PubMed] [Google Scholar]
- 29.Campbell KB, Chandra M, Kirkpatrick RD, Slinker BK, Hunter WC. Interpreting cardiac muscle force-length dynamics using a novel functional model. Am J Physiol Heart Circ Physiol. 2004;286(4):H1535–45. doi: 10.1152/ajpheart.01029.2003. [DOI] [PubMed] [Google Scholar]
- 30.Hunter PJ, McCulloch AD, ter Keurs HE. Modelling the mechanical properties of cardiac muscle. Prog Biophys Mol Biol. 1998;69(2–3):289–331. doi: 10.1016/s0079-6107(98)00013-3. [DOI] [PubMed] [Google Scholar]
- 31.Kawai M, Brandt PW. Sinusoidal analysis: a high resolution method for correlating biochemical reactions with physiological processes in activated skeletal muscles of rabbit, frog and crayfish. J Muscle Res Cell Motil. 1980;1(3):279–303. doi: 10.1007/BF00711932. [DOI] [PubMed] [Google Scholar]
- 32.Kawai M, Halvorson HR. Role of MgATP and MgADP in the cross-bridge kinetics in chemically skinned rabbit psoas fibers. Study of a fast exponential process (C) Biophys J. 1989;55(4):595–603. doi: 10.1016/S0006-3495(89)82857-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stuyvers BD, Miura M, ter Keurs HE. Dynamics of viscoelastic properties of rat cardiac sarcomeres during the diastolic interval: involvement of Ca2+ J Physiol. 1997;502 (Pt 3):661–77. doi: 10.1111/j.1469-7793.1997.661bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wannenburg T, Heijne GH, Geerdink JH, Van Den Dool HW, Janssen PM, De Tombe PP. Cross-bridge kinetics in rat myocardium: effect of sarcomere length and calcium activation. Am J Physiol Heart Circ Physiol. 2000;279(2):H779–90. doi: 10.1152/ajpheart.2000.279.2.H779. [DOI] [PubMed] [Google Scholar]
- 35.Campbell KB, Razumova MV, Kirkpatrick RD, Slinker BK. Nonlinear myofilament regulatory processes affect frequency-dependent muscle fiber stiffness. Biophys J. 2001;81(4):2278–96. doi: 10.1016/S0006-3495(01)75875-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Palmer BM, Suzuki T, Wang Y, Barnes WD, Miller MS, Maughan DW. Two-state model of acto-myosin attachment-detachment predicts C-process of sinusoidal analysis. Biophys J. 2007;93(3):760–9. doi: 10.1529/biophysj.106.101626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reiser PJ, Kline WO. Electrophoretic separation and quantitation of cardiac myosin heavy chain isoforms in eight mammalian species. Am J Physiol. 1998;274(3 Pt 2):H1048–53. doi: 10.1152/ajpheart.1998.274.3.H1048. [DOI] [PubMed] [Google Scholar]
- 38.Suzuki T, Palmer BM, James J, Wang Y, Chen Z, VanBuren P, et al. Effects of cardiac myosin isoform variation on myofilament function and crossbridge kinetics in transgenic rabbits. Circulation Heart failure. 2009;2(4):334–41. doi: 10.1161/CIRCHEARTFAILURE.108.802298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rossmanith GH, Hoh JF, Kirman A, Kwan LJ. Influence of V1 and V3 isomyosins on the mechanical behaviour of rat papillary muscle as studied by pseudo-random binary noise modulated length perturbations. J Muscle Res Cell Motil. 1986;7(4):307–19. doi: 10.1007/BF01753651. [DOI] [PubMed] [Google Scholar]
- 40.Rovner AS, Fagnant PM, Trybus KM. The two heads of smooth muscle myosin are enzymatically independent but mechanically interactive. The Journal of biological chemistry. 2003;278(29):26938–45. doi: 10.1074/jbc.M303122200. [DOI] [PubMed] [Google Scholar]
- 41.Marston SB, Taylor EW. Comparison of the myosin and actomyosin ATPase mechanisms of the four types of vertebrate muscles. J Mol Biol. 1980;139(4):573–600. doi: 10.1016/0022-2836(80)90050-9. [DOI] [PubMed] [Google Scholar]
- 42.Jakab G, Kiss E, Kranias EG, Edes I. Effect of thyroid status on basal phosphorylation of cardiac myofibrillar phosphoproteins in rats. Cardioscience. 1994;5(1):19–24. [PubMed] [Google Scholar]
- 43.Mahdavi V, Lompre AM, Chambers AP, Nadal-Ginard B. Cardiac myosin heavy chain isozymic transitions during development and under pathological conditions are regulated at the level of mRNA availability. Eur Heart J. 1984;5 (Suppl F):181–91. doi: 10.1093/eurheartj/5.suppl_f.181. [DOI] [PubMed] [Google Scholar]
- 44.Lompre AM, Nadal-Ginard B, Mahdavi V. Expression of the cardiac ventricular alpha- and beta-myosin heavy chain genes is developmentally and hormonally regulated. The Journal of biological chemistry. 1984;259(10):6437–46. [PubMed] [Google Scholar]
- 45.Narolska NA, van Loon RB, Boontje NM, Zaremba R, Penas SE, Russell J, et al. Myocardial contraction is 5-fold more economical in ventricular than in atrial human tissue. Cardiovasc Res. 2005;65(1):221–9. doi: 10.1016/j.cardiores.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 46.Alpert NR, Hasenfuss G, Mulieri LA, Blanchard EM, Leavitt BJ, Ittleman F. The reorganization of the human and rabbit heart in response to haemodynamic overload. Eur Heart J. 1992;13 (Suppl D):9–16. doi: 10.1093/eurheartj/13.suppl_d.9. [DOI] [PubMed] [Google Scholar]
- 47.Moss RL, Razumova M, Fitzsimons DP. Myosin crossbridge activation of cardiac thin filaments: implications for myocardial function in health and disease. Circ Res. 2004;94(10):1290–300. doi: 10.1161/01.RES.0000127125.61647.4F. [DOI] [PubMed] [Google Scholar]
- 48.Narolska NA, Eiras S, van Loon RB, Boontje NM, Zaremba R, Spiegelen Berg SR, et al. Myosin heavy chain composition and the economy of contraction in healthy and diseased human myocardium. J Muscle Res Cell Motil. 2005;26(1):39–48. doi: 10.1007/s10974-005-9005-x. [DOI] [PubMed] [Google Scholar]
- 49.Nakao K, Minobe W, Roden R, Bristow MR, Leinwand LA. Myosin heavy chain gene expression in human heart failure. J Clin Invest. 1997;100(9):2362–70. doi: 10.1172/JCI119776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miyata S, Minobe W, Bristow MR, Leinwand LA. Myosin heavy chain isoform expression in the failing and nonfailing human heart. Circ Res. 2000;86(4):386–90. doi: 10.1161/01.res.86.4.386. [DOI] [PubMed] [Google Scholar]
- 51.Turnbull L, Hoh JF, Ludowyke RI, Rossmanith GH. Troponin I phosphorylation enhances crossbridge kinetics during beta-adrenergic stimulation in rat cardiac tissue. J Physiol. 2002;542(Pt 3):911–20. doi: 10.1113/jphysiol.2002.022707. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.